

Abstract
The recently described junctional adhesion molecules (JAMs) in man and mice are involved in homotypic and heterotypic intercellular interactions. Here, a third member of this family, human JAM-3, was identified and described as a novel counterreceptor on platelets for the leukocyte β2-integrin Mac-1 (αMβ2, CD11b/CD18). With the help of two monoclonal antibodies, Gi11 and Gi13, against a 43-kD surface glycoprotein on human platelets, a full-length cDNA encoding JAM-3 was identified. JAM-3 is a type I transmembrane glycoprotein containing two Ig-like domains. Although JAM-3 did not undergo homophilic interactions, myelo-monocytic cells adhered to immobilized JAM-3 or to JAM-3–transfected cells. This heterophilic interaction was specifically attributed to a direct interaction of JAM-3 with the β2-integrin Mac-1 and to a lower extent with p150.95 (αXβ2, CD11c/CD18) but not with LFA-1 (αLβ2, CD11a/CD18) or with β1-integrins. These results were corroborated by analysis of K562 erythroleukemic cells transfected with different heterodimeric β2-integrins and by using purified proteins. Moreover, purified JAM-3 or antibodies against JAM-3 blocked the platelet-neutrophil interaction, indicating that platelet JAM-3 serves as a counterreceptor for Mac-1 mediating leukocyte–platelet interactions. JAM-3 thereby provides a novel molecular target for antagonizing interactions between vascular cells that promote inflammatory vascular pathologies such as in atherothrombosis.
Keywords: monoclonal antibody, adhesion molecule, integrins, neutrophils, athero- thrombosis
Introduction
Multicellular interactions between leukocytes and the vessel wall play a pivotal role in inflammatory processes or vascular remodeling. Leukocyte recruitment to vascular endo-thelium requires multistep adhesive and signaling events including selectin-mediated rolling, leukocyte activation, and integrin-mediated firm adhesion and diapedesis (1). On the leukocytes, members of the β2-integrin family, LFA-1 (αLβ2, CD11a/CD18), Mac-1 (αMβ2, CD11b/CD18), and p150.95 (αXβ2, CD11c/CD18), as well as β1-integrins interact with endothelial counterligands such as intercellular adhesion molecule 1 (ICAM-1)* (2), surface-associated fibrinogen (3), or vascular cell adhesion molecule (VCAM)-1, to mediate the described paracellular interactions. At sites of vascular injury, where the endothelial cell lining has been denuded, leukocyte infiltration can occur through interactions with deposited platelets and fibrin (4). In vivo studies indeed demonstrated colocalization of both cell types within atherosclerotic and postangioplasty restenotic lesions, or in areas of ischemia-reperfusion injury (5, 6). Reminiscent of the leukocyte–endothelium interactions, a sequential adhesion process including leukocyte attachment to and transmigration across surface-adherent platelets, has been proposed: Platelet-P-selectin–mediated initial tethering and rolling of leukocytes through their P-selectin glycoprotein ligand-1 (PSGL-1) is followed by Mac-1–dependent firm adhesion and transplatelet migration (7). Recent data indicated glycoprotein (GP) Ibα on platelets as a potential counterreceptor for Mac-1 (8). Furthermore, ICAM-2 and αIIbβ3-associated fibrinogen have also been proposed to mediate Mac-1–dependent platelet–leukocyte interactions (9, 10); however, the exact contribution of each system remains to be elucidated.
Recently, new members of the Ig-superfamily named junctional adhesion molecules (JAMs), which mediate cell-to-cell adhesion have been identified on several blood cell populations. JAM-1 is a transmembrane protein which possesses two V-type immunoglobulin domains and is found on endothelial and epithelial cells at sites of cell–cell contacts in particular within tight junctions (11). JAM-1 was shown to participate in monocyte transmigration across endothelial cells, and blocking mAbs against JAM-1 were able to inhibit monocyte infiltration and attenuate cytokine-induced meningitis in mice (12–14). Platelet JAM-1 might also be involved in primary hemostasis (15–17), as cross-linking of JAM-1 with FcγRII resulted in platelet aggregation. Interestingly, human platelet antibodies against JAM-1 were reported in patients with thrombocytopenia (16). Human JAM-1 is also constitutively expressed on circulating monocytes, neutrophils, erythrocytes, and lymphocyte subsets (18–20). Finally, JAM-1 has been described as a receptor for reovirus (21). In contrast, JAM-2 (also referred to as VE-JAM) is prominently expressed at intercellular boundaries of the endothelium, particularly in venules (22). JAM-2 is also involved in adhesive processes of lymphocytes (23).
In this study, we describe the isolation and characterization of a new member of the JAM family, namely JAM-3, expressed on human platelets, and provide evidence that JAM-3 is a novel counterreceptor for the integrin Mac-1 (CD11b/CD18) thereby playing a critical role for platelet–leukocyte interactions.
Materials and Methods
Antibodies and Purified Proteins.
The following mAbs were used in this study: mAb Gi11 and Gi13 were characterized previously (24); mAb w6/32 against HLA class I molecule (provided by Dr. S. Ferrone, New York Medical College, New York, NY); mAb Gi5, Gi9, and Gi21, specific for αIIbβ3, α2β1 integrins, and DAF (CD55), respectively, (25); mAb AP1 against GPIbα (provided by Dr. P. Newman, Blood Research Institute, Milwaukee, WI), mAb 60.3 against β2 integrin subunit (CD18) (provided by Dr. J. Harlan, University of Washington, Seattle, WA), mAb L15 against LFA-1 (provided by Dr. C. Figdor, University Medical Center, Nijmegen, Netherlands), mAb CBRM1/5 against Mac-1 (provided by Dr. T. Springer, Harvard Medical School, Boston, MA); mAb LPM19c against Mac-1 (provided by Dr. A. May, Deutsches Herzzentrum, Munich, Germany); mAb 3.9 against p150.95 (Ancell; reference 26); β2-integrin stimulating mAb Kim127 and Kim185 (provided by Dr. M. Robinson, Celltech, Slough, UK); mAbs K20 against β1-chain (CD29), HP2.1 specific for α4-integrin (CD49d) and 3G8 against CD16 (Immunotech); mAbs against ICAM-1 and fibrinogen (Dako); mAb MY4 specific for anti-CD14 (Beckman Coulter); blocking mAb GA6* against CD62P (P-selectin) and normal mouse IgG (Becton Dickinson). Polyclonal antibody against JAM-3 was produced in rabbits using standard procedures. Purified Mac-1, LFA-1, and ICAM-1 were a gift from Dr. S. Bodary (Genentech, San Francisco, CA). There was no contamination of the recombinant Mac-1 preparation with JAM-3. Purified glycocalicin was provided by Dr. K. Clemetson (Theodor Kocher Institute, Bern, Switzerland) and purified I-domain of Mac-1 was provided by Dr. D. Tuckwell (School of Biological Sciences, Manchester, UK). The stable thromboxane A2 mimetic U46619 was a gift Dr. Stegmeier, Roche (Mannheim, Germany) and fMLP was from Sigma-Aldrich. Mab Gi11 was labeled with NHS-LC biotin (Paesel) as recommended by the manufacturer.
Isolation of Gi11 Antigen from Human Platelets.
Outdated platelet concentrates were used as source for purification of Gi11 antigen. Washed platelets were centrifuged with 3,700 g at 4°C for 45 min, and the pellet was resuspended to 1010 platelets/ml in lysis buffer (20 mM Tris, 2 mM EDTA, 150 mM NaCl, 2% NP-40, 2 mM Pefabloc, pH 7.8). After solubilization (at 4°C for 1 h) cell debris was removed by centrifugation with 17,000 g at 4°C for 45 min. From the supernatant the solubilized Gi11 antigen was isolated by sequential antibody adsorption steps on nonimmune mouse IgG, mAb w6/32, and mAb Gi11, coupled to Sepharose. Affinity columns were washed with 100 ml washing buffer (0.1 M Tris, 0.1% NP-40, pH 7.8; flow rate 50 ml/h) and 270 ml cell supernatant was applied onto the first column with a flow rate of 5–10 ml/h. After sequential washing with (a) 50 ml 0.1 M Tris, 0.5 M NaCl, 0.1% NP-40, pH 7.8, (b) 50 ml 1.0 M Tris, 0.1% NP-40, pH 7.8, and (c) 50 ml 0.1 M Tris, 0.1% NP-40, pH 8.0, antigens were eluted from the mAb Gi11 affinity column with 0.05 M diethylamine, 0.1% NP-40, pH 11.5, in 3.0 ml fractions into tubes containing 1 ml 1M Tris, 0.1% NP-40, pH 6.8. Eluted antigens were identified by silver staining and immunoblot. Gi11 antigen containing fractions were pooled, dialyzed in PBS containing 0.1% NaN3, 0.1% NP-40, and concentrated using ultrafiltration technique (Centriplus YM 10; Millipore) to ∼1 ml. The protein concentration of the purified Gi11 antigen was determined using bicinchoninic assay (BCA; Perbio).
Protein and Peptide Analysis.
Protein and peptides (20 to 100 pmol) were sequenced by Edman degradation on an Applied Biosystems pulsed-liquid-phase sequencer 477A. For analysis of amino terminal sequences, gels were electroblotted onto polyvinyliden-difluoride (PVDF) membrane. For internal sequences bands were excised, and the proteins were digested in situ with trypsin. Peptides were eluted and separated by capillary HPLC, and individual fractions were selected for sequencing.
Immunoprecipitation and Western Blot Analysis.
Cells were surface labeled with 5 mM NHS-LC biotin (Paesel), lysed and subjected to immunoprecipitation as described previously (27). Immunprecipitates were analyzed on 10% SDS-PAGE under nonreducing and reducing conditions, transferred onto nitrocellulose, and visualized using peroxidase labeled streptavidin (dilution 1:3,000) and chemiluminescence substrate (Amersham Biosciences).
Purified Gi11 antigen (0.5 –1 μg) or 25 μl platelet lysate were run on 10% SDS-PAGE under nonreducing and reducing conditions. Proteins were then transferred onto nitrocellulose membrane and visualized by silver staining or incubated with mAb Gi11 (2.5 μg/ml). Bound IgG was detected using peroxidase labeled rabbit anti mouse IgG (dilution 1:100,000, Dianova) and chemiluminescence substrate (Amersham Biosciences).
Isolation of Platelet mRNA and Rapid Amplifications of 3′ and 5′ cDNA Ends-PCR.
Total platelet mRNA was isolated as described previously (27). Rapid amplifications of 3′ and 5′ cDNA ends (RACE) was performed using SMART RACE cDNA Amplification Kit (CLONTECH Laboratories, Inc.). PCR primers GSP-1 (5′-783GGGCATCTGTGCATACAGACGTG808-3′) and GSP-2 (5′-96CAGTGTTTCTGCACGACCCGCCAAGTC71-3′) were constructed based on the nucleotide sequence accessible in the GenBank (see below) and were numbered according to the JAM-3 cDNA sequence (see Fig. 2) .
Figure 2.


JAM-3 cDNA sequence and deduced amino acid sequence. (A) The NH2-terminal amino acid sequence and internal sequences derived from amino acid sequence analysis are underlined. The transmembrane region is indicated by a double line. Putative N- (Asn104, Asn192) and O-glycosylation sites (Thr60) are shown in bold. The protein kinase C phosphorylation site at Ser281 is circled and PDZ binding motif is boxed. The GenBank/EMBL/DDBJ accession no. for the nucleotide sequence of the human platelet JAM-3 is AF448478. (B) Phylogenetic tree of homologous human (h) JAM-3 and murine (m) JAM-2. Phylogenetic distances obtained with the Clustal W program are indicated as values above the branches. (C) Alignment of hJAM-3 and mJAM-2 was done using the SMART program. Immunoglobulin C-2 domains and the transmembrane domain are indicated by single and double line, respectively. Cysteine residues are shown in bold and nonidentical amino acids are shaded in dark. The mJAM-2 sequence is accessible from the database (AJ300304).
Generation of JAM-3 Construct.
30 μl of platelet mRNA were heated to 68°C for 10 min and quickly cooled on ice water. First-strand cDNA was synthesized using 1 μl pd(N)6 primer and first-strand beads (Amersham Biosciences) according to the manufacturer's protocol. Aliquots of 20 μl were preheated to 90°C for 5 min and digested with 2U RNaseH enzyme (GIBCO BRL) at 37°C. 20 μl cDNA were diluted with 10× PCR buffer, and 3 μl (5 μM) GSP-3 (5′-1ATGGCGCTGAGGCGGCCA18-3′), 3 μl (5 μM) GSP-4 (5′-933TCAGATCACAAACGATGAC915-3′), 8 μl (1.25 mM) dNTP and 2 U Taq Gold polymerase (PerkinElmer) were added in a total volume of 50 μl. Amplification was performed on a DNA thermal cycler 480 (PerkinElmer) for 32 cycles. Each cycle consisted of denaturation at 95°C for 30 s, annealing at 50°C for 30 s, and extension at 72°C for 60 s. In the final cycle, the samples were kept at 72°C for 10 min and then chilled to 4°C. Amplified JAM-3 cDNA was purified using QIAquick gel extraction kit (QIAGEN) and subcloned into the T/A cloning site of the plasmid vector pcDNA4His/Max-TOPO (Invitrogen) and then transformed into the DH5α high-efficiency competent Escherichia coli (GIBCO BRL). Recombinant colonies were selected using Zeocin (25 μg/ml; Invitrogen). Plasmid DNA from seven clones was purified using QIAprep (QIAGEN), sequenced using Big Dye Terminator Cycle Sequencing Kit according to the manufacturer's protocol and analyzed on an ABI Prism Genetic Analyzer 310 (Applied Biosystems).
Expression of JAM-3 Construct in CHO Cells.
CHO cells were grown in Ham's F12 medium supplemented with 10% fetal calf serum and 0.5% penicillin/streptomycine and were transfected with JAM-3 construct by the use of Lipofectamine (GIBCO BRL). In brief, 1 μg of the JAM-3 construct was mixed with 15 μl lipofectamine in 200 μl OptiMEM and then added to subconfluent CHO cells. After 5 h, 2 ml Ham's F12 medium containing 10% FCS was added and the incubation was continued for 48 h. After splitting, stable transfectants were selected with Zeocin (final concentration 200 μg/ml; Invitrogen) for 2 wk and subcloned. Transfectants expressing recombinant JAM-3 were identified by flow cytometry analysis using mAb Gi11. A positive clone was enriched by the use of MACS MicroBeads coupled with goat anti–mouse IgG (Miltenyi Biotec).
Isolation of Cells from Peripheral Blood.
Platelets were isolated from acid-citrate-dextrose (ACD) anticoagulated blood and washed with Tyrode's buffer containing prostaglandin E1 and apyrase as described previously (27). For the isolation of white blood cells, blood was diluted with an equal volume of PBS, layered onto Ficoll-Plaque™ Plus (Amersham Biosciences) and centrifuged with 17,000 g, at 20°C for 35 min. Mononuclear cells were recovered from the interface and washed twice with PBS. Monocytes were then separated from lymphocytes as described previously (28). Granulocytes were isolated from the red blood cell fraction by lysis of erythrocytes with ammonium chloride.
Determination of JAM-3 Expression by Flow Cytometry.
100 μl cell suspensions were incubated with 20 μl mAb Gi11 (0.02 mg/ml) at 4°C for 30 min, washed twice, and labeled with 100 μl fluorescein isothiocyanate-conjugated rabbit anti–mouse IgG (1:80 dilution; Dako). Cells were resuspended in 500 μl PBS containing 0.5% BSA and analyzed by flow cytometry on a FACSCalibur™ (Becton Dickinson). Antibody binding was quantified using DAKO QIFIKIT Calibrations Beads (Dako).
Cell Lines.
Myelomonocytic cells (U937) were from American Type Culture Collection and were cultured in RPMI-1640 containing 10% FCS. K562 cells (nontransfected, and transfected with LFA-1 or p150.95) were provided by Dr. Y. van Kooyk (University Medical Center, Nijmegen, Netherlands) and were cultured in a mixture of 75% RPMI-1640 containing 10% FCS and 25% ISCOVE's medium supplemented with 5% FCS. K562 cells transfected with Mac-1 were provided by Dr. M. Smith (Celltech, Slough, UK). All culture media were from GIBCO BRL.
Cell Adhesion Assay.
Cell adhesion to JAM-3, fibrinogen (Kabivitrum) or BSA (as control) coated plates was tested as described previously (29). Briefly, microtiter plates were coated with 10 μg/ml JAM-3 or fibrinogen (in bicarbonate buffer, pH 9.6), respectively, and blocked with 3% BSA solution. U937 cells, which had been differentiated for 24 h in the presence of vitamin D3 (100 nM; Biomol) and transforming growth factor-β (2 ng/ml; R&D Systems), or K562 cells were washed in serum-free RPMI and plated onto the precoated wells (105/well) at 37°C for 60 min in the absence or presence of inhibitors. After the incubation period, the wells were washed and adherent cells were fixed with methanol/acetone (1:1) at 4°C for 30 min. Adherent cells were then stained with crystal violet and quantified by measuring absorbance at 590 nm.
Fluorescence Adhesion Assay.
Adhesion of neutrophils, U937 cells or K562 cells to immobilized ligands, surface-adherent platelets or to CHO cells was investigated as described (30). Briefly, nontransfected CHO or JAM-3–transfected CHO cells were grown to confluency onto 96-well plates. Human platelets (1.5 × 107/well) were added to microtiter plates precoated with 0.2% gelatin. After 1 h at 37°C unbound platelets were removed. Fluorescence labeled neutrophils (105/well), differentiated U937 cells or K562 cells were washed twice followed by no pretreatment or stimulation with phorbol myristate acetate (PMA; 50 ng/ml) or the β2-integrin stimulating mAb Kim127 (10 μg/ml). Cells were washed and added to immobilized JAM-3, platelets or to CHO cells at 37°C for 60 min in the absence or presence of inhibitors. After washing, adhesion of neutrophils, U937, or K562 cells was quantified as the percentage of total cells added using a fluorescence microplate reader (Bio-Tek). When purified JAM-3 was used as a competitor in both adhesion assays the final concentration of NP-40 in the buffer was less than 0.005% and did not affect the assay at all.
ELISA for Ligand–Receptor Interactions.
A previously outlined procedure was used (30). Briefly, maxisorp plates (Greiner) were coated with Mac-1 or LFA-1 (5 μg/ml) dissolved in 20 mM HEPES, 150 mM NaCl, 1 mM Mn2+, pH 7.2 (HEPES buffer), and then blocked with 3% BSA in HEPES buffer. Binding of ICAM-1 (10 μg/ml), JAM-3 (0–15 μg/ml), or biotinylated fibrinogen (2 μg/ml) to the immobilized integrin was performed in a final volume of 50 μl (HEPES buffer supplemented with 0.05% Tween-20) in the absence or presence of inhibitors. After incubation at 22°C for 2 h and a washing step, bound JAM-3 was detected by biotinylated mAb Gi11 and peroxidase-conjugated streptavidin (1:2,000 dilution; Dako), bound ICAM-1 was detected by appropriate mAb and peroxidase-conjugated goat anti–mouse immunoglobulin (1:2,000 dilution; Dako). Biotinylated fibrinogen was detected directly with peroxidase-conjugated streptavidin (see above). Bound antibodies or streptavidin were quantified using 2,2′-azino-di-[3-ethylbenzthiazoline sulfonate(6)] as substrate (Boehringer Mannheim) in a microplate reader (Molecular Devices) at 405 nm. Nonspecific binding to BSA-coated wells was used as blank and was subtracted to calculate specific binding.
Determination of Platelet Adhesion to Neutrophils in Whole Blood.
Whole blood adhesion experiments were performed in a micro-couette as described (31–33). ACD anticoagulated blood was obtained from healthy volunteers, diluted 1:1 with Tyrode buffer and subjected to shear (20 and 2,000 s−1) for 10 min at 37°C in the presence of 10 μl inhibitors. The samples were fixed with a fixative containing methacroleine (32). Leukocyte-platelet aggregates were determined by two color flow cytometry in FACScan™ (Becton Dickinson) using FITC-labeled mAb against CD41a (Beckman Coulter) and PE-labeled mAb against CD66 (Beckman Coulter).
Data Analysis and Statistical Analysis.
Data analysis was performed using Internet-based tools BLAST2.0 (http://www.ch. embnet.org), CLUSTAL (http://genius.embnet.dkfz-heidelberg. de), and SMART (http://smart.EMBL-Heidelberg.de). Statistical analysis was performed using analysis of variance (ANOVA) and the Student's t test; P values of <0.05 were regarded as significant.
Results
Detection, Purification, and Molecular Identification of Platelet Gi11 and Gi13 Antigen.
To isolate the Gi11 antigen aliquots of 500 ml platelet lysates derived from ∼5 × 1012 outdated platelets were applied to normal mouse IgG, mAb w6/32 and mAb Gi11 affinity columns. Approximately 1.1 mg purified Gi11-protein could be isolated from a single affinity adsorption/desorption run. Silver staining of the major fractions showed a band with M r of 43 kD which reacted with mAb Gi11 as well as with mAb Gi13 in immunoblotting under reducing conditions (Fig. 1) . Both mAb recognized different epitopes on Gi11-antigen as was deduced from binding competition experiments (unpublished data).
Figure 1.

Purification and identification of Gi11 antigen from platelets. Purified Gi11 antigen was subjected to 10% SDS-PAGE under reducing conditions and was visualized by Coomasie blue staining (lane 2) in comparison to indicated molecular weight standards (lane 1). Gi11-antigen was then transferred onto nitrocellulose membrane, stained with mAb Gi11 or Gi13 (lanes 3 and 4) and visualized using streptavidin–chemiluminescence system.
The sequence analysis of the immunoreactive 43 kD protein yielded 14 NH2-terminal residues (VNLKSSNRTPVVQEF). In addition, the amino terminal sequence of three major internal proteolytic fragments IQDEQTTYVFF, SLKIWNVT, and MATLHCQESEGHPRPHYSWYR could be identified (Fig. 2). Alignment of the NH2-terminal sequence and the proteolytic fragments with the protein database of the National Center for Biotechnology Information using BLAST 2.0 showed significant similarity with a protein sequence derived from human fetal brain (GenBank/EMBL/DDBJ accession no. AF356518.1) that was recently described by Arrate et al. as JAM-3 (34). The complete nucleotide sequence of human platelet JAM-3 cDNA derived from RACE-PCR and the deduced amino acid sequence is shown in Fig. 2 A.
Analysis of the Platelet JAM-3 Amino Acid Sequence.
Platelet JAM-3 is encoded by a mRNA of 1740 bases that encodes a protein of 310 amino acids with a signal sequence corresponding to amino acids 1–31. JAM-3 is a type I integral membrane protein with an extracellular amino-terminus (residues 32–310), a transmembrane domain (residues 245–267), and a cytoplasmic oriented carboxy-terminus (residues 268–310). Furthermore, the protein contains two potential N-glycosylation (Asn104, Asn192) sites and one potential O-glycosylation site (Thr60). The cytoplasmic tail contains three potential phosphorylation sites (Tyr270,282, 293) and a putative phosphorylation site for casein kinase II (Thr296) or protein kinase C (Ser281). At the extreme carboxy-terminus, a potential binding motif for PDZ domains is apparent. JAM-3 exhibits two regions (amino acids 44–122 and 151–226) of high homology to members of the immunoglobulin superfamily. Based on the analysis using the SMART program these two regions represent C2-type immunoglobulin domains.
Alignment of Human JAM-3 with Human and Murine JAMs.
Multiple sequence alignment of human JAM-3 revealed 36% identity with human JAM-2 and 32% with human JAM-1, whereas comparison with the known murine JAM-1, -2, and –3 showed amino acid identities of 34, 86, and 35%, respectively. Human JAM-3 is more closely related to murine JAM-2 than to murine JAM-1 or murine JAM-3. Murine JAM-3 is related to human JAM-2 (79% homology), whereas murine JAM-1 is related to human JAM-1 (67% homology). The phylogenetic tree of human and murine JAMs and the alignment between human JAM-3 and murine JAM-2 are shown in Fig. 2, B and C.
Cellular Distribution of JAM-3.
To examine the expression of JAM-3 in various peripheral blood cells, the surface expression of JAM-3 on platelets, granulocytes, monocytes, lymphocytes, and erythrocytes isolated from healthy blood donors was analyzed by flow cytometry using mAb Gi11 (Fig. 3) . Significant JAM-3 expression was only detected on platelets, but not on other blood cells. Quantitative determination demonstrated that platelets possess 1594 ± 303 copies (n = 6) of JAM-3 on their surface. Analysis with myeloid cell lines (K562, U937, HL-60, HEL VJ20, MEG-01, KG1a, and Dami) indicated that JAM-3 is only expressed on megakaryocytic cell lines MEG-01, Dami, and KG1a cells, but not on K562 and U937 cells (35). The absence of JAM-3 surface expression on U937 and K562 used in our study is shown in Fig. 3.
Figure 3.

Flow cytometry analysis of JAM-3 expression on blood cells, U937 and K562 cell lines with mAb Gi11. The closed curves represent the reaction with mAb Gi11, the thin lines with normal mouse IgG (negative control) and the bold lines with different mAb specific for the indicated cell types: Gi9 (α2β1 integrin; platelets), 3G8 (CD16; granulocytes), MY4 (CD14; monocytes), w6/32 (HLA class I; lymphocytes), and Gi21 (CD55; erythrocytes, K562 cell line) and K20 (β1 integrin; U937 cell line).
JAM-3 Is a Counterreceptor for Mac-1.
The described homophilic or heterophilic interactions of JAMs responsible for cell–cell adhesion (17, 36) prompted us to investigate the binding properties of human platelet JAM-3 in this respect. Full-length JAM-3 was transfected into CHO cells and stable transfectants were isolated and analyzed by flow cytometry using mAb Gi11 (Fig. 4 A). Immunoblotting analysis demonstrated that the recombinant JAM-3 revealed the expected protein band with the molecular mass of 43 kD compared with authentic platelet JAM-3 (Fig. 4 B). When the interaction of JAM-3–transfected CHO cells to immobilized JAM-3 was tested, only very weak binding was noted, indistinguishable from binding of nontransfected cells (unpublished data).
Figure 4.
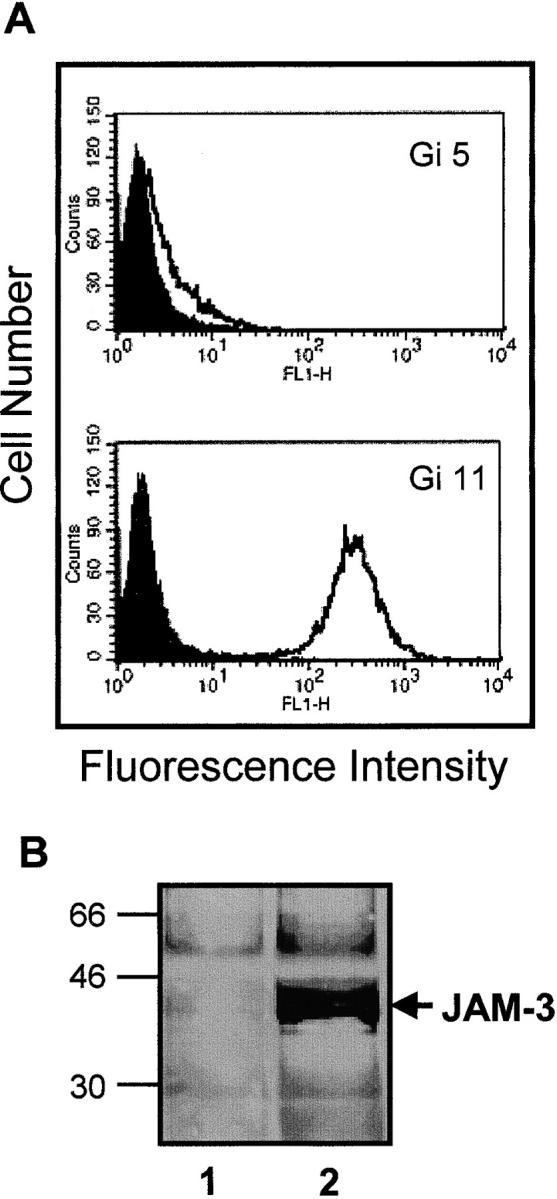
Characterization of CHO cells expressing stable JAM-3. (A) Flow cytometry analysis of JAM-3 transfectants using mAb Gi11 (anti-JAM3) and mAb Gi5 (anti-GPIIb/IIIa) is shown. The closed curves represent the reaction of antibodies with untransfected CHO cells. (B) Immunoblotting analysis using mAb Gi11 of lysates from untransfected cells (lane 1) or JAM-3 transfectants (lane 2) is presented.
In contrast, myelomonocytic U937 cells, which do not express JAM-3, adhered strongly to JAM-3–coated plates. This adhesion was abolished by an antibody against JAM-3 and a blocking mAb against β2-integrin, whereas a blocking mAb against β1-integrin had no effect. Moreover, U937 cell adhesion to immobilized JAM-3 could be inhibited by a blocking mAb against Mac-1 or by the Mac-1 ligand fibrinogen, whereas a blocking mAb against LFA-1 or p150.95 did not affect cell adhesion (Fig. 5 A). In addition, the Mac-1–mediated adhesion of U937 cells to fibrinogen was inhibited by purified JAM-3 (Fig. 5 B). The same pattern of interaction between JAM-3 and Mac-1 was identified when the adhesion of U937 cells to JAM-3 transfected CHO cells was tested. The efficiency of cell-to-cell adhesion was almost doubled in the presence of PMA, a known activator of integrins, and adhesion was inhibited by mAb against the common β2-subunit or Mac-1, but not by mAbs against LFA-1, p150.95, or β1-integrins (Fig. 5 C). In any case, nontransfected CHO cells did not present any appreciable adhesion. In these experiments, cell adhesion was not only inhibited by the blocking Mac-1 mAb LPM19c, but also by the mAb CBRM1/5, as the CBRM1/5 activation-dependent epitope was expressed on differentiated and PMA-stimulated U937 cells (unpublished data). Finally, neutrophils isolated from peripheral blood, also adhered specifically to immobilized JAM-3 in a Mac-1–dependent manner (unpublished data).
Figure 5.

Leukocyte adhesion to JAM-3 is Mac-1 dependent. (A) The adhesion of PMA-stimulated (50 ng/ml) U937 cells to immobilized JAM-3 is shown in the absence (−) or presence of blocking mAb against the integrin β1-chain, β2-chain, LFA-1, Mac-1, p150.95, JAM-3 (each 10 μg/ml), or soluble fibrinogen (FBG, 20 μg/ml). (B) The adhesion of PMA-stimulated (50 ng/ml) U937 cells to immobilized fibrinogen is shown in the absence (−) or presence of blocking mAbs against integrins (each 10 μg/ml) or purified JAM-3 (20 μg/ml). Cell adhesion is expressed as absorbance at 590 nm. All data are mean ± SD (n = 3) of a typical experiment; similar results were obtained in at least three separate experiments. (C) U937 cell adhesion to nontransfected CHO cells (white bars) or JAM-3–transfected CHO cells (black bars) was studied without or with PMA (50 ng/ml) pretreatment as indicated, and in the absence (−) or presence of blocking mAb against integrins or against JAM-3 (each 10 μg/ml). The number of adherent cells is expressed as percentage of total added cells. All data are mean ± SD (n = 3) of a typical experiment; similar results were obtained in at least three separate experiments; *: P < 0.01 compared with respective control; ns, not significant.
To further define a direct interaction between JAM-3 and members of the β2-integrin family, the adhesion of K562 erythroleukemic cells transfected either with LFA-1, Mac-1, or p150.95 to immobilized JAM-3 was investigated. Whereas nontransfected K562 cells or LFA-1 transfectants did not show any significant adhesion to JAM-3 both under unstimulated or stimulated conditions (with PMA or the stimulating antibody Kim127), K562 cells expressing Mac-1 or p150.95 adhered to immobilized JAM-3 especially in the presence of the β2-integrin stimulating antibody Kim127 (Fig. 6 A). Similarly, only Mac-1– and p150.95-transfected K562 cells adhered to JAM-3–transfected CHO cells, whereas LFA-1 transfectants or nontransfected cells did not adhere. Moreover, none of the K562 cells interacted with nontransfected CHO cells (Fig. 6 C). Adhesion of Mac-1– or p150.95-transfected cells to immobilized JAM-3 or JAM-3–transfected CHO cells was inhibited by blocking mAb against Mac-1 and p150.95, respectively, and also by a mAb against JAM-3 (Fig. 6, B and D). Taken together, these data strongly indicate that JAM-3 acts as a novel counterreceptor for both β2-integrins Mac-1 and p150.95 and is capable of mediating heterophilic cell–cell interactions.
Figure 6.


Characterization of the adhesion of K562 cells to JAM-3. (A) Adhesion of untransfected K562 cells (nt), or K562 cells transfected with LFA-1-, Mac-1-, or p150.95 to immobilized JAM-3 was studied in the absence (white bars) or presence of PMA (50 ng/ml; gray bars) or of the β2-integrin stimulating mAb Kim127 (10 μg/ml; black bars). (B) Adhesion of Mac-1- or p150.95-transfectant to immobilized JAM-3 after stimulation with mAb Kim127 in the absence (white bars) or presence of mAb against Mac-1 (dark gray bar), mAb against p150.95 (light gray bar), or mAb against JAM-3 (black bars; each 10 μg/ml). (C) Adhesion of K562 cells (nt), of LFA-1-, Mac-1-, or p150.95-transfectant to CHO cells or JAM-3–transfected CHO cells was studied without stimulation (open bars) or after stimulation with PMA (gray bars) or with mAb Kim127 (black bars). (D) Adhesion of Mac-1- or p150.95-transfectant to JAM-3–transfected CHO cells was studied in the absence (−) or presence of PMA or mAb Kim127 without (white bars) or with mAb against Mac-1 (dark gray bars), mAb against p150.95 (light gray bars) or mAb against JAM-3 (black bars; each 10 μg/ml). The number of adherent cells is expressed as percent of total added cells. All data are mean ± SD (n = 3) of a typical experiment; similar results were observed in at least three separate experiments.
JAM-3 Directly Binds to Mac-1.
As the previous observations suggested a direct molecular interaction between the β2-integrin Mac-1 and JAM-3, the direct binding of JAM-3 to immobilized purified Mac-1 in comparison to LFA-1 was analyzed. JAM-3 directly bound to Mac-1, and this binding was similar in its extent to the binding of ICAM-1 or fibrinogen, which both are known ligands for Mac-1 (Fig. 7 A). Moreover, binding of JAM-3 to Mac-1 presented a saturable dose-dependency (Fig. 7 B) and was inhibited by a mAb against Mac-1 and a mAb against JAM-3 (Fig. 7 C). In contrast, no specific binding of JAM-3 to immobilized LFA-1 was identified, whereas the expected binding of ICAM-1 to LFA-1 was apparent (Fig. 7 A). As fibrinogen was shown to inhibit the Mac-1–dependent adhesion of U937 cells to immobilized JAM-3 (see Fig. 5 A), the possibility that the two Mac-1 ligands, fibrinogen, and JAM-3, may compete for each other was tested. Fibrinogen inhibited the binding of JAM-3 to immobilized Mac-1, and vice versa, JAM-3 blocked the interaction between fibrinogen and Mac-1, indicative for overlapping binding sites of both ligands on Mac-1 (Fig. 7 C). This observation was further strengthened by demonstrating a direct interaction between JAM-3 and the purified I-domain of Mac-1 (unpublished data), which also constitutes the major binding site for fibrinogen.
Figure 7.
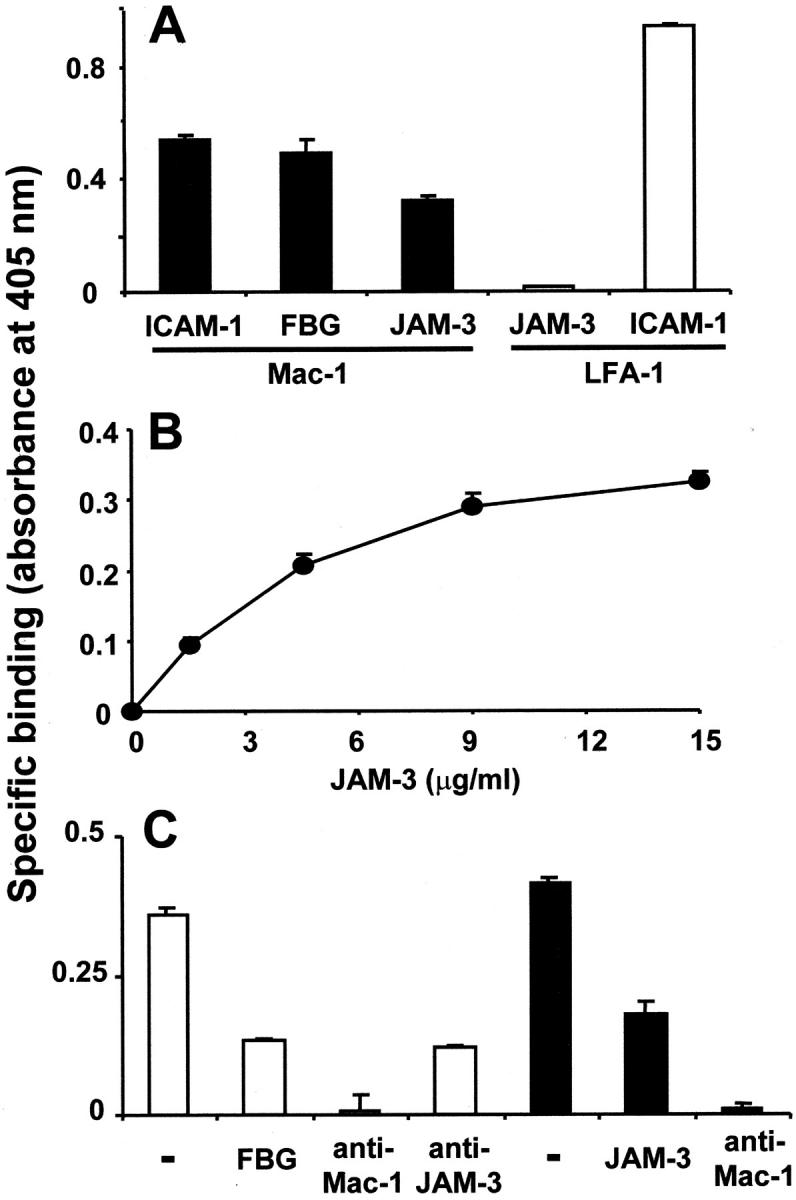
Interaction between purified JAM-3 and Mac-1 proteins. (A) The binding of ICAM-1, fibrinogen (FBG), or JAM-3 to immobilized Mac-1 (black bars) and the binding of JAM-3 or ICAM-1 to immobilized LFA-1 (white bars) was studied. (B) Dose-dependent specific binding of JAM-3 to immobilized Mac-1 is shown. (C) The binding of JAM-3 (white bars) to immobilized Mac-1 was analyzed in the absence (−) or presence of fibrinogen (FBG, 20 μg/ml), mAb against Mac-1 or mAb against JAM-3 (each 10 μg/ml). The binding of fibrinogen (black bars) to immobilized Mac-1 was studied in the absence (−) or presence of purified JAM-3 (20 μg/ml) or of mAb against Mac-1 (10 μg/ml). Specific binding is expressed as absorbance at 405 nm. Data are mean ± SD (n = 3) of a typical experiment; similar results were observed in at least three separate experiments.
JAM-3 Facilitates Platelet–Leukocyte Interactions.
In a recent report, Simon et al. (8) demonstrated that platelet GPIbα serves as a potential counter-receptor for leukocyte Mac-1. However, the presence of additional Mac-1 ligands on the surface of platelets was not ruled out. To establish that JAM-3 can facilitate the interaction between platelets and leukocytes, the adhesion of neutrophils isolated from healthy volunteers or U937 cells to surface-adherent platelets was analyzed. PMA- or fMLP-stimulated neutrophil adhesion to surface-adherent platelets was abolished in the presence of a mAb against Mac-1 but not mAb against LFA-1 or p150.95 (Fig. 8 A and Table I). Individual antibodies against JAM-3, mAb Gi13, or rabbit anti–JAM-3, or anti-GPIbα antibody partially reduced adhesion of neutrophils to platelets, and the combination of both antibodies completely abolished binding of neutrophils to platelets, suggesting that JAM-3 and GPIbα on the platelet surface together serve as the predominant counterreceptors for Mac-1. Moreover, the addition of purified JAM-3 could completely inhibit binding of neutrophils to surface-adherent platelets (Fig. 8 A and Table I), an effect that appeared to be comparable to the inhibitory capacity of soluble glycocalicin in the same system. Blocking other potential Mac-1 ligands such as αIIbβ3-bound fibrinogen or ICAM-2 had no effect on the neutrophil–platelet interaction (unpublished data). Similar results were obtained when adhesion of U937 cells to platelets was examined (unpublished data).
Figure 8.

JAM-3 mediates platelet–leukocyte adhesive interactions. (A) PMA-stimulated adhesion of human neutrophils to surface-adherent platelets was studied in the absence (−) or presence of mAb against Mac-1, LFA-1, JAM-3, or GPIbα as well as a mixture of mAbs against JAM-3 and GPIbα (each 10 μg/ml) or in the presence of purified JAM-3 or soluble glycocalicin (GPIbα; each 10 μg/ml), respectively. Data are mean ± SD (n = 3) of a typical experiment; similar results were observed in at least three separate experiments. (B) Adhesion of human neutrophils to surface-adherent platelets from a patient with Bernard-Soulier-Syndrome was studied in the absence (−) or presence of mAb against Mac-1, GPIbα, or JAM-3 (10 μg/ml), or of purified JAM-3 (10 μg/ml). The number of adherent cells is expressed as percent of total added cells. Data are mean ± SD (n = 3) of a typical experiment; similar results were observed in 2 separate experiments; *: P < 0.01 compared with control; ns, not significant.
Table I.
JAM-3 Mediates Platelet–Leukocyte Interactions
| Additives | Cell adhesion (% adherent cells) |
|---|---|
| None | 53.2 ± 1.8 |
| Anti–Mac-1 | 5.2 ± 1.2a |
| Anti-p150.95 | 48.4 ± 2.3 |
| Anti-JAM-3 | 24.9 ± 2.7a |
| Anti-GPIbα | 27.1 ± 1.9a |
| Anti–JEM-3 + anti-GPIbα | 5.8 ± 0.9a |
| JAM-3 | 4.8 ± 0.9a |
| GPIbα | 8.2 ± 1.7a |
FMLP-stimulated adhesion of human neutrophils to surface-adherent platelets was studied in the absence (−) or presence of mAb against Mac-1, p150.95, JAM-3, or GPIbα as well as a mixture of mAbs against JAM-3 and GPIbα (each 10 μg/ml) or in the presence of purified JAM-3 or soluble glycocalicin (GPIbα; each 10 μg/ml), respectively. The number of adherent cells is expressed as percent of total added cells. Data are mean ± SD (n = 3) of a typical experiment; similar results were observed in at least three separate experiments.
P < 0.01 as compared to control.
To further define the contribution of JAM-3 in mediating platelet–leukocyte interactions, the binding of neutrophils or U937 cells to surface-adherent platelets from a patient with Bernard-Soulier-Syndrome (BSS) was assessed. The particular patient has a homozygous Asn45Ser mutation in the GPIX gene leading to only 20% GPIb/IX surface expression compared with normal subjects (unpublished data). Adhesion of neutrophils to BSS-platelets was significantly reduced as compared with normal platelets (Fig. 8 B). In this case the inhibitory capacity of mAb against GPIbα was hardly discernible, whereas the blocking mAb against JAM-3 abolished the Mac-1–dependent adhesion of neutrophils. Also, purified JAM-3 blocked the neutrophil interaction with BSS-platelets. Similar results were obtained with U937 cells (unpublished data). Taken together, these data indicate that JAM-3, by binding to Mac-1, facilitates the heterotypic interaction between leukocytes and platelets. Thus, JAM-3 and GPIbα together serve as the major Mac-1 counterreceptors on the platelet surface.
Relevance of JAM-3 in Platelet–Leukocyte Aggregates in Whole Blood.
To further substantiate the physiological relevance of the interaction between JAM-3 and Mac-1 the ex vivo detection of platelet–neutrophil aggregates from whole blood was performed in a micro-couette both under conditions of low and high shear. The formation of these aggregates was almost completely abolished in the presence of antibody against P-selectin. Soluble glycocalicin reduced the aggregate formation under both conditions of low and high shear rate, whereas purified JAM-3 provided a significant inhibition only under conditions of low shear (Fig. 9) .
Figure 9.

Role of JAM-3 in platelet–neutrophil aggregate formation in whole blood. The platelet–neutrophil adhesion in whole blood stimulated with the stable thromboxane A2 mimetic U46619 (5 μM) is shown in the absence or presence of purified JAM-3 (20 μg/ml), soluble glycocalicin (20 μg/ml), or Fab-fragments of blocking mAb GA6* against P-selectin (PS; 10 μg/ml). Adhesion experiments were performed in a micro-couette at low (20 s−1) and high (2,000 s−1) shear rates. The data represent mean ± SD of four independent experiments.
Discussion
Leukocyte recruitment to the site of inflammation or infection depends on the interactions of these mobile cells with the vessel wall, reactions which are largely controlled by the degree and duration of adhesive contacts of the involved cell types. In particular, leukocytes interact with the intact endothelium or with deposited platelets and fibrin at sites of vascular injury after denudation of the endothelial cell lining. Upon certain pathological conditions leukocytes and platelets colocalize in the vessel wall (4–6, 37). A sequential adhesion model of leukocyte attachment to and transmigration across surface-adherent platelets has been proposed, including P-selectin-mediated initial rolling (7) and the subsequent Mac-1–dependent firm adhesion and transplatelet migration (38–40). However, the potential platelet counterreceptors for Mac-1 are only partially identified. In this study, a third member of the family of junctional adhesion molecules, JAM-3, was characterized as a novel counterreceptor on platelets for leukocyte Mac-1. Together with GPIbα, JAM-3 serves as the major heterotypic adhesion receptor responsible for cell-to-cell adhesion between platelets and leukocytes.
In particular, two mAb Gi11 and Gi13 had been raised against a 43-kD glycoprotein on human platelets and these tools allowed the identification of a full-length cDNA encoding a 279-amino acid mature protein. This type I integral membrane glycoprotein reveals two domains with intrachain disulfide bonds at the extracellular region, typical for Ig-loops of the immunoglobulin superfamily and is identical with the very recently described human JAM-3 (34). Multiple sequence alignments of the protein revealed the highest amino acid identity (86%) with murine JAM-2 (41), whereas the homology between murine JAM-3 and human JAM-2 was 79%. Thus, murine JAM-2 represents the mouse counterpart of human JAM-3 and vice versa, murine JAM-3 is the mouse counterpart of human JAM-2. The analysis of different peripheral blood cell populations and cell lines indicated that JAM-3 is exclusively expressed on platelets or megakaryoblastic cell lines.
A possible engagement of JAM-3 in homophilic cellular interactions could hardly be identified, as opposed to the properties of two previously described members of the JAM family (17, 23). However, the following features of JAM-3 are consistent with a specific heterophilic interaction with leukocyte β2-integrin Mac-1: (a) Myelomonocytic U937 cells adhered to immobilized JAM-3 or JAM-3 transfected cells and this interaction was attributed to Mac-1. (b) Analyzing adhesion of K562 cells transfected with different β2-integrins to either immobilized JAM-3 or JAM-3-transfected cells further established that JAM-3 interacts with Mac-1 but not with LFA-1. K562 cells bearing a third β2-integrin, p150.95, also interacted with JAM-3, which is not surprising as p150.95 and Mac-1 share many similarities in ligand binding (42). Although U937 cells and neutrophils express p150.95 integrin, the adhesion of U937 cells to immobilized JAM-3 (Fig. 5), the adhesion of neutrophils to purified JAM-3 (unpublished data), or the adhesion of neutrophils to surface-adherent platelets (Table I) was not affected by a blocking mAb against p150.95. Similarly, anti-p150.95 did not impair adhesion of U937 cells to fibrinogen (Fig. 5). These observations indicate that p150.95 is not sufficient to mediate adhesion to JAM-3 when coexpressed with Mac-1. Thus, Mac-1 serves as the predominant counter-receptor of JAM-3. (c) In a purified system, a direct binding between JAM-3 and Mac-1 was observed. JAM-3 was particularly found to recognize the I-domain of Mac-1 (unpublished data) and JAM-3 and fibrinogen competed with each other for binding to Mac-1, suggesting overlapping binding sites on the β2-integrin. Conversely, the exact binding epitope(s) for Mac-1 on JAM-3 remain to be identified. (d) A major consequence for JAM-3 binding to Mac-1 is a firm platelet–leukocyte interaction. In this respect, antibodies to both JAM-3 and GPIbα, each partially inhibited the interaction of human neutrophils with surface-adherent platelets, whereas a combination of both antibodies totally abolished the cell–cell interaction. In addition, either purified JAM-3 or GPIbα alone almost completely inhibited the platelet-leukocyte interaction, probably because each ligand may block binding sites on Mac-1 for both JAM-3 and GPIbα. Furthermore, neutrophil interaction with platelets derived from a BSS-patient was markedly reduced, compared with normal platelets, and interestingly, neutrophil adhesion to BSS-platelets was almost completely blocked by the antibody against JAM-3. Thus, under our experimental conditions, JAM-3 and GPIbα are the major mediators of neutrophil-platelet adhesive contacts. (e) The physiological relevance of our findings was substantiated by identifying a role for JAM-3 in mediating platelet–leukocyte aggregates in whole blood. Whereas soluble glycocalicin blocked aggregate formation under both low and high shear rate, purified JAM-3 was effective only under low shear rate. Hence, JAM-3 mediates adhesion of neutrophils to platelets under low shear conditions; i.e., it might strengthen the neutrophil-platelet interaction already mediated by other Mac-1 ligands on platelets. However, a potential contribution of JAM-3 in the accumulation of neutrophils to surface-adherent platelets under physiological flow conditions cannot be excluded from the data presented here.
Although we found no participation of other adhesion–receptor systems in firm adhesion of neutrophils to platelets, the possibility of additional Mac-1–dependent and –independent binding sites on platelets cannot be ruled out. Other potential Mac-1 ligands present on the platelet membrane include fibrinogen (bound to GPIIb-IIIa; reference 10), ICAM-2 (43), high molecular weight kininogen (30, 44), or glycosaminoglycans (45). In particular, the functional relevance of fibrinogen and ICAM-2 as Mac-1 counterreceptors appears to increase with higher shear (46). Further detailed functional analysis of the contribution of each platelet associated Mac-1 ligand in platelet/neutrophil interactions under different shear conditions will provide clarification in this respect. Interactions contributing to Mac-1–independent leukocyte–platelet aggregate formation include thrombospondin bridging between GPIV receptors on platelets and monocytes (47) or binding of P-selectin on activated platelets to leukocyte PSGL-1 (48, 49).
In addition to mediating the accumulation of leukocytes at sites of platelet deposition within regions of the injured vasculature, the juxtacrine interaction between neutrophils and platelets can induce upregulation of neutrophil adhesion molecules (50) or promote integrin activation (39), chemokine synthesis (51, 52), and induction of the respiratory burst (50). Interestingly, both neutrophil–platelet and monocyte–platelet aggregates have been identified in the peripheral blood of patients with coronary artery disease and may be markers of disease activity (50, 53). Thus, the possibility that the described interaction between JAM-3 and Mac-1 contributes to such processes is intriguing and is currently under investigation.
Recently, evidence is accumulating that JAM family members possess important features for cellular communications between endothelium and leukocytes during the inflammatory process. For example, JAM-1 was shown to facilitate the transmigration of neutrophils and monocytes across the vessel wall (12, 13, 36). JAM-1 is also proposed to modulate paracellular permeability (14, 20), as it associates with the tight junction components ZO-1 and AF-6 via their PDZ domains (54, 55). As the PDZ binding motif is conserved in JAM-3, future investigations will focus on a putative role for JAM-3 in endothelial barrier function. In contrast to JAM-1, which has already been implicated in single patients with immune thrombocytopenia, JAM-3 does not represent a major target for platelet antibodies as almost 1,000 patients with various forms of immune thrombocytopenia were negative for JAM-3–reactive antibodies (unpublished data).
Due to the fact that JAM-1 has been shown to undergo homophilic binding interactions, the propensity of dimerization (or multimerization) has been suggested. However, a recent report points to the role of JAM-1 also in heterophilic interactions by demonstrating its binding to the β2-integrin LFA-1 (36). The observation that JAM-1 and JAM-3 can independently function as counterreceptors for the β2-integrins LFA-1 and Mac-1, respectively, provides a completely new perspective in understanding alternative processes responsible for the (patho-)physiology of inflammatory cell recruitment.
Taken together, this study reports the characterization of a novel member of the JAM family, JAM-3 from human platelets, which constitutes a specific counterreceptor for leukocyte Mac-1 and thereby contributes as a major adhesion molecule in the platelet–leukocyte interaction. Future studies will also aim to further characterize the molecular anatomy of the JAM-3/Mac-1 interaction and to identify novel strategies that could disrupt platelet–leukocyte complexes in order to device therapeutic interventions in atherothrombosis and postangioplasty restenosis.
Acknowledgments
We appreciate the excellent technical assistance of O. Eva, S. Werth, H. Berghöfer, A. Giptner, H.G. Welker, and T. Schmidt-Wöll. We are grateful to Dr. D. Linder for performing the amino acid sequence analysis, and Dr. S.M. Kanse for helpful discussions. The authors thank Dr. A. Matzdorff, who kindly referred the BSS case to us and also to the members of the BSS family for their cooperation.
This work was supported by a grant (SFB 547) of the Deutsche Forschungsgemeinschaft (Bonn, Germany) to S. Santoso and a grant from the Novartis Foundation for Therapeutic and Clinical Research (Nürnberg, Germany) to K.T. Preissner and T. Chavakis.
Footnotes
Abbreviations used in this paper: BSS, Bernard-Soulier-Syndrome; GP, glycoprotein; ICAM, intercellular adhesion molecule; JAM, junctional adhesion molecule; RACE, rapid amplifications of 3′ and 5′ cDNA ends.
References
- 1.Springer, T.A. 1994. Traffic signals for lymphocyte recirculation and leukocyte emigration: a multistep paradigm. Cell. 76:301–314. [DOI] [PubMed] [Google Scholar]
- 2.Smith, C.W., S.D. Marlin, R. Rothlein, C. Toman, and D.C. Anderson. 1989. Cooperative interactions of LFA-1 and Mac-1 with intercellular adhesion molecule-1 in facilitating adherence and transendothelial migration of human neutrophils in vitro. J. Clin. Invest. 83:2008–2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Languino, L.R., A. Duperray, K.J. Joganic, M. Fornaro, G.B. Thornton, and D.C. Altieri. 1995. Regulation of leukocyte-endothelial interactions and leukocyte transendothelial migration by intercellular adhesion molecule 1-fibrinogen recognition. Proc. Natl. Acad. Sci. USA. 92:7734–7738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marcus, A.J. 1994. Thrombosis and inflammation as multicellular processes: significance of cell-cell interactions. Semin. Hematol. 31:261–269. [PubMed] [Google Scholar]
- 5.Rinder, C.S., J.L. Bonan, H.M. Rinder, J. Matthew, R. Hines, and B.R. Smith. 1992. Cardiopulmonary bypass induces leukocyte-platelet adhesion. Blood. 79:1201–1205. [PubMed] [Google Scholar]
- 6.Ross, R. 1999. Atherosclerosis-an inflammatory disease. N. Engl. J. Med. 340:115–126. [DOI] [PubMed] [Google Scholar]
- 7.McEver, R.P. 2001. Adhesive interactions of leukocytes, platelets and the vessel wall during hemostasis and inflammation. Thromb. Haemost. 86:746–756. [PubMed] [Google Scholar]
- 8.Simon, D.I., C.P. Chen, H. Xu, C.Q. Li, J.F. Dong, L.V. McIntire, C.M. Ballantyne, L. Zhang, M.I. Furman, M.C. Berndt, and J.A. Lopez. 2000. Platelet glycoprotein Ibα is a counterreceptor for the leukocyte integrin Mac-1 (CD11b/CD18). J. Exp. Med. 192:193–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Diacovo, T.G., A.R. de Fougerolles, D.F. Bainton, and T.A. Springer. 1994. A functional integrin ligand on the surface of platelets: intercellular adhesion molecule-2. J. Clin. Invest. 94:1243–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weber, C., and T.A. Springer. 1997. Neutrophil accumulation on activated, surface-adherent platelets in flow is mediated by interaction of Mac-1 with fibrinogen bound to αIIbβ3 and stimulated by platelet-activating factor. J. Clin. Invest. 100:2085–2093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lampugnani, M.G., and E. Dejana. 1997. Interendothelial junctions: structure, signalling and functional roles. Curr. Opin. Cell Biol. 9:674–682. [DOI] [PubMed] [Google Scholar]
- 12.Martin-Padura, I., S. Lostaglio, M. Schneemann, L. Williams, M. Romano, P. Fruscella, C. Panzeri, A. Stoppacciaro, L. Ruco, A. Villa, et al. 1998. Junctional adhesion molecule, a novel member of the immunoglobulin superfamily that distributes at intercellular junctions and modulates monocyte transmigration. J. Cell Biol. 142:117–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Del Maschio, A., A. De Luigi, I. Martin-Padura, M. Brockhaus, T. Bartfai, P. Fruscella, L. Adorini, G. Martino, R. Furlan, M.G. De Simoni, and E. Dejana. 1999. Leukocyte recruitment in the cerebrospinal fluid of mice with experimental meningitis is inhibited by an antibody to junctional adhesion molecule (JAM). J. Exp. Med. 190:1351–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ozaki, H., K. Ishii, H. Horiuchi, H. Arai, T. Kawamoto, K. Okawa, A. Iwamatsu, and T. Kita. 1999. Cutting edge: Combined treatment of TNF-α and IFN-γ causes redistribution of junctional adhesion molecule in human endothelial cells. J. Immunol. 163:553–557. [PubMed] [Google Scholar]
- 15.Naik, U.P., Y.H. Ehrlich, and E. Kornecki. 1995. Mechanism of platelet activation by a stimulatory antibody: cross linking of a novel platelet receptor for mab F11 with the FcγR11 receptor. Biochem. J. 310:155–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sobocka, M.B., T. Sobocki, P. Banerjee, C. Weiss, J.I. Rushbrook, A.J. Norin, J. Hartwig, M.O. Salifu, M.S. Markell, A. Babinska, et al. 2000. Cloning of the human platelet F11 receptor: a cell adhesion molecule member of the immunoglobulin superfamily involved in platelet aggregation. Blood. 95:2600–2609. [PubMed] [Google Scholar]
- 17.Naik, U.P., M.U. Naik, K. Eckfeld, P. Martin-Deleon, and J. Spychala. 2001. Characterization and chromosomal localization of JAM-1, a platelet receptor for a stimulatory monoclonal antibody. J. Cell Sci. 114:539–547. [DOI] [PubMed] [Google Scholar]
- 18.Malergue, F., F. Galland, F. Martin, P. Mansuelle, M. Aurrand-Lions, and P. Naquet. 1998. A novel immunoglobulin superfamily junctional adhesion molecule expressed by antigen presenting cells, endothelial cells and platelets. Mol. Immunol. 35:1111–1119. [DOI] [PubMed] [Google Scholar]
- 19.Williams, L.A., I. Martin-Padura, E. Dejana, N. Hogg, and D.L. Simmons. 1999. Identification and characterisation of human junctional adhesion molecule (JAM). Mol. Immunol. 36:1175–1188. [DOI] [PubMed] [Google Scholar]
- 20.Liu, Y., A. Nusrat, F.J. Schnell, T.A. Reaves, S. Walsh, M. Pochet, and C.A. Parkos. 2000. Human junction adhesion molecule regulates tight junction resealing in epithelia. J. Cell Sci. 113:2363–2374. [DOI] [PubMed] [Google Scholar]
- 21.Barto, E.S., J.C. Forrest, J.L. Connoly, J.D. Chappell, Y. Liu, F.J. Schnell, A. Nusrat, C.A. Parkos, and T.S. Dermody. 2001. Junctional adhesion molecule is a receptor for reovirus. Cell. 104:441–451. [DOI] [PubMed] [Google Scholar]
- 22.Palmeri, D., A. van Zante, C.C. Huang, S. Hemmerich, and S.D. Rosen. 2000. Vascular endothelial junction-associated molecule, a novel member of the immunoglobulin superfamily, is localized to intercellular boundaries of endothelial cells. J. Biol. Chem. 275:19139–19145. [DOI] [PubMed] [Google Scholar]
- 23.Cunningham, S.A., M.P. Arrate, J.M. Rodriguez, R.J. Bjercke, P. Vanderslice, A.P. Morris, and T.A. Brock. 2000. A novel protein with homology to the junctional adhesion molecule. Characterization of leukocyte interactions. J. Biol. Chem. 275:34570–34756. [DOI] [PubMed] [Google Scholar]
- 24.Santoso, S., J. Lohmeyer, H. Rennich, K.J. Clemetson, and C. Mueller-Eckhardt. 1984. Platelet surface antigens: analysis by monoclonal antibodies. Blut. 48:161–170. [DOI] [PubMed] [Google Scholar]
- 25.Santoso, S., R. Kalb, M. Walka, V. Kiefel, C. Mueller-Eckhardt, and P.J. Newman. 1993. The human platelet alloantigens Bra and Brb are associated with a single amino acid polymorphism on glycoprotein Ia (integrin subunit α2). J. Clin. Invest. 92:2427–2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marx, N., F.J. Neumann, D. Zohlnhofer, T. Dickfeld, A. Fischer, S. Heimerl, and A. Schomig. 1998. Enhancement of monocyte procoagulant activity by adhesion on vascular smooth muscle cells and intercellular adhesion molecule-1-transfected Chinese hamster ovary cells. Circulation. 98:906–911. [DOI] [PubMed] [Google Scholar]
- 27.Santoso, S., J. Amrhein, H.A. Hofmann, U.J.H. Sachs, M.M. Walka, H. Kroll, and V. Kiefel. 1999. A point mutation Thr799Met on the α2 integrin leads to the formation of new human platelet alloantigen Sita and affects collagen-induced aggregation. Blood. 94:4103–4111. [PubMed] [Google Scholar]
- 28.Hölschermann, H., C. Rascher, C. Oelschläger, G. Stapfer, A. Langenstein, A. Staubitz, U. Maus, H. Tillmans, H. Bang, and W. Haberbosch. 2001. Opposite regulation of tissue factor expression by calcineurin in monocytes and endothelial cells. J. Immunol. 1666:7112–7120. [DOI] [PubMed] [Google Scholar]
- 29.Chavakis, T., S.M. Kanse, F. Lupu, H.P. Hammes, W. Muller-Esterl, R.A. Pixley, R.W. Colman, and K.T. Preissner. 2000. Different mechanisms define the antiadhesive function of high molecular weight kininogen in integrin- and urokinase receptor dependent interactions. Blood. 96:514–522. [PubMed] [Google Scholar]
- 30.Chavakis, T., S.M. Kanse, R.A. Pixley, A.E. May, I. Isordia-Salas, R.W. Colman, and K.T. Preissner. 2001. Regulation of leukocyte recruitment by polypeptides derived from high molecular weight kininogen. FASEB J. 15:2365–2376. [DOI] [PubMed] [Google Scholar]
- 31.Xia, Z., and M.M. Frojmovic. 1994. Aggregation efficiency of activated normal and fixed platelets in a simple shear field: Effect of shear and fibrinogen occupancy. Biophys. J. 66:2190–2201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ruf, A., M. Pick, V. Deutsch, A. Goldfarb, E.A. Rachmilewitz, A. Bezeaud, M.C. Gullin, and A. Eldor. 1997. The procoagulant activity of red blood cells (RBC) from patients with β thalassemia, determined by annexin V binding, correlates with “in-vivo” platelet activation. Br. J. Haematol. 98:51–56. [DOI] [PubMed] [Google Scholar]
- 33.Kasirer-Friede, A., and M.M. Frojmovic. 1998. Ristocetin- and thrombin-induced platelet aggregation at physiological shear rates: differential roles for GPIb and GPIIb-IIIa receptor. Thromb. Haemost. 80:428–436. [PubMed] [Google Scholar]
- 34.Arrate, M.P., J.M. Rodriguez, T.M. Tran, T.A. Brock, and S.A. Cunningham. 2001. Cloning of human junctional adhesion molecule 3 (JAM3) and its identification as the JAM2 counter-receptor. J. Biol. Chem. 276:45826–45832. [DOI] [PubMed] [Google Scholar]
- 35.Bettaieb, A., M. Titeaux, P. Fromont, W. Vainchenker, and P. Bierling. 1995. Characterization of unclustered workshop platelet panel mAb. Leukocyte Typing V. White Cell Differentiation Antigens. S.F. Schlossmann, L. Boumsell, W. Gilks, J.M. Harlan, T. Kishimoto, C. Morimoto, J. Rita, S. Shaw, R. Silverstein, T. Springer, T.F. Tedder, and R.F. Todd, editors. Oxford University Press, Oxford. 1220–1222.
- 36.Ostermann, G., K.S. Weber, A. Zernecke, A. Schroeder, and C. Weber. 2002. JAM-1 is a ligand of the beta-2 integrin LFA-1 involved in transendothelial migration of leukocytes. Nat. Immunol. 3:151–158. [DOI] [PubMed] [Google Scholar]
- 37.Rogers, C., E.R. Edelman, and D.I. Simon. 1998. A monoclonal antibody to the β2-leukocyte integrin Mac-1 (CD11b/CD18) reduces intimal thickening after angioplasty or stent implantation in rabbits. Proc. Natl. Acad. Sci. USA. 95:10134–10139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Diacovo, T.G., S.J. Roth, J.M. Buccola, D.F. Bainton, and T.A. Springer. 1996. Neutrophil rolling, arrest, and transmigration across activated, surface adherent platelets via sequential of P-selectin and the beta 2-integrin CD11b/CD18. Blood. 88:146–157. [PubMed] [Google Scholar]
- 39.Evangelista, V., S. Manarini, S. Rotondo, N. Martelli, R. Polischuk, J.L. McGregor, G. de Gaetano, and C. Cerletti. 1996. Platelet/polymorphonuclear leukocyte interaction in dynamic conditions: evidence of adhesion cascade and cross talk between P-selectin and the β2-integrin CD11b/CD18. Blood. 88:4183–4194. [PubMed] [Google Scholar]
- 40.Kuijper, P.H.M., H.I. Gallardo Torres, J.-W.J. Lammers, J.J. Sixma, L. Koendermann, and J.J. Zwanginga. 1997. Platelet and fibrin deposition at the damaged vessel wall: cooperative substrates for neutrophil adhesion under flow conditions. Blood. 89:166–175. [PubMed] [Google Scholar]
- 41.Aurrand-Lions, M., C. Johnson-Leger, C. Wong, L. Du Pasquier, and B.A. Imhof. 2001. Heterogeneity of endothelial junctions is reflected by differential expression and specific cellular localization of the three JAM family members. Blood. 98:3699–3707. [DOI] [PubMed] [Google Scholar]
- 42.Isacke, C.M., and M.A. Horton. 2000. The Adhesion Molecule. Facts Book. Academic Press, London. 152 pp.
- 43.Xie, J., R. Li, P. Kotovuori, C. Vermont-Desroches, J. Wijdenes, M.A. Arnout, P. Nortamo, and C.G. Gahmberg. 1995. Intercellular adhesion molecule-2 (CD102) binds to the leukocyte integrin CD11b/CD18 through the A domain. J. Immunol. 155:3619–3628. [PubMed] [Google Scholar]
- 44.Gustafson, E.J., A.H. Schmaier, Y.T. Wachtfogel, N. Kaufman, U. Kucich, and R.W. Colman. 1989. Human neutrophils contain and bind molecular weight kininogen. J. Clin. Invest. 84:28–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Diamond, M.S., R. Alon, C.A. Parkos, M.T. Quinn, and T.A. Springer. 1995. Heparin is an adhesive ligand for the leukocyte integrin Mac-1 (CD11b/CD18). J. Cell Biol. 130:1473–1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kuijper, P.H., H.I. Gallardo Tores, J.W. Lammers, J.J. Sixma, L. Koenderman, and J.J. Zwanginga. 1998. Platelet associated fibrinogen and ICAM-2 induce firm adhesion of neutrophils under flow conditions. Thromb. Haemost. 80:443–448. [PubMed] [Google Scholar]
- 47.Silverstein, R., A.S. Asch, and R.L. Nachman. 1989. Glycoprotein IV mediates thrombospondin-dependent platelet-monocyte and platelet-U937 cell adhesion. J. Clin. Invest. 84:546–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Skinner, M.P., C.M. Lucas, G.F. Burns, C.N. Chesterman, and M.C. Berndt. 1991. GMP-140 binds to neutrophils is inhibited by sulphated glycans. J. Biol. Chem. 266:5371–5374. [PubMed] [Google Scholar]
- 49.Moore, K.L., A. Varki, and R.P. McEver. 1991. GMP-140 binds to a glycoprotein receptor on human neutrophils: evidence for a lectin-like interaction. J. Cell Biol. 112:491–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ott, I., F.-J. Neumann, M. Gawaz, M. Schmitt, and A. Schomig. 1996. Increased neutrophil-platelet adhesion in patients with unstable angina. Circulation. 94:1239–1246. [DOI] [PubMed] [Google Scholar]
- 51.Weyrich, A.S., T.M. McIntyre, R.P. McEver, S.M. Prescott, and G.A. Zimmermann. 1995. Monocyte tethering by P-selectin regulates monocyte chemotatic protein-1 and tumor necrosis factor-α-secretion. J. Clin. Invest. 95:2297–2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Weyrich, A.S., M.R. Elstad, R.P. McEver, T.M. McIntyre, K.L. Moore, J.H. Morrissey, S.M. Prescott, and G.A. Zimmermann. 1996. Activated platelets signal chemokine synthesis by human monocytes. J. Clin. Invest. 97:1525–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Furman, M.I., S.E. Benoit, M.R. Barnard, C.R. Valeri, M.L. Borbone, R.C. Becker, H.B. Hechtman, and A.D. Michelson. 1998. Increased platelet reactivity and circulating monocyte-platelet aggregates in patients with stable coronary artery disease. J. Am. Coll. Cardiol. 31:352–358. [DOI] [PubMed] [Google Scholar]
- 54.Bazzoni, G., O.M. Martinez-Estrada, F. Orsenigo, M. Cordenonsi, S. Citi, and E. Decana. 2000. Interaction of junctional adhesion molecule with the tight junction components ZO-1, cingulin, and occluding. J. Biol. Chem. 275:20520–20526. [DOI] [PubMed] [Google Scholar]
- 55.Ebnet, K., C.U. Schulz, M.K. Meyer zu Brickwedel, G.G. Pendl, and D. Vestweber. 2000. Junctional adhesion molecule interacts with PDZ domain-containing proteins AF-6 and ZO-1. J. Biol. Chem. 275:27979–27988. [DOI] [PubMed] [Google Scholar]