

Abstract
Many tumors overexpress members of the inhibitor of apoptosis protein (IAP) family. IAPs contribute to tumor cell apoptosis resistance by the inhibition of caspases, and are degraded by the proteasome to allow further progression of apoptosis. Here we show that tumor cells can alter the specificity of cytosolic proteolysis in order to acquire apoptosis resistance, which promotes formation of rapidly growing tumors. Survival of tumor cells with low proteasomal activity can occur in the presence of high expression of Tri-peptidyl-peptidase II (TPP II), a large subtilisin-like peptidase that complements proteasomal activity. We find that this state leaves tumor cells unable of effectively degrading IAPs, and that cells in this state form rapidly growing tumors in vivo. We also find, in studies of apoptosis resistant cells derived from large in vivo tumors, that these have acquired an altered peptidase activity, with up-regulation of TPP II activity and decreased proteasomal activity. Importantly, we find that growth of subcutaneous tumors is limited by maintenance of the apoptosis resistant phenotype. The apoptosis resistant phenotype was reversed by increased expression of Smac/DIABLO, an antagonist of IAP molecules. Our data suggest a reversible mechanism in regulation of apoptosis resistance that drives tumor progression in vivo. These data are relevant in relation to the multitude of therapy-resistant clinical tumors that have increased levels of IAP molecules.
Keywords: apoptosis, proteasome, tri-peptidyl-peptidase II, IAP, caspase
Introduction
Ubiquitinated proteins are degraded by the 26S proteasome, a large multi-catalytic protease complex present in the cytosol and nucleus of mammalian cells (1–3). This complex contains a catalytic core (20S) and an accessory complex (19S) that recognizes ubiquitinated substrates and translocates these to the catalytic sites in the 20S particle (4). The proteasome is responsible for most of cytosolic protein degradation in eukaryotic cells, of both short-lived and long-lived proteins, and some of the degradation fragments are presented bound to MHC class I molecules (1). However, substantially less than full proteasomal activity is required for sustained viability of mammalian cells, thus indicating the presence of an over-capacity for proteasomal proteolysis (5–8). This could be used in situations of increased demand for protein degradation such as heat shock, or alternatively it may be necessary to assure a distinct timing in degradation of regulatory factors. However, it is not clear why this excess in proteolytic capacity exists and whether it is necessary for control of cellular pathways regulated by proteasomal protein degradation.
Proteasomal inhibition (>80%), and other types of cellular stress, causes release of cytochrome c from mitochondria into the cytosol and triggers assembly of the apoptosome (5, 9–11). Regulatory factors controlled through the ubiquitin–proteasome pathway control these events, including mitochondrial release of cytochrome c and subsequent activation of caspases (9–13). Inhibitor of apoptosis proteins (IAPs)* are endogenous caspase inhibitors, which must be degraded by the proteasome to allow further progression of apoptosis (12, 13). Increased in vivo expression of IAPs leads to suppression of normally occurring cell death in both mammals and insects (14, 15). Proteins of the IAP family contain baculoviral IAP repeat (BIR)-domains by which they can bind to and inhibit caspases and thereby rescue cells from apoptosis. Several members of this family, e.g., XIAP, c-IAP-1, and Survivin, are frequently up-regulated in tumor cells, where they cause resistance to cancer therapy due to inhibition of tumor cell apoptosis (16–18). Inhibitors of the proteasome can transiently inhibit apoptosis due to accumulation of IAPs, but cell cycle arrest and apoptosis eventually follows (11, 13). As cells may grow with reduced proteasomal activity, and even with much reduced activity when compensatory proteolysis is induced (6–8), it appears possible that this may be involved in the control of IAP expression. Cellular viability in the presence of inhibited proteasomal activity may be allowed by increased activity of Tri-peptidyl-peptidase II (TPP II, 19), a large subtilisin-like peptidase that is believed to degrade polypeptides downstream of proteasomal protein degradation (6–8). Increased activity of TPP II is sufficient to maintain proliferation of EL-4 lymphoma cells when the proteasome is inhibited, but it is not clear whether this state has any consequences for pathways controlled by ubiquitin-dependent proteolysis (8). This state is also observed in Epstein Barr virus–transformed B cells overexpressing c-myc, but it is unknown whether this has any impact on tumor progression (20).
In this paper we have studied apoptosis control in tumor cells with high TPP II activity, which can grow despite proteasomal inhibition. Several studies have implicated a high expression of 20S proteasomes in tumor cells, in line with a high demand for proteolysis in rapidly proliferating cells (21, 22). However, death by apoptosis balances cellular proliferation, and resistance to apoptosis may be at least as important as the rate of proliferation for growth of the tumor tissue (23). We find that EL-4 lymphoma cells that can grow in the presence of low proteasomal activity acquire apoptosis resistance due to a failure in degradation of IAPs. The rate of in vivo tumor growth of such cells was strongly increased. Rapid tumor growth, as well as a delayed degradation of IAPs, could be induced by transfection of TPP II. In addition, in cells derived from large in vivo tumors we observe a slower degradation of IAPs, as well as well as a reduced activity of the proteasome in combination with up-regulated TPP II activity. Our data suggest a novel mechanism for apoptosis resistance in tumors.
Materials and Methods
Cells and Transfections.
EL-4 is a Benzopyrene-induced mouse T cell lymphoma derived from C57Bl/6. EL-4 control and EL-4ad cells were maintained in vitro in RPMI 1640 supplemented with 5% FCS, and 50 μM of nitro-phenol-tri-leucine-vinyl-sulphone (NLVS; reference 24) for EL-4ad cells. EL-4ad cells were obtained by seeding control EL-4 cells at 105/ml in 12-well plates and continuously incubated in 10 μM NLVS. This treatment blocks all active β-subunits except for Z/MECL-1, and kills most cells within 24–48 h. However, a subpopulation adapts to grow progressively within 2–3 wk (6). The concentration of NLVS is then gradually increased to 50 μM. EL-4 cells adapted to growth in starvation medium were first incubated in medium containing RPMI 1640 supplemented with 2.5% FCS and 50% PBS; and the cells were then gradually adapted to resist culture in 75% PBS, similarly to what has been described previously (25). EL-4ad transfectants with pEF control vector or pEF/DIABLO were generated by incubation with 5 μg/ml Lipofectamine® (Life Technologies) and transfectants were selected by 8 μg/ml Puromycin. The pcDNA3 and pcDNA3-TPP II-transfectants of EL-4 are described previously (8).
Tumor Growth Experiments.
Tumor cells were washed in PBS and resuspended in a volume of 200 μl per inoculate. The cells were then inoculated into the right flank at 105 or 106 per syngeneic C57Bl/6 mouse and growth of the tumor was monitored by measurement 1–2 times per week. The mice were irradiated with 400R before tumor inoculation in order to inhibit antitumor immune responses. The tumor volume was calculated as the mean volume in mice with tumors growth, according to (a1 × a2 × a3)/2 (the numbers ai denote tumor diameter, width and depth). The data represent tumor volume in mice with growing tumors, and mice that rejected the tumor cells at inoculation were thus excluded. EL-4/tumor cells were derived from tumors of at least 1 cm3 and were cultured in RPMI supplemented with 5% FCS a few days in vitro before experiments to exclude confounding factors such as rate of growth, nutritional state and contaminating nontumor cells.
Apoptosis Induction and DNA Fragmentation Assays.
For induction of apoptosis we used serum starvation (RPMI/0.1% FCS), 10 nM TNF-α, 25 μM etoposide, or 1 μM Nocodazole. Cells were seeded at 106 cells/ml in 12-well plates and incubated for 18 h. DNA from EL-4 control and adapted cells were purified by standard Chloroform extraction and 2.5 μg of DNA was loaded on 1.8% agarose gel for detection of DNA from apoptotic cells. For analysis of caspase activation we used the following short reporter peptide substrates: DEVD-AFC (caspase 3), LETD-AFC (caspase 8), and LEHD-AFC (caspase 9; Enzyme Systems Products). All apoptosis assays were made several times and representative data were chosen for display. EL-4ad− are EL-4ad cells that were washed from inhibitor and culture for three weeks in vitro, in RPMI1640 supplemented with 5% FCS, in the absence of NLVS.
Antibodies and Western Blot Analysis.
We used the following reagents for detection with standard Western blot analysis techniques: rabbit anti-cytochrome c serum (Santa Cruz Biotechnology, Inc.); 4G10 monoclonal anti-SMAC/DIABLO (Alexis Corporation); anti-ubiquitin (DakoCytomation); affinity-purified rabbit anti-mouse XIAP (R&D Systems); CH9 anti-proteasomal subunit α3 (Affiniti Research Products Limited). Chicken anti-TPP II serum (Immunsystem). Protein concentration was determined with BCA Protein Assay Reagent (Pierce Chemical Co.), and 5 μg of protein was loaded per lane for separation by SDS/PAGE.
Heat Shock Incubation and Degradation of Ubiquitinated Proteins.
EL-4/tumor cells were obtained from killed C57Bl/6 mice with large (>1 cm3) tumors, by excising pieces and that were put into single cell suspension, and were cultured in vitro for 2–4 d. Larger tumor sizes were chosen for analysis, as this increases the requirements for nutrion and oxygen supply in growing tumor tissue in vivo. 106 EL-4/tumor, EL-4, EL-4ad cells, and C57Bl/6 Con A blasts were exposed to 42°C for 30 min and aliquots of cells were either lysed directly or after 30 min, 4 and 16 h of incubation at 37°C. The levels of ubiquitin-conjugates were evaluated by Western blot analysis of cell lysates with anti-ubiquitin anti-serum (DakoCytomation).
Protein Purification and Peptidase Assays.
100 × 106 cells were sedimented and lysed by vortexing in glass beads and homogenization buffer (50 mM Tris Base, pH 7.5, 250 mM Sucrose, 5 mM MgCl2, 1 mM DTT). Cellular lysates were submitted to differential centrifugation where a supernatant from a 1 h centrifugation at 100,000 g (cytosol) was submitted to 100,000 g centrifugation for 5 h, which sedimented high molecular weight cytosolic proteins/protein complexes, ∼5% of the cytosolic protein. The resulting pellet dissolved in 50 mM Tris Base, pH 7.5, 30% Glycerol, 5 mM MgCl2, and 1 mM DTT, and 1 μg of high molecular weight protein was used as enzyme in peptidase assays. To test the activity of the proteasome and TPP II we used the substrates succinyl-LLVY-AMC and AAF-AMC (Sigma-Aldrich), respectively, at 100 μM concentration in 100 μl of test buffer composed of 50 mM Tri Base pH 7.5, 5 mM MgCl2, and 1 mM DTT. Cleavage activity was measured by emission at 460 nm in a LS50B Luminescence Spectrometer (PerkinElmer). Peptidase activity of high molecular weight proteins was tested three times and representative data were chosen for display.
Results
Cells that Grow with Low Proteasomal Activity Are Resistant to Apoptosis.
We used EL-4ad, a variant cell line of EL-4, that grow in the presence of a covalent proteasome inhibitor (NLVS; references 6 and 24). We tested whether EL-4ad cells were able to properly control apoptosis induction, as this normally follows when cells are grown in the presence of proteasomal inhibitors (11). We observed that EL-4ad cells failed to undergo apoptosis when exposed to serum starvation for 36 h, and even continued low levels of proliferation (Fig. 1, b–d) . Furthermore, EL-4ad responded very poorly to TNF-α and etoposide in comparison to EL-4 control cells, as shown by inefficient activation of caspases 3, 8, and 9 as well as absence of DNA fragmentation (Fig. 2 , a-c). However, after culture of EL-4ad in the absence of proteasomal inhibitor the apoptosis resistance was reversed, and DNA fragmentation was again observed in response to TNF-α and etoposide (Fig. 2 d). This reversal was observed 2–3 wk after removal of the proteasomal inhibitor, and coincided with a regained dependence of proteasomal proteolysis in EL-4ad (unpublished data). These data suggest that EL-4 cells may acquire apoptosis resistance through an altered specificity of cytosolic proteolysis.
Figure 1.
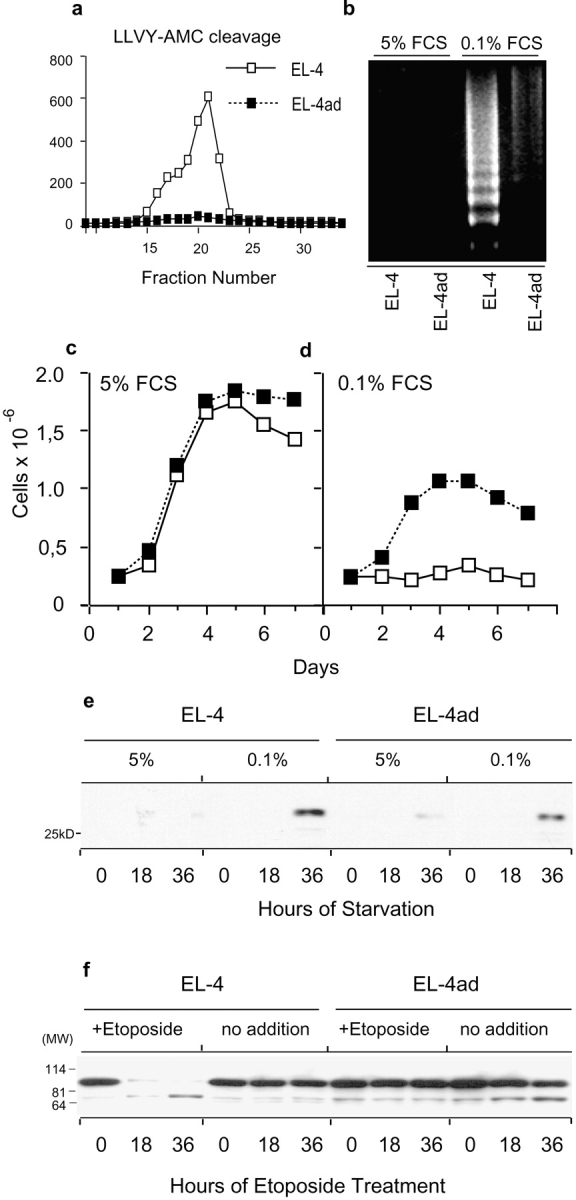
Resistance to serum starvation-induced apoptosis in cells adapted to reduced proteasomal activity. (a) Chymotryptic proteasomal activity in EL-4 and EL-4ad measured by cleavage of succinyl-LLVY-AMC in fractions of high molecular weight cytosolic protein eluted from a Superose 6 column. (b) DNA fragmentation measured during growth of EL-4 control and EL-4ad cells in normal cell culture medium (5% FCS) and in serum starvation medium (0.1% FCS). (c and d) Proliferation of EL-4 and EL-4ad cells in normal cell culture medium (c) versus serum starvation medium (d). (e) Western blot analysis for cytochrome c using 5 μg of cytosols from EL-4 and EL-4ad cells growing in normal (5% FCS) or serum starvation medium (0.1% FCS). (f) Western blot analysis for PARP in EL-4 and EL-4ad cells treated with etoposide.
Figure 2.

Reversible apoptosis resistance induced by adaptation to reduced proteasomal activity. (a) DNA fragmentation in EL-4 control and EL-4ad cells exposed to 10 nM TNF-α (left panel) or 25 μM Etoposide (right panel). (b and c) The activities of caspases 8, 9, and 3 in response to treatment with 10 nM TNF-α (b) or 25 μM Etoposide (c) were tested in parallel. The data represent the mean out of three independent experiments, where the background in EL-4 control cells was normalized to 100%. Standard deviation is indicated. (d) Loss of apoptosis resistance after culture in the absence of proteasomal inhibitor, as measured by DNA fragmentation in response to TNF-α or Etoposide. EL-4 control (lanes labeled 1), EL-4ad (2), and EL-4ad−, i.e., EL-4ad cultured without NLVS (3) were compared in indicated lanes.
A multitude of ubiquitin-dependent regulatory factors control apoptosis, either upstream or downstream of mitochondrial cytochrome c-release (9, 10, 12, 13). To test whether cytochrome c was released from mitochondria during cellular stress in EL-4ad cells we exposed these to serum starvation for up to 36 h. We detected cytosolic cytochrome c after 36 h of serum starvation in both EL-4 control and EL-4ad cells, by Western blot analysis of purified cytosols (Fig. 1 e). The ability of EL-4ad to maintain some proliferation despite initiation of the apoptotic program bears resemblance to observations made in mice deficient for caspase 9 (26). Our results suggest the presence of factors that inhibit apoptosis downstream of mitochondrial cytochrome c-release in EL-4ad cells. As cleavage of PARP was inhibited in EL-4ad, these results also suggest that these factors work upstream to nuclear DNA fragmentation (Fig. 1 f).
Failure to Degrade IAP Molecules Causes Apoptosis Resistance in Growing EL-4ad Cells.
Many reports describe that IAPs must be degraded by the proteasome subsequently to mitochondrial cytochrome c release to allow activation of caspase 9, and further progression into apoptosis (12–18). We tested if EL-4ad could degrade XIAP, a 56 kD endogenous antagonist of caspase 9, in response to etoposide treatment. We found degradation of XIAP in EL-4 cells, whereas this was very slow in EL-4ad cells (Fig. 3 a). In EL-4ad cells, a substantial accumulation of XIAP was observed already before apoptosis, in comparison to EL-4 control cells, which may further inhibit caspase activation (Fig. 2, b and c). Culture of EL-4ad cells in the absence of NLVS makes EL-4ad dependent of proteasomal proteolysis. This accelerated degradation of XIAP (Fig. 3 a). We further found that c-IAP-1 was similarly induced and degradation was slow after etoposide treatment of EL-4ad cells (unpublished data). Thus, we find that the altered specificity of cytosolic proteolysis causes slow degradation of IAP molecules in EL-4ad cells.
Figure 3.

Inhibited proteasomal degradation of IAP molecules contributes to apoptosis resistance in EL-4ad cells. (a) XIAP expression in EL-4, EL-4ad, and EL-4ad− cells after treatment with 25 μM Etoposide. EL-4ad− denotes EL-4ad cells that were cultured in the absence of NLVS. XIAP molecules were detected by Western blot analysis of 5 μg of cellular lysates from the indicated cell lines. As loading controls, β-actin was probed. (b) Smac/DIABLO-expression in EL-4ad cells transfected with either pEF control vector or pEF-DIABLO. (c and d) Reversal of EL-4ad apoptosis resistance by Smac/DIABLO as measured by DNA fragmentation (c) and Caspase activation (d).
To test if the apoptosis resistance of EL-4ad cells depended on IAP molecules we used Smac/DIABLO, an IAP-inactivator (27–29). Smac/DIABLO is released through the outer mitochondrial membrane together with cytochrome c, and binds to IAP molecules to prevent their inhibitory effect on caspase activation (29, 30). Further, Smac/DIABLO is also reported as necessary for apoptosis induction in prostate cancer cells (31). EL-4ad cells transfected with pEF-Smac/DIABLO obtained increased Smac/DIABLO-expression, as detected by the specific mAb 4G10, using empty pEF vector-transfected cells as control (Fig. 3 b). During normal culture conditions (5% FCS) this increased Smac/DIABLO expression had minor effects on EL-4ad, as the rate of proliferation could not be distinguished from pEF control vector-transfected EL-4ad cells. However, a substantial effect was observed during serum starvation, showing a reduced proliferation of Smac/DIABLO-transfected EL-4ad cells in comparison to pEF control vector-transfected cells (Fig. 4 a). During triggering of apoptosis with Etoposide we found that caspase 9 and 3-activation as well as DNA fragmentation was restored in EL-4ad cells transfected with pEF-Smac/DIABLO, but not pEF empty vector (Fig. 3, c and d). A set of three additional EL-4ad lines transfected with either pEF empty vector or pEF-Smac/DIABLO were analyzed for DNA fragmentation and caspase 9 activity, with similar results (Fig. 4). By using Smac/DIABLO, our data suggest that the inadequate degradation of IAP molecules inhibit the transduction of apoptotic signals in EL-4ad cells.
Figure 4.

Reversal of apoptosis resistance in an expanded set of Smac/DIABLO-transfectant EL-4ad lines. (a) Proliferation of EL-4ad-pEF and EL-4ad-pEF/DIABLO cells was measured during normal (5% FCS) or conditions of serum starvation (0.1% FCS). The data represent the mean of proliferation of four independently transfected lines. (b and c). Reversal of EL-4ad apoptosis resistance in three independently transfected Smac/DIABLO-transfected lines, as measured by DNA fragmentation (b) and Caspase 3 activation following exposure to Etoposide (c).
EL-4 Cells Adapted to Low Proteasomal Activity Form Rapidly Growing In Vivo Tumors.
Apoptosis resistance mediated by IAP molecules is frequently observed in tumor cells, and is regarded as a major problem during tumor therapy (16–18, 32–35). We did not observe an increased growth rate for EL-4ad cells, whereby IAP-mediated apoptosis resistance does not appear to be a significant growth advantage during in vitro cell culture (Fig. 1, c and d). Subcutaneous tumor growth in vivo may expose growing cells to more stress (e.g., competition for nutrients and oxygen), and we therefore tested in vivo growth of EL-4 control and EL-4ad cells by inoculation into irradiated syngeneic C57Bl/6 mice (Fig. 5 , a and b). We found that tumors of EL-4ad cells were rapidly growing and reached a size of ∼1 cm3 in 30 d (Fig. 5 a). In contrast, growth of EL-4 tumors was significantly slower and did not reach more than 0.2–0.3 cm3 during this time period. Using a lower cell dose than initially used, the difference in growth rate between EL-4 and EL-4ad tumors was even more evident (Fig. 5 b). Although the differences between EL-4 and EL-4ad tumors were clear, at later stages the growth rate of EL-4 control tumors reached the same as observed for tumors of EL-4ad (Fig. 5 a). Therefore we killed animals and excised cells from large (>1 cm3, as indicated in Fig. 5 a) in vivo tumors for later analysis. Importantly, we found that rapid growth of subcutaneous EL-4 tumors was linked to apoptosis resistance, as Smac/DIABLO-transfection of EL-4ad cells reduced tumor growth rate, in comparison to tumors of pEF-control vector transfected cells (Fig. 5 c). Increased growth of EL-4ad tumors, compared with EL-4 control, was present also in immunodeficient mice (PKOB/RAG−/−; Fig. 5 d). Thus, EL-4 lymphoma cells with an altered specificity of cytosolic proteolysis have a growth advantage in vivo over control EL-4 cells.
Figure 5.

Rapid tumor growth by EL-4 cells adapted to low proteasomal activity. (a-d) EL-4 and EL-4ad cells were grafted at 106 or 105 cells, as indicated in figure, in syngeneic C57Bl/6 mice and tumor size was monitored. (a and b) Tumor growth of EL-4 and EL-4ad cells in syngeneic C57Bl/6 mice. EL-4/tumor in panel a denotes cells removed from killed mice to be analyzed further in Figs. 8 and 9, (c) Tumor growth of EL-4ad cells transfected with either pEF control vector or with pEF-Smac/DIABLO. (d) Tumor growth of EL-4 and EL-4ad cells in immunodeficient PKOB/RAG1−/− mice, deficient for T and B cells as well as NK cell cytotoxic activity. The data represent the mean of at least eight mice per group in a-c, whereas four mice per group were used in d.
Increased TPP II Activity Reduces IAP Degradation and Induces Rapid Tumor Growth In Vivo.
We next studied whether proteolytic pathways accessory to the proteasome, that are up-regulated in EL-4ad cells, could alter the degradation of IAP molecules. For this purpose we used EL-4 wild type cells transfected with either control vector, pcDNA3, or with pcDNA3-TPP II, described previously (8). TPP II transfection conferred a reduced reliance of proteasomal proteolysis, since EL-4.TPP II proliferated in the presence of NLVS, whereas as EL-4pcDNA3 cells did not (Fig. 6 a). In addition we observed that EL-4.TPP II proliferated in the presence of 50 μM AdaAhx3L3VS, a novel amino-terminally extended vinyl sulphone that inhibits all β-subunits (B1, B2, and B5) with comparable efficiency (36, 37; Fig. 6 b). These data confirm previous findings that up-regulation of TPP II allows EL-4 cells to use minimal proteasomal β-subunit activity and still manage cellular proliferation (6–8, 37).
Figure 6.

TPP II transfection confers resistance to proteasomal inhibition and reduces IAP degradation. (a and b) EL-4 cells transfected with pcDNA3 control vector or pcDNA3-TPP II, were treated either with NLVS (a) or with AdaAhx3Leu3VS (b). Whereas NLVS (reference 24) inhibits predominantly the chymotryptic activity of the proteasome, AdaAhx3Leu3VS (reference 37) inhibits all proteasomal β-subunits with comparable efficiency. Cellular proliferation was measured daily by counting live cells by trypan blue exclusion, and the data represent the mean from two independent experiments. (c) EL-4 cells transfected with either pcDNA3 or pcDNA3-TPP II were treated with Etoposide and DNA fragmentation was followed for up to 36 h. (d) Tumor growth of 106 EL-4pcDNA3 and EL-4.TPP II cells in vivo in syngeneic C57Bl/6 mice. Growth was monitored weekly, and the data represent the mean out of two independent experiments with a total of six mice per group.
We next tested if TPP II affected the response to an apoptotic stimulus by exposing EL-4pcDNA3 and EL-4.TPP II cells to etoposide. We found by Western blot analysis of cellular lysates that EL-4.TPP II failed to fully degrade both XIAP and c-IAP-1 compared with EL-4pcDNA3 cells during etoposide treatment (Fig. 7 , a and b). Further, whereas DNA fragmentation was induced in EL-4pcDNA3 cells this effect was not present in EL-4.TPP II 36 h after onset of apoptosis. Thus, adequate apoptosis control was not maintained in EL-4.TPP II. Further, treatment of EL-4.TPP II with 50 μM NLVS, a treatment that allows survival of most EL-4.TPP II cells (Fig. 6 a), led to a substantially increased level of XIAP and c-IAP-1 (Fig. 7 a). The same treatment of EL-4pcDNA3 control cells, lacking up-regulation of accessory proteolysis, caused a transient increase of XIAP and c-IAP-1, but failed to protect from apoptosis over an extended period of time (Figs. 7 b and 6 c).
Figure 7.

Increased expression of TPP II allows accumulation of IAP molecules. (a) EL-4.pcDNA3 and (b) EL-4.TPP II were either treated with 50 μM NLVS or not. Then, these two groups were exposed to Etoposide and degradation of XIAP and c-IAP-1 was followed by Western blot analysis of 5 μM of cellular lysate. β-actin was Western blotted as loading controls.
To explore whether increased TPP II activity regulated the growth of EL-4 tumors in vivo we tested growth of EL-4.pcDNA3 and EL-4.TPP II tumors in syngeneic C57Bl/6 mice after subcutaneous inoculation. Although no differences in proliferation rate were found in vitro, EL-4 cells with increased TPP II-expression (EL-4.TPP II) had a strongly increased rate of in vivo growth compared with tumors transfected with pcDNA3 control vector (Fig. 6 d). This supports the notion that apoptosis resistance linked to TPP II was responsible for the observed effects on growth of EL-4 tumors in vivo.
These results are in line with previous studies showing that proteasomal inhibition can confer a transient apoptosis resistance by a failure to degrade IAPs, although subsequent apoptosis is observed due to requirement for efficient proteasomal β-subunit activity for cellular viability (11, 13). In addition, these results show that increased TPP II expression interferes with efficient XIAP and c-IAP-1 degradation in EL-4 cells, a state not compatible with efficient transduction of apoptotic signals (12, 13). Inhibition of the proteasome with NLVS in EL-4.TPP II leads to a further accumulation of IAPs without subsequent apoptosis.
Selection of Altered Peptidase Activity and Delayed IAP Degradation during In Vivo Tumor Growth.
Previous results have shown that tumor cells, as well as immature blasts, express high levels of 20S proteasomes, in line with the high demand for proteolysis in rapidly growing cells (21, 22). This is also in line with rapid cell cycle progression in tumor cells by efficient degradation of regulatory factors at cell cycle checkpoints. This may be one reason for the therapeutic effect of proteasomal inhibitors to some forms of cancer (38, 39). However, it could be possible that growth conditions that are more in favor for apoptosis resistant cells could show selection of a phenotype that more resembles the one observed in EL-4ad cells. We have observed that an altered specificity of cytosolic proteolysis causes apoptosis resistance in EL-4 cells (Figs. 1 and 6), and also that rapid in vivo growth of EL-4 tumors favored growth of such cells (Fig. 5). Therefore we examined the activity of the proteasome and TPP II in EL-4 control cells before and after growth in subcutaneous tumors in vivo.
Partial purifications of high molecular weight proteins from EL-4 control cells, EL-4ad, and EL-4 cells derived from large (>1 cm3) in vivo tumors (denoted EL-4/tumor) were prepared, and these were tested for cleavage of peptide substrates preferred by either TPP II (AAF-AMC) or the proteasome (succ-LLVY-AMC). We found that high molecular weight peptidases in EL-4/tumor cells had increased cleavage of AAF-AMC and reduced cleavage of succ-LLVY-AMC, compared with EL-4 control cells (Fig. 8 a). This activity profile corresponded to increased TPP II activity and reduced proteasomal activity, as determined by specific inhibitors of either the proteasome or TPP II (24, 40). Interestingly, we found the same pattern of peptidase activities in cells derived from subcutaneous tumors of ALC lymphoma and B16 melanoma (Fig. 8, b and c). Thus, we observed an altered activity of the proteasome and TPP II in cells derived from large in vivo tumors, although the alterations of peptidase activities were not as pronounced as in EL-4ad cells (Fig. 8, a-c). In line with this, EL-4 cells derived from these in vivo tumors were still dependent on proteasomal activity since they did not grow in the presence of NLVS (Fig. 8 e). The levels of proteasomal β-subunit protein in the tumor-derived cells (EL-4, ALC, and B16) were not significantly different, in comparison to in vitro grown cells, suggesting a post-translational regulation of 20S proteasomal activity (Fig. 8 f; reference 41).
Figure 8.

Altered activity of high molecular weight peptidases during tumor growth. (a–c) Measurements of high molecular weight peptidase activity in cell lines derived from in vitro cultures and in vivo tumors derived from (a) EL-4, (b) ALC lymphoma, and (c) B16 melanoma. Inhibitors specific for either the proteasome (NLVS; reference 24) or TPP II (Butabindide; reference 40), were used to identify the activities cleaving succinyl-LLVY-AMC and AAF-AMC. (d) Activity of high molecular weight peptidases measured in EL-4.pcDNA3 and EL-4.TPP II, experiments performed as in a–c. (e) In vitro growth rate of EL-4, EL-4ad versus EL-4/tumor cells in the presence or absence of 5 μM NLVS, as measured by counting live cells by trypan blue exclusion. (f) Western blotting of high molecular weight proteins for HC9, a proteasomal α-subunit (α3). 2, 5, or 10 μg of protein was loaded in each lane, as indicated in figure.
To test the efficiency of IAP degradation following initiation of apoptosis in EL-4/tumor cells, we treated these with etoposide and compared the removal of XIAP and c-IAP-1 to that present in EL-4 control and EL-4ad cells. In line with the observed peptidase activities, we observed an inefficient degradation of both XIAP and c-IAP-1 in EL-4/tumor cells whereas these IAPs were effectively degraded in EL-4 control cells. We further observed a weak induction of DNA fragmentation in EL-4/tumor cells in comparison to EL-4 control cells, showing an ineffective transduction of the apoptotic signal (Fig. 9 a). These data support the notion that an altered specificity of cytosolic proteolysis is selected in apoptosis resistant tumors in vivo.
Figure 9.

Selection of apoptosis resistance and delayed degradation of XIAP and c-IAP-1 during tumor growth (a) EL-4 control, EL-4ad, and EL-4/tumor cells were treated with 1 μM Nocodazole and induction of apoptosis was monitored by the detection of DNA fragmentation. DNA was purified from the cells and separated by a 1.8% agarose gel. (b) Western blotting of XIAP and c-IAP-1 in cells treated with etoposide for the indicated length of time. 5 μg of protein was loaded in each lane. Western blotting of β-actin was used as loading controls. (c), EL-4 control, EL-4ad, EL-4/tumor (derived from in vivo tumors), or C57Bl/6 ConA blasts were incubated for 30 min at 42°C and degradation of ubiquitin-conjugates was followed for up to 16 h with Western blot with anti-ubiquitin.
We next tested the ability of EL-4ad and EL-4/tumor cells to remove ubiquitin conjugates, since ubiquitin conjugation of regulatory factors controls transduction of apoptotic signals. We exposed these cells to heat shock (42°C, 30 min) and followed removal of ubiquitin conjugates by Western blot analysis of cellular lysates, in comparison to EL-4 control cells treated similarly. We found that both EL-4/tumor cells and EL-4ad cells had great difficulties in clearing heat shock-induced ubiquitin conjugates, in comparison to EL-4 control cells. Thus, although compensatory proteolysis is induced in EL-4ad and EL-4/tumor cells, this appears to be less efficient in removing ubiquitin-conjugates than the proteasome. In contrast, C57Bl/6 Con A blasts readily cleared the ubiquitin conjugates within 16 h, showing that this feature was specific for malignant but not for normal lymfoblasts (Fig. 8 d). We note that hyperthermia is a method that is sometimes used in clinical treatment of tumors (42).
Cellular Growth with Insufficient Nutrition Increases TPP II Activity and Resistance to Apoptosis.
It has been proposed that poor nutritional conditions in the tumor microenvironment contributes to selection of apoptosis resistant cells (43). As increased expression of TPP II conferred a partial protection from apoptosis of EL-4 cells, and induced rapid tumor growth in vivo (Fig. 6), we set out to test if poor nutritional conditions may affect the activity of TPP II in tumor cells. For this purpose we adapted EL-4 cells for long-term survival in starvation medium with reduced content of amino acids and growth factors (25). EL-4 control cells and EL-4 cells growing in starvation medium were lysed and high molecular weight proteins were prepared for Western blot analysis and enzymatic activity assays. By analysis with anti-TPP II serum we found high levels of TPP II protein among cytosolic high molecular weight proteins in EL-4 cells growing in starvation medium, and this induction was observed already after short–term incubation (20 h, Fig. 10 a). This also correlated with a strong increase in enzymatic activity cleaving AAF-AMC (Fig. 10 c). Further, EL-4 cells adapted to proliferation in starvation medium were resistant to apoptosis and expressed high levels of c-IAP-1; and this was not properly degraded upon treatment with etoposide (Fig. 10 b, unpublished data). These data fit well with the notion that apoptosis resistance mediated by IAP molecules allows a limited proliferation during serum starvation (Fig. 4 a).
Figure 10.

Induction of TPP II activity and rapid tumor growth by adaptation to cellular starvation. (a) EL-4 cells incubated in cell culture medium diluted with PBS were lysed, and high molecular weight cytosolic proteins were analysed by Western blot for TPP II expression. (b) EL-4 cells and EL-4 cells growing in starvation medium were exposed to etoposide and degradation of c-IAP-1 was followed by Western blotting analysis. (c) The high molecular weight cytosolic fractions in (a) were analyzed for cleavage of AAF-AMC. The inhibitors NLVS and AAF-CMK were included as controls. (d-e) EL-4 cells and EL-4 cells adapted to growth in starvation medium were grafted to irradiated C57Bl/6 mice at 106 (d) or 104 (e) cells per mouse.
Finally, in order to further test if adaptation to growth in starvation medium affected tumor growth of these EL-4 cells in vivo, we grafted 106 or 104 EL-4 control cells versus EL-4 cells adapted to growth in starvation medium to C57Bl/6 mice. We found that EL-4 cells adapted to starvation medium had an increased rate of tumor growth, especially in inoculates with lower cell numbers (104), further substantiating that this adaptation is used to increase the growth of EL-4 tumors in vivo (Fig. 10 d). Thus, TPP II has a substantial influence on EL-4 tumor growth in vivo, and is also regulated in response to the nutritional state of the microenvironment.
Discussion
These data suggest that an alteration in the activity of proteolytic pathways that are responsible for cellular protein turn-over contributes to apoptosis resistance in tumor cells. EL-4 cells adapted to low proteasomal activity, denoted EL-4ad, were resistant to apoptosis, at least in part due to a failure in efficient degradation of IAP molecules, and EL-4 cells with this phenotype grew rapidly as tumors in vivo. Our data also show that up-regulation of TPP II, a peptidase that allows EL-4 cells to grow with low proteasomal activity, leads to rapid tumor growth in vivo. The observed EL-4 apoptosis resistance phenotype was reversible, and was not present when not selected for, such as during optimal in vitro culture conditions. However, a partial induction of this phenotype (an altered specificity of cytosolic proteolysis and a slow degradation of IAP molecules) was observed in apoptosis resistant EL-4 cells derived from large subcutaneous in vivo tumors.
As growth of EL-4 tumors in vivo was limited by apoptosis susceptibility, our data are in line with the previously reported high expression of 20S proteasomes in rapidly proliferating cells (21, 22, 44). Apoptosis resistance may become even more important than rapid proliferation during growth under suboptimal conditions, such as low nutrition and limited oxygen supply (23, 43). It was recently proposed that the tumor microenvironment may drive the selection of apoptosis resistant cells, especially in the presence of hypoxia (43). We find that adaptation to growth in starving culture conditions in vitro causes a phenotype in EL-4 cells including TPP II up-regulation, poor degradation of c-IAP-1, and rapid tumor growth in vivo (Fig. 10). This may suggest a link between the microenvironment and signals that alter the specificity of intracellular proteolysis. Although mouse tumors grafted experimentally can proliferate to kill an animal within weeks, clinical tumors develop over the course of years, and the problems to apply certain promising tumor therapies may be related to this difference in growth profile (45, 46). Thus, the study of tumor survival mechanisms, such as the one presented in this paper, may be crucial to understand tumor progression, and especially when studied over long periods of time. Mammalian cells have a considerable over-capacity for proteasomal proteolysis when compared with what is necessary for proliferation in vitro. Up to 80% of the rate-limiting chymotryptic activity of the proteasome can be inhibited in wild-type HeLa cells without any accumulation of ubiquitinated proteins (5). This excess capacity may be present for rapid removal of mis-folded proteins during cellular stress, or potentially for rapid degradation of regulatory factors. Our findings indicate that modulation of this over-capacity alters the transduction of signals that depend on ubiquitin-dependent protelysis. More specifically we found that a reduction of this capacity is a signal that leads to apoptosis resistance in EL-4 tumor cells.
A large number of tumors overexpress IAP-family members and these are believed to be involved in failure of tumor therapy by causing resistance to cellular stress (16–18, 32–35). Overexpression of Survivin is observed in most tumor cells and inhibits apoptosis during the G2/M phase by inhibiting caspases, and is also involved in chromosomal segregation and exit from mitosis (16, 47). We have observed increased chromosome numbers in several independent lines of EL-4ad cells, in comparison to EL-4 control cells (unpublished data). Although the apoptosis resistance and adaptation to low proteasomal activity in EL-4ad cells is reversible it may thus predispose for the accumulation of genetic, and therefore irreversible, damage. This may also be explained by the involvement of N-end rule degradation of chromosomal cohesins, as the absence of this pathway causes a loss of chromosomes in yeast mutants (48). Although EL-4ad cells maintain the turn-over of proteins, the presence of low proteasomal activity makes full control of cellular physiology more difficult to achieve. The precise control of ubiquitin-dependent pathways observed in normal cells may not be preferred by tumor cells, which could instead benefit from an imprecise control during selection of malignant characteristics.
The most well studied system with respect IAP molecules may be Drosophila, which express DIAP-1, DIAP-2, and a homologue of surririn (49). Results from this system show that the genes controlling IAP stability (Grim, Hid, and Reaper; reference 49) have a profound impact on apoptosis susceptibility, perhaps more profound compared with ectopic expression of the IAPs themselves (15, 50). Thus, although a high steady-state level of IAPs before apoptosis may cause a delay in transduction of the apoptotic signal, this may not prevent eventual removal of these molecules. Similarly, in several studies where XIAP have been transfected into mammalian tumor cells this has created only a partial protection from apoptosis, which probably reflects that XIAP was still rapidly degraded upon apoptosis triggering (13, 51). An increase in IAP stability may therefore be essential for apoptosis resistance also in naturally occurring tumor cells. Thus, although comparisons between insect and mammalian IAPs should be made with care, the IAP biology in Drosophila may give important information of relevance for apoptosis resistance in mammalian cells. In reverse, a similar shift in specificity of cytosolic proteolysis, as reported here in mouse tumor cells, is possible also in Drosophila since TPP II is reported and has been cloned from this species (52).
Proteasomal inhibition has previously been linked to disease pathogenesis, either as a pharmacological side-effect or as a the consequence of a failure of protein degradation. A partial proteasomal inhibition is reported as a possible side effect causing hyper-lipidemia in patients treated with HIV protease inhibitors (53, 54). Further, in neuro-degenerative disease, proteins containing poly-Glutamine repeats assemble into aggregates that also contain ubiquitin and 20S proteasomes (55, 56). These aggregates are believed to cause inhibition of the ubiquitin-proteasome pathway with apoptosis as a consequence (56). In addition, Burkitt lymphoma cells have reduced proteasomal activity and increased TPP II activity, a phenotype controlled by c-myc overexpression in lymphoblastoid cell lines (20). Our results suggest that this phenotype may be linked to tumor malignancy, which is interesting in view of the frequent up-regulation of c-myc in naturally occurring tumors (57). We show that an altered specificity of cytosolic proteolysis disturbs the transduction of signals transmitted by ubiquitin-dependent proteolysis. Our data point to the involvement of IAP molecules in suppression of apoptosis in tumors that reduce proteasomal activity. The participation of other regulatory factors in this apoptosis resistance is however not excluded. Future studies may address whether an altered specificity of cytosolic proteolysis contributes to the acquisition of malignant characteristics in other tumor cells. More specifically, the status of the ubiquitin-proteasome pathway of tumors with up-regulation of IAP family molecules merits further study.
Acknowledgments
We thank Georg Klein, Maria Masucci, Hans-Gustaf Ljunggren, and Mayte Bejarano for discussions and suggestions on the manuscript; Hidde Ploegh and Benedikt Kessler generously provided proteasomal inhibitors. David Vaux and Carina Magnusson provided the pEF-DIABLO construct.
This work was funded by grants from the Swedish Foundation for Strategic Research, Swedish Research Council, Cancerfonden and Magnus Bergwalls Stiftelse (Stockholm, Sweden).
Footnotes
Abbreviations used in this paper: IAP, inhibitor of apoptosis protein; NLVS, nitro-phenol-tri-leucine-vinyl-sulphone; TPP II, tri-peptidyl-peptidase II.
References
- 1.Rock, K.L., and A.L. Goldberg. 1999. Degradation of cell proteins and the generation of MHC class I-presented peptides. Annu. Rev. Immunol. 17:739–779. [DOI] [PubMed] [Google Scholar]
- 2.Schwartz, A.L., and A. Ciechanover. 1999. The ubiquitin-proteasome pathway and pathogenesis of human diseases. Annu. Rev. Med. 50:57–74. [DOI] [PubMed] [Google Scholar]
- 3.Voges, D., P. Zwickl, and W. Baumeister. 1999. The 26S proteasome: a molecular machine designed for controlled proteolysis. Annu. Rev. Biochem. 68:1015–1068. [DOI] [PubMed] [Google Scholar]
- 4.Horwich, A.L., E.U. Weber-Ban, and D. Finley. 1999. Chaperone rings in protein folding and degradation. Proc. Natl. Acad. Sci. USA. 96:11033–11040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dantuma, N.P., K. Lindsten, R. Glas, M. Jellne, and M.G. Masucci. 2000. Short-lived green fluorescent proteins for quantifying ubiquitin/proteasome-dependent proteolysis in living cells. Nat. Biotechnol. 18:538–543. [DOI] [PubMed] [Google Scholar]
- 6.Glas, R., M. Bogyo, J.S. McMaster, M. Gaczynska, and H.L. Ploegh. 1998. A proteolytic system that compensates for loss of proteasome function. Nature. 392:618–622. [DOI] [PubMed] [Google Scholar]
- 7.Geier, E., G. Pfeifer, M. Wilm, M. Lucchiari-Hartz, W. Baumeister, K. Eichmann, and G. Niedermann. 1999. A giant protease with potential to substitute for some functions of the proteasome. Science. 283:978–981. [DOI] [PubMed] [Google Scholar]
- 8.Wang, E.W., B.M. Kessler, A. Borodovsky, B.F. Cravatt, M. Bogyo, H.L. Ploegh, and R. Glas. 2000. Integration of the ubiquitin-proteasome pathway with a cytosolic oligopeptidase activity. Proc. Natl. Acad. Sci. USA. 97:9990–9995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kroemer, G., and J.C. Reed. 2000. Mitochondrial control of cell death. Nat. Med. 6:513–519. [DOI] [PubMed] [Google Scholar]
- 10.Hengartner, M.O. 2000. The biochemistry of apoptosis. Nature. 407:770–776. [DOI] [PubMed] [Google Scholar]
- 11.Drexler, H.C. 1997. Activation of the cell death program by inhibition of proteasome function. Proc. Natl. Acad. Sci. USA. 94:855–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goyal, L. 2001. Cell death inhibition: keeping caspases in check. Cell. 104:805–808. [DOI] [PubMed] [Google Scholar]
- 13.Yang, Y., S. Fang, J.P. Jensen, A.M. Weissman, and J.D. Ashwell. 2000. Ubiquitin protein ligase activity of IAPs and their degradation in proteasomes in response to apoptotic stimuli. Science. 288:874–877. [DOI] [PubMed] [Google Scholar]
- 14.Conte, D., P. Liston, J.W. Wong, K.E. Wright, and R.G. Korneluk. 2001. Thymocyte-targeted overexpression of xiap transgene disrupts T lymphoid apoptosis and maturation. Proc. Natl. Acad. Sci. USA. 98:5049–5054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hay, B.A., D.A. Wassarman, and G.M. Rubin. 1995. Drosophila homologs of baculovirus inhibitor of apoptosis proteins function to block cell death. Cell. 83:1253–1262. [DOI] [PubMed] [Google Scholar]
- 16.Altieri, D.C. 2001. The molecular basis and potential role of survivin in cancer diagnosis and therapy. Trends. Mol. Med. 7:542–547. [DOI] [PubMed] [Google Scholar]
- 17.Holcik, M., H. Gibson, and R.G. Korneluk. 2000. XIAP: apoptotic brake and promising therapeutic target. Apoptosis. 6:253–261. [DOI] [PubMed] [Google Scholar]
- 18.Imoto, I., Z.Q. Yang, A. Pimkhaokham, H. Tsuda, Y. Shimada, M. Imamura, M. Ohki, and J. Inazawa. 2001. Identification of cIAP1 as a candidate target gene within an amplicon at 11q22 in esophageal squamous cell carcinomas. Cancer Res. 61:6629–6634. [PubMed] [Google Scholar]
- 19.Tomkinson, B. 1999. Tripeptidyl peptidases: enzymes that count. Trends Biochem. Sci. 24:355–359. [DOI] [PubMed] [Google Scholar]
- 20.Gavioli, R., T. Frisan, S. Vertuani, G.W. Bornkamm, and M.G. Masucci. 2001. c-myc overexpression activates alternative pathways for intracellular proteolysis in lymphoma cells. Nat. Cell Biol. 3:283–288. [DOI] [PubMed] [Google Scholar]
- 21.Kumatori, A., K. Tanaka, N. Inamura, S. Sone, T. Ogura, T. Matsumoto, T. Tachikawa, S. Shin, and A. Ichihara. 1990. Abnormally high expression of proteasomes in human leukemic cells. Proc. Natl. Acad. Sci. USA. 87:7071–7075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shimbara, N., E. Orino, S. Sone, T. Ogura, M. Takashina, M. Shono, T. Tamura, H. Yasuda, K. Tanaka, and A. Ichihara. 1992. Regulation of gene expression of proteasomes (multi-protease complexes) during growth and differentiation of human hematopoietic cells. J. Biol. Chem. 267:18100–18109. [PubMed] [Google Scholar]
- 23.Holmgren, L., M.S. O'Reilly, and J. Folkman. 1995. Dormancy of micrometastases: balanced proliferation and apoptosis in the presence of angiogenesis suppression. Nat. Med. 1:149–153. [DOI] [PubMed] [Google Scholar]
- 24.Bogyo, M., J.S. McMaster, M. Gaczynska, D. Tortorella, A.L. Goldberg, and H.L. Ploegh. 1997. Covalent modification of the active site threonine of proteasomal beta subunits and the Escherichia coli homolog HslV by a new class of inhibitors. Proc. Natl. Acad. Sci. USA. 94:6629–6634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yaman, I., J. Fernandez, B. Sarkar, R.J. Schneider, M.D. Snider, L.E. Nagy, and M. Hatzoglou. 2002. Nutritional control of mRNA stability is mediated by a conserved AU-rich element that binds the cytoplasmic shuttling protein HuR. J. Biol. Chem. 277:41539–41546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kuida, K., T.F. Haydar, C.Y. Kuan, Y. Gu, C. Taya, H. Karasuyama, M.S. Su, P. Rakic, and R.A. Flavell. 1998. Reduced apoptosis and cytochrome c-mediated caspase activation in mice lacking caspase 9. Cell. 94:325–337. [DOI] [PubMed] [Google Scholar]
- 27.Verhagen, A.M., P.G. Ekert, M. Pakusch, J. Silke, L.M. Connolly, G.E. Reid, R.L. Moritz, R.J. Simpson, and D.L. Vaux. 2000. Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell. 102:43–53. [DOI] [PubMed] [Google Scholar]
- 28.Du, C., M. Fang, Y. Li, L. Li, and X. Wang. 2000. Smac, a mitochondrial protein that promotes cytochrome c–dependent caspase activation by eliminating IAP inhibition. Cell. 102:33–42. [DOI] [PubMed] [Google Scholar]
- 29.Srinivasula, S.M., et al. 2001. A conserved XIAP-interaction motif in caspase-9 and Smac/DIABLO regulates caspase activity and apoptosis. Nature. 410:112–116. [DOI] [PubMed] [Google Scholar]
- 30.Verhagen, A.M., and D.L. Vaux. 2002. Cell death regulation by the mammalian IAP antagonist Diablo/Smac. Apoptosis. 7:163–166. [DOI] [PubMed] [Google Scholar]
- 31.Carson, J.P., M. Behnam, J.N. Sutton, C. Du, X. Wang, D.F. Hunt, M.J. Weber, and G. Kulik. 2002. Smac is required for cytochrome c-induced apoptosis in prostate cancer LNCaP cells. Cancer Res. 62:18–23. [PubMed] [Google Scholar]
- 32.Jaattela, M. 1999. Escaping cell death: survival proteins in cancer. Exp. Cell Res. 248:30–43. [DOI] [PubMed] [Google Scholar]
- 33.Sasaki, H., Y. Sheng, F. Kotsuji, and B.K. Tsang. 2000. Down-regulation of X-linked inhibitor of apoptosis protein induces apoptosis in chemoresistant human ovarian cancer cells. Cancer Res. 60:5659–5666. [PubMed] [Google Scholar]
- 34.Grossman, D., and D.C. Altieri. 2001. Drug resistance in melanoma: mechanisms, apoptosis, and new potential therapeutic targets. Cancer Metastasis Rev. 20:3–11. [DOI] [PubMed] [Google Scholar]
- 35.Yang, L., T. Mashima, S. Sato, M. Mochizuki, H. Sakamoto, T. Yamori, T. Oh-Hara, and T. Tsuruo. 2003. Predominant suppression of apoptosome by inhibitor of apoptosis protein in non-small cell lung cancer H460 cells: therapeutic effect of a novel polyarginine-conjugated Smac peptide. Cancer Res. 63:831–837. [PubMed] [Google Scholar]
- 36.Kessler, B.M., D. Tortorella, M. Altun, A.F. Kisselev, E. Fiebiger, B.G. Hekking, H.L. Ploegh, and H.S. Overkleeft. 2001. Extended peptide-based inhibitors efficiently target the proteasome and reveal overlapping specificities of the catalytic beta-subunits. Chem. Biol. 8:913–929. [DOI] [PubMed] [Google Scholar]
- 37.Kessler, B., X. Hong, J. Petrovic, A. Borodovsky, N.P. Dantuma, M. Bogyo, H.S. Overkleeft, H.L. Ploegh, and R. Glas. 2003. Pathways accessory to proteasomal proteolysis are less efficient in MHC class I antigen production. J. Biol. Chem. 278:10013–10021. [DOI] [PubMed] [Google Scholar]
- 38.Adams, J. 2002. Proteasome inhibition: a novel approach to cancer therapy. Trends Mol. Med. 8:S49–54. [DOI] [PubMed] [Google Scholar]
- 39.Garber, K. 2002. Cancer research. Taking garbage in, tossing cancer out? Science. 295:612–613. [DOI] [PubMed] [Google Scholar]
- 40.Rose, C., F. Vargas, P. Facchinetti, P. Bourgeat, R.B. Bambal, P.B. Bishop, S.M. Chan, A.N. Moore, C.R. Ganellin, and J.C. Schwartz. 1996. Characterization and inhibition of a cholecystokinin-inactivating serine peptidase. Nature. 380:403–409. [DOI] [PubMed] [Google Scholar]
- 41.Glickman, M.H., D.M. Rubin, H. Fu, C.N. Larsen, O. Coux, I. Wefes, G. Pfeifer, Z. Cjeka, R. Vierstra, W. Baumeister, et al. 1999. Functional analysis of the proteasome regulatory particle. Mol. Biol. Rep. 26:21–28. [DOI] [PubMed] [Google Scholar]
- 42.van der Zee, J. 2002. Heating the patient: a promising approach? Ann. Oncol. 13:1173–1184. [DOI] [PubMed] [Google Scholar]
- 43.Yu, J.L., B.L. Coomber, and R.S. Kerbel. 2002. A paradigm for therapy-induced microenvironmental changes in solid tumors leading to drug resistance. Differentiation. 70:599–609. [DOI] [PubMed] [Google Scholar]
- 44.Kanayama, H., K. Tanaka, M. Aki, S. Kagawa, H. Miyaji, M. Satoh, F. Okada, S. Sato, N. Shimbara, and A. Ichihara. 1991. Changes in expressions of proteasome and ubiquitin genes in human renal cancer cells. Cancer Res. 51:6677–6685. [PubMed] [Google Scholar]
- 45.Twombly, R. 2002. First clinical trials of endostatin yield lukewarm results. J. Natl. Cancer Inst. 94:1520–1521. [DOI] [PubMed] [Google Scholar]
- 46.Senderowicz, A.M. 2002. The cell cycle as a target for cancer therapy: basic and clinical findings with the small molecule inhibitors flavopiridol and UCN-01. Oncologist. 7(Suppl 3):12–19. [DOI] [PubMed] [Google Scholar]
- 47.Kallio, M.J., M. Nieminen, and J.E. Eriksson. 2001. Human inhibitor of apoptosis protein (IAP) survivin participates in regulation of chromosome segregation and mitotic exit. FASEB J. 15:2721–2723. [DOI] [PubMed] [Google Scholar]
- 48.Rao, H., F. Uhlmann, K. Nasmyth, and A. Varshavsky. 2001. Degradation of a cohesin subunit by the N-end rule pathway is essential for chromosome stability. Nature. 410:955–959. [DOI] [PubMed] [Google Scholar]
- 49.Martin, S.J. 2002. Destabilizing influences in apoptosis: sowing the seeds of IAP destruction. Cell. 109:793–796. [DOI] [PubMed] [Google Scholar]
- 50.Vucic, D., W.J. Kaiser, and L.K. Miller. 1998. Inhibitor of apoptosis proteins physically interact with and block apoptosis induced by Drosophila proteins HID and GRIM. Mol. Cell. Biol. 18:3300–3309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nomura, T., H. Mimata, Y. Takeuchi, H. Yamamoto, E. Miyamoto, and Y. Nomura. 2003. The X-linked inhibitor of apoptosis protein inhibits taxol-induced apoptosis in LNCaP cells. Urol. Res. 31:37–44. [DOI] [PubMed] [Google Scholar]
- 52.Renn, S.C., B. Tomkinson, and P.H. Taghert. 1998. Characterization and cloning of tripeptidyl peptidase II from the fruit fly, Drosophila melanogaster. J. Biol. Chem. 273:19173–19182. [DOI] [PubMed] [Google Scholar]
- 53.Liang, J.S., O. Distler, D.A. Cooper, H. Jamil, R.J. Deckelbaum, H.N. Ginsberg, and S.L. Sturley. 2001. HIV protease inhibitors protect apolipoprotein B from degradation by the proteasome: a potential mechanism for protease inhibitor-induced hyperlipidemia. Nat. Med. 7:1327–1331. [DOI] [PubMed] [Google Scholar]
- 54.Andre, P., M. Groettrup, P. Klenerman, R. de Giuli, B.L. Booth, Jr., V. Cerundolo, M. Bonneville, F. Jotereau, R.M. Zinkernagel, and V. Lotteau. 1998. An inhibitor of HIV-1 protease modulates proteasome activity, antigen presentation, and T cell responses. Proc. Natl. Acad. Sci. USA. 95:13120–13124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wyttenbach, A., J. Carmichael, J. Swartz, R.A. Furlong, Y. Narain, J. Rankin, and D.C. Rubinsztein. 2000. Effects of heat shock, heat shock protein 40 (HDJ-2), and proteasome inhibition on protein aggregation in cellular models of Huntington's disease. Proc. Natl. Acad. Sci. USA. 97:2898–2903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bence, N.F., R.M. Sampat, and R.R. Kopito. 2001. Impairment of the ubiquitin-proteasome system by protein aggregation. Science. 292:1552–1555. [DOI] [PubMed] [Google Scholar]
- 57.Lutz, W., J. Leon, and M. Eilers. 2002. Contributions of Myc to tumorigenesis. Biochim. Biophys. Acta. 1602:61–71. [DOI] [PubMed] [Google Scholar]