

Abstract
The major histocompatibility complex (MHC)-dependent presentation of processed tissue-specific self-antigens can contribute to either peripheral (extrathymic) tolerance or the differentiation of autoreactive T cells. Here, we have studied the MHC class II molecule presentation of gastric parietal cell (PC)-specific H+/K+-ATPase, which induces a destructive autoimmune gastritis in BALB/c mice lacking CD4+ CD25+ regulatory T cells. Immunofluorescence microscopy showed physical association of CD11c+ dendritic cells (DCs) with PCs in the gastric mucosa. H+/K+-ATPase protein was found within vesicular compartments of a few CD11c+ DCs only in the draining gastric lymph node (LN) and these antigen-containing DCs increased markedly in number with the onset of tissue destruction in autoimmune animals. Both CD8αhi and CD8αlo gastric DCs, but not peripheral or mesenteric DCs, showed evidence of constitutive in vivo processing and presentation of H+/K+-ATPase. These data provide direct support for a widely held model of local tissue antigen uptake and trafficking by DCs in normal animals and demonstrate that DCs in the draining LN can present a tissue-specific self-antigen under noninflammatory conditions without fully deleting autoreactive T cells or inducing active autoimmunity.
Keywords: antigen-presenting cells, autoimmune disease, gastritis, autoantigen, CD11c antigen
Introduction
The immune system recognizes and efficiently eliminates invading pathogens without typically causing extensive damage due to effector responses to self-antigens. Such functional discrimination between foreign and self depends in part on processed antigen displayed by thymic presenting cells (APC) whose recognition purges the repertoire of strongly reactive T cells (1, 2). Even so, peripheral mechanisms of tolerance induction are required to eliminate or limit the activation of T cells with specificities for those peripheral self-antigens not available to developing thymocytes (3, 4).
Recent experiments indicate that (CD11c+) dendritic cells (DCs)*that have not been activated by pathogen-related or endogenous inflammatory stimuli can make a major contribution to peripheral tolerance by inducing the deletion of specific T cells (5). Based on these findings, it has been proposed that under nonpathologic conditions, DCs continuously sample self-antigens from normal peripheral tissues and delete the relevant T cells by presenting processed forms of these antigens after migration to the draining LN (6, 7). However, only a few studies have provided direct evidence for such a sequence of events involving normal tissue antigens (for review see reference 7). In addition, reports that nonactivated DCs tend to store proteins that enter the endocytic pathway rather than process them for MHC class II molecule presentation raise some questions about this model in its simplest form (8–10).
A better understanding of the role of DC antigen presentation in self-tolerance and autoimmunity thus requires a clearer definition of the cells that acquire tissue antigens under normal or pathologic conditions and the extent to which they actively process and present these antigens. We have chosen the gastric proton pump H+/K+-ATPase as a model system for the study of these questions. H+/K+-ATPase expression is restricted to gastric parietal cells (PCs) and this protein is a primary target autoantigen for the experimental autoimmune gastritis (AIG) that develops after the thymectomy of BALB/c mice at day 3 of life or after the transfer of CD4+ CD25− T cells to lymphopenic BALB/cnu/nu mice (for review see references 11 and 12).
Here, we report direct visualization of CD11c+ CD8α− DCs as the cells responsible for the uptake of this self-antigen in the stomach, as well as the selective transport to and constitutive presentation of processed H+/K+-ATPase by DCs within the draining gastric LN of healthy animals. Because such antigen-presenting DCs are found in animals still possessing functional autoaggressive T cells, these results suggest that “quiescent” DCs do not always fully deplete the repertoire of T cells reactive with some tissue-specific antigens. This study also provides evidence that tissue destruction by immune cells markedly augments the number of antigen-containing DCs in the draining LN as well as the antigen presentation function of these DCs, providing a possible explanation for disease amplification and epitope spreading (13) during autoimmune processes.
Materials and Methods
Antigen-specific T Cells and Animals.
An OVA 323–339-specific, I-Ad–restricted T cell line was established from DO11.10 transgenic mice by repetitive cycles of antigen stimulation and rest in vitro. The generation of the CD4+ H+/K+-ATPase α chain peptide 630–641-reactive, I-Ad–restricted T cell clone TXA-23 has been previously described (14).
4–6-wk-old BALB/c mice were obtained from the National Cancer Institute. DO11.10 transgenic mice expressing a TCR specific for OVA peptide 323–339 presented by I-Ad (15) were obtained from Taconic. 4–6-wk-old BALB/cnu/nu mice were purchased from Charles River Laboratories, housed in sterile cages, and given autoclaved food and acidified water. TCR transgenic mice expressing the receptor of the TXA-23 clone were generated as previously described (16) and backcrossed eight generations onto the BALB/c background. All animals were maintained in our animal facility and cared for in accordance with institutional guidelines for animal welfare.
Medium, Reagents, and Antibodies.
Cell cultures were performed in RPMI 1640 supplemented with 10% heat-inactivated FCS, 100 U/ml penicillin, 100 μg/ml streptomycin, 2 mM l-glutamine, 10 mM Hepes, 0.1 mM nonessential amino acids, and 1 mM sodium pyruvate (cRPMI 1640; all from Biofluids). mAbs and/or fluorochrome conjugates against CD2 (RM2-5), CD3 (2C11), CD4 (RM4-4), CD8α (Ly-2), CD11b (M1/70), CD11c (HL3), CD19 (1D3), CD40 (3/23), CD45R/B220 (RA3-6B2), CD80 (16-10A1), CD86 (GL-1), MHC class II I-Ad/I-Ed (2G9), and IFN-γ (XMG1.2) were obtained from BD Biosciences. mAb against CD28 was obtained from Harlan and cells producing mAbs against CD11c (N418; reference 17) were obtained from American Type Culture Collection (ATCC). Ab to DEC-205 (NLDC 145) was from Bachem. Mouse mAb 2G11 against the β subunit of the H+/K+-ATPase was provided by J.G. Forte, University of California, Berkeley, CA.
The H+/K+-ATPase α chain peptide (PITAKAIAASVG, amino acid [aa] 630–641) and OVA peptide (ISQAVHAAHAEINEAGR, aa 323–339) were obtained through the NIAID Peptide Synthesis Unit and determined to be free of LPS contamination using a chromogenic Limulus amebocyte lysate test kit from BioWhittaker. Native H+/K+-ATPase protein was prepared from rabbit stomach as previously described (18).
Chloroquine (Sigma-Aldrich) was prepared as a 100-μM stock solution in RPMI 1640.
Induction and Evaluation of AIG.
AIG was induced in BALB/cnu/nu mice as previously described (19) with CD4+ T cells isolated using mouse CD4 subset columns (R&D Systems) from which CD25+ CD4+ T cells were depleted using the VARIOMACS technique (Miltenyi Biotec). Recovered CD4+ CD25− T cells were resuspended in PBS and injected intravenously into BALB/cnu/nu mice (3 − 5 × 106 cells/animal). After 4 wk, the animals were killed and gastric LNs, peripheral LNs, and stomachs were collected. For histological evaluation of gastritis, stomachs were fixed in 4% paraformaldehyde and sectioned. The sections were stained with hematoxylin and eosin (American Histolabs).
Fluorescence Immunohistochemistry.
LNs, spleens, and stomach fragments were frozen in embedding medium (Cryoform; International Equipment Co.). 7-μm frozen sections were cut, air-dried, and fixed in acetone for 10 min at −20°C, and then rehydrated in Tris-buffered saline containing 0.05% Tween 20. After blocking for 30 min with PBS/10% FCS/5% milk powder, the sections were incubated with N-418 (anti-CD11c) hybridoma supernatant (ATCC) followed by incubation with Texas red (TXRD)-conjugated goat anti–Armenian hamster IgG (Jackson ImmunoResearch Laboratories). For double labeling, the sections were first stained as described above and then additionally stained with FITC-labeled Ab or with rat anti–mouse Ab followed by FITC-conjugated mouse anti–rat IgG (Jackson ImmunoResearch Laboratories). After staining, the sections were washed, mounted with ProLong Antifade Kit (Molecular Probes), and allowed to dry overnight. Irrelevant isotype-matched Ab (BD Biosciences) were used as controls to validate the specificity of staining.
Enzymatic Immunohistochemistry.
After blocking endogenous peroxidase in 0.3% H2O2, the sections were incubated with 2G11-FITC or an isotype-matched control, followed by mouse anti–FITC horseradish peroxidase (HRPO)-labeled Ab (Chimicon International Inc.). HRPO localization was revealed using a metal-enhanced diaminobenzidine substrate (Pierce Chemical Co.). Pictures were taken with the bright field setting of the microscope and additionally analyzed using the public domain NIH image program (developed at the United States NIH and available at http://rsb.info.nih.gov/nih-image/). Images were scanned and analyzed as two-dimensional arrays of pixels (picture elements). Pixels are represented by 8-bit unsigned integers, ranging in value from 0–255. 0 pixels are displayed as white and those with a value of 255 as black. Positive staining events were displayed in a surface plot view and in a threshold histogram plot profile. In the threshold histogram plot profile only gray levels are displayed beyond a defined background level that had been set to 0.
Microscopy.
The stained sections were photographed on a Leica compound microscope (Leica Camera AG) with a 10× air objective or a 40×, 63×, or 100× oil immersion objective connected to a digital video camera and analyzed using Openlab software (Improvision Inc.), Adobe Photoshop 6.0 (Adobe Systems), and the public domain NIH image program. For confocal microscopy, images were collected on a Leica TCS-NT/SP confocal microscope with a 40× or 63× oil immersion objective (Leica), zoom4. Fluorochromes were excited using an argon laser at 488 nm for FITC and a krypton laser at 568 nm for TXRD. Detector slits were configured to minimize any cross talk between the channels. Z-stacks (optical sections) of the images were collected with an optical thickness of 0.2 mm. Images were processed using the Leica TCS-NT/SP software (version 1.6.587), Imaris 3.1.1 (Bitplane AG), Adobe Photoshop 6.0 (Adobe Systems), and Canvas 7 Standard Edition (Deneba Systems, Inc.).
Cell Preparation.
Gastric, peripheral (inguinal, brachial, and axillary), and mesenteric LNs were removed and pooled separately. Single cell suspensions were prepared by collagenase/DNase digestion for 30 min at 37°C and washed with PBS/2.5mM EDTA containing 0.2% BSA. CD11c+ DCs were isolated from these suspensions using the VARIOMACS technique and microbeads coated with Ab against CD11c (Miltenyi Biotec). The positively selected CD11c+ DCs were collected and immediately used for additional phenotypic or functional analysis. Alternatively, for the isolation of distinct APC types, cells were first incubated with directly labeled MACS® microbeads against CD19 and both the positively selected CD19+ B cells and the negatively selected cell fraction were collected. On average, 98 ± 0.5% of positively selected cells were B cells as determined by FACS® analysis. CD11c+ DCs were subsequently isolated from the negatively selected fraction as described above and again both the positively selected CD11c+ DCs and the negatively selected cell fraction were collected. On average, 91 ± 2% of positively isolated cells were CD11c+. The remaining negatively selected fraction consisted mainly of T cells as determined by the expression of CD3ɛ on >95% of the cells. Distinct DC subsets were isolated by incubating single cell suspensions with biotinylated mAb against CD2, CD3, and CD19 followed by the incubation with streptavidin MACS® microbeads. The negatively selected cells were additionally incubated with anti-CD8α MACS® microbeads. The positively selected CD8αhi cells were collected and found to consist of >85% CD8αhi CD11c+ DCs. The negatively selected cells were additionally incubated with anti-CD11c MACS® microbeads and this time the positively selected CD8αlo CD11c+ cells were collected. Alternatively, the negatively selected cells were stained with mAb against CD11c and CD8α. CD11c+ CD8αhi and CD11c+ CD8αlo cells were sorted on a FACSVantage™ (Becton Dickinson).
To produce gastric PCs, the stomach was removed and after separating the gastric LN, a single cell suspension was prepared by passing the mucosa through a wire mesh into PBS containing 1 mM EDTA. Cells were washed twice and layered over a 30% Percoll gradient, and then centrifuged for 20 min at 2,000 rpm. Cells at the interface were harvested and washed twice in PBS with 0.2% BSA and 1 mM EDTA. The majority of cells consisted of PCs as confirmed by the intracellular staining of cytospin preparations for H+/K+-ATPase using FITC-2G11.
TXA-23 Activation Assay.
APCs were either left without antigen or incubated for the indicated time with H+/K+-ATPase peptide, OVA peptide, or H+/K+-ATPase protein at the indicated concentrations, or with a PC preparation at a ratio of 1:2 for the indicated time period. TXA-23 or DO.11.10 T cells were then added to APCs at a ratio of 5:1 and the cells were cultured for 16–18 h at 37°C in a humidified atmosphere in the presence of 5% CO2. During the last 4 h of culture, 2 μM monensin was added to the culture. Cells were harvested, washed, and immediately used for additional analysis.
To inhibit antigen processing during cell cultures, DCs were treated with 75 μM chloroquine for 30 min and then incubated for 3 h without the addition of antigen or with 5 μg/ml H+/K+-ATPase peptide, 50 μg/ml native rabbit H+/K+-ATPase protein, or a PC preparation (DC/PC ratio = 1:2) at 37°C. After the incubation, TXA-23 T cells were added and the cells were cocultured for an additional 16–18 h in the continued presence of chloroquine.
In Vivo Proliferation Assay.
Thymocytes from 4-wk-old TXA-23 TCR transgenic female mice were prepared as a single cell suspension in HBSS buffer containing 5% FCS and 10 mM Hepes. The cells were then washed three times in PBS, resuspended to 107 cells/ml, and incubated for 8 min at room temperature in 1 μM carboxyfluorescein succinimidyl ester (CFSE). The incorporation of dye was stopped by incubation in an equal volume of FCS followed by washing in HBSS buffer. 5 × 106 labeled cells were transferred intravenously into BALB/c wild-type females. 3 d later, gastric and mesenteric LNs were removed and single cell suspensions were prepared. Live cells gated on CD4 were analyzed by flow cytometry for the intensity of CFSE staining as an indication of the extent of cell division.
Flow Cytometry.
Cell suspensions were incubated with anti-Fc receptor Ab (2.4G2; BD Biosciences) and stained with the indicated FITC-, PE-, CyChrome-, allophycocyanin-labeled or biotinylated Ab for 15 min at 4°C in PBS containing 2.5 mM EDTA and 0.2% BSA. For staining with biotin-conjugated primary Ab, the cells were washed twice with PBS/EDTA/BSA and subsequently incubated with the second step reagent streptavidin-allophycocyanin. For intracellular cytokine staining, monensin-treated cells were fixed in 4% paraformaldehyde for 5 min at 37°C, washed, and stained in buffer containing 0.1% saponin. Cells were analyzed on a FACSCalibur® cytometer (Becton Dickinson) using CellQuest™ (Becton Dickinson) and FlowJo® (TreeStar) software. Irrelevant isotype-matched Ab (BD Biosciences) were used as controls to validate the specificity of staining.
Online Supplemental Material.
Triple stainings of sections of a gastric LN from a healthy BALB/c mouse are shown in Fig. S1. Sections were stained for both H+/K+-ATPase (green) and CD11c (red) as described above as well as for CD3, CD19, MHC class II, DEC-205, CD11b, or CD8α (blue) using CyChrome 5–conjugated donkey anti–rat IgG (Jackson ImmunoResearch Laboratories) as second step reagent. H+/K+-ATPase–containing CD11c+ DCs were found to reside in the T cell areas (Fig. S1 a, left) but not B cell areas (Fig. S1 a, right) of the gastric LN. H+/K+-ATPase+ CD11c+ DCs expressed MHC class II (Fig. S1 b, first and second panels) molecules and the majority was DEC-205+ (Fig. S1 b, third and fourth panels). H+/K+-ATPase+ CD11c+ DCs could either be CD8α+ (Fig. S1 c, first and second panels) or CD8α− (Fig. S1 c, third and fourth panels) as well as CD11b+ (Fig. S1 d, first and second panels) or CD11b− (Fig. S1 d, third and fourth panels). The online supplemental figure is available at http://www.jem.org/cgi/content/full/20020991/DC1.
Results
H+/K+-ATPase Association with DCs in the Gastric LNs.
DCs have been suggested to continuously transport tissue-specific self-antigens to the draining LNs and present processed forms of these antigens to both CD4+ (20, 21) and CD8+ (22, 23) T cells. Presently, reagents suitable for direct visualization of specific peptide–MHC class I or II combinations are only available for a limited set of determinants (24–29). In the absence of such reagents for the H+/K+-ATPase/I-Ad determinant, we first attempted to identify cells that present the H+/K+-ATPase self-antigen based on the presence of the source antigen itself. For this purpose, we used a mouse mAb (2G11) against the H+/K+-ATPase β subunit (30). Fig. 1 a shows hematoxylin and eosin staining of the gastric mucosa as well as immunofluorescence staining with 2G11 mAb. 2G11 selectively stained cells in the PC layer. No H+/K+-ATPase expression could be detected in sections of the duodenum or the colon (not depicted).
Figure 1.
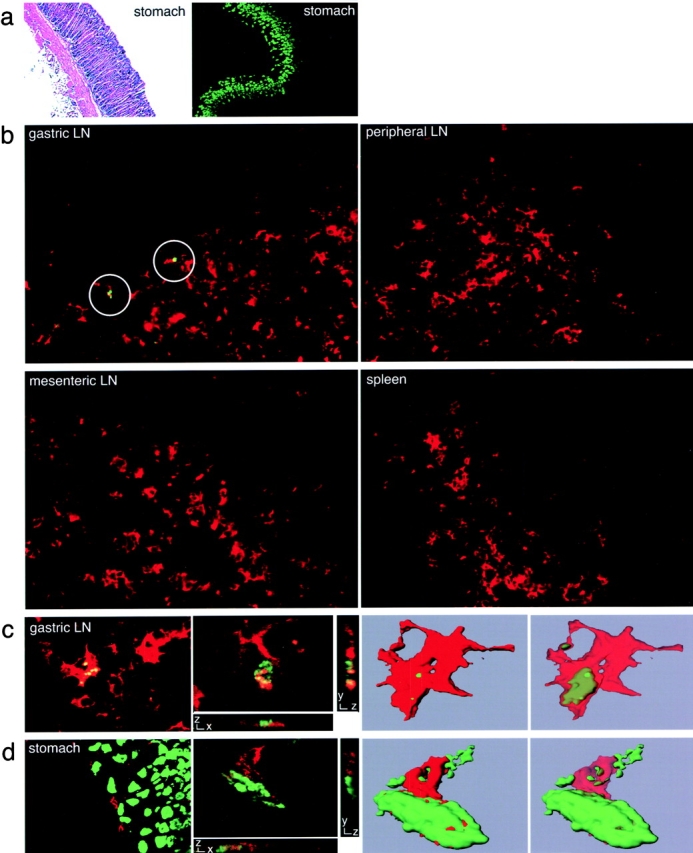
Immunofluorescence staining of H+/K+-ATPase and DCs in gastric LN and stomach sections. (a) Sections of gastric tissue from healthy, untreated BALB/c mice were stained with hematoxylin and eosin (left) or with FITC-2G11, specific for the β subunit of gastric H+/K+-ATPase (right). (b) Sections of gastric LN, peripheral LN, mesenteric LN, and spleen from healthy BALB/c mice were stained with FITC-2G11 (green) with anti–CD11c-TXRD (red). Positive staining with the anti–H+/K+-ATPase mAb was detected in sections of gastric LN (circles). (c) Sections of a gastric LN from a healthy BALB/c mouse stained for both H+/K+-ATPase (green) and CD11c (red) (first panel). Laser scanning confocal microscopy was conducted on a similarly stained gastric LN section. One individual optical section in the z-plane is shown with cross-sectional profiles in the y and x axes (second panel). A 3D reconstruction of the whole cell was produced from these image data (third panel) and the intracellular localization of the H+/K+-ATPase staining (green) was visualized by decreasing the opacity of the CD11c staining (red; fourth panel). (d) A stomach section from a healthy BALB/c mouse stained for both H+/K+-ATPase (green) and CD11c (red; first panel). Laser scanning confocal microscopy was conducted on a similarly stained stomach section. One individual optical section in the z-plane is shown with cross-sectional profiles in the y and x axes (second panel). A 3D reconstruction of the whole cell was produced from these image data, which demonstrates the juxtaposition of H+/K+-ATPase+ PCs (green) and a CD11c+ DCs (red; third panel). By decreasing the opacity of the CD11c staining (red), the intracellular localization of H+/K+-ATPase staining (green) was visualized (fourth panel).
Using the 2G11 mAb, we examined whether H+/K+-ATPase could be detected in the draining gastric LN and whether it was associated with DCs. Localized reactivity was seen in gastric LN sections from untreated animals, but not in sections from mesenteric or peripheral LN or spleen. Although detectable, the frequency of H+/K+-ATPase signals was low in the gastric LN of normal healthy mice (on average 5 to 6 positive staining events were detected in 60 tissue sections). This H+/K+-ATPase reactivity was exclusively associated with CD11c+ DCs (Fig. 1 b) that resided in the T cell areas of the gastric LN (Fig. S1). Their low frequency precluded multiplex staining analysis by flow cytometry (unpublished data). Microscopic analysis, however, did show that these rare antigen-containing cells also expressed MHC class II molecules and that most were DEC-205+. CD8α+ and CD8α− as well as CD11b+ and CD11b− DCs contained the H+/K+-ATPase antigen (Fig. S1). To gain more information about the physical relationship between the H+/K+-ATPase self-antigen and DCs, laser scanning confocal microscopy was performed (Fig. 1 c). This analysis showed an intracellular staining pattern for the H+/K+-ATPase (Fig. 1 c, first and second panels). A 3D reconstruction of the whole cell was created from the image information. By decreasing the opacity of the CD11c surface staining (red), the intracellular cytoplasmic localization of the H+/K+-ATPase staining (green) could be clearly visualized (Fig. 1 c, third and fourth panels).
Physical Contact between DCs and H+/K+-ATPase–expressing PCs in the Stomach.
Gastric LN DCs containing H+/K+-ATPase could acquire this antigen locally in the stomach tissue and subsequently migrate to the draining LN or H+/K+-ATPase could reach afferent lymphatic vessels and be taken up by DCs resident in the draining LN. To address this question, stomach sections were costained with anti–H+/K+-ATPase mAb and mAb against DCs (CD11c). CD11c+ DCs were found between the PC layer and the muscularis mucosa in close association with 2G11+ PCs. These MHC class II+ cells could also be stained with mAb to CD11b but not to CD8α (unpublished data). Laser scanning confocal microscopy provided evidence that DCs in the stomach can acquire H+/K+-ATPase from PCs (Fig. 1 d). Thus, at least some DCs constitutively residing in the stomach tissue appear to acquire the H+/K+-ATPase self-antigen in this organ and subsequently transport it to the draining LN where it can be visualized in intracellular vesicles.
Gastric but Not Peripheral DCs Induce the Activation of TXA-23 T Cells.
The preceding studies provide clear evidence of tissue antigen acquisition by DCs and the selective accumulation of these antigen-containing cells in the draining LN. Whether these DC actually process and present such endocytosed antigen under noninflammatory conditions is an unresolved question (8–10, 31). To address this issue, we took advantage of a CD4+ H+/K+-ATPase–specific T cell clone, TXA-23, which produces Th1 cytokines in response to I-Ad presentation of H+/K+-ATPase α chain peptide aa 630–641 (14). Adopting the method originally used by Crowley et al. (32) to determine which cells presented in vivo administered soluble antigen, the TXA-23 T cell clone was used in in vitro antigen presentation assays with cells isolated from the draining gastric and peripheral LN of healthy BALB/c mice. The activation of TXA-23 cells was assessed by intracellular staining for IFN-γ. As a control, DO11.10 T cells specific for OVA presented by I-Ad were cultured and analyzed in parallel. In the absence of added antigen, gastric but not peripheral DCs induced IFN-γ production by the TXA-23 T cell clone (Fig. 2 a). In contrast, in the absence of added antigen, neither gastric DCs nor peripheral DCs induced IFN-γ production by the DO11.10 T cell line. All DC populations induced IFN-γ production from both T cell lines upon the addition of the appropriate peptide.
Figure 2.

Gastric but not peripheral DCs induce the activation of TXA-23 T cells. (a) Intracellular IFN-γ staining of TXA-23 and DO11.10 cells T cells. TXA-23 and DO11.10 T cells were cultured for 16–18 h alone (no DC) in the presence of isolated gastric or peripheral DCs (+DC), and in the presence of DCs with added H+/K+-ATPase peptide (final concentration 50μg/ml) or OVA peptide (+DC +peptide; final concentration 10μM). For TXA-23 cells, one representative experiment out of ten, and for DO11.10 one out of four, is shown. Markers were set according to isotype-matched negative control staining in this and all similar experiments. (b) The stimulatory capacity of isolated gastric DCs compared with that of peripheral DCs pulsed with graded amounts of H+/K+-ATPase peptide. (c) Surface phenotype of gastric, peripheral, and mesenteric DCs (filled) in comparison to isotype-matched negative control staining (open). IFN-γ responses of TXA-23 cells that were cultured for 16–18 h in the presence of gastric, peripheral, or mesenteric DCs in the absence of added antigen are also shown (one representative experiment out of three).
To estimate the level of H+/K+-ATPase presentation by gastric DCs, we compared the stimulatory capacity of isolated gastric DCs to that of peripheral DCs pulsed with graded amounts of H+/K+-ATPase peptide. The activation of TXA-23 T cells by unpulsed gastric DCs gave an IFN-γ response equal to that seen using peripheral DCs pulsed with 0.2 μM H+/K+-ATPase peptide, which is at least 10 times more than the minimal stimulatory concentration (Fig. 2 b).
LNs draining the airways or the intestine are constantly exposed to environmental antigens and irritants. This might result in greater tonic activation of DCs in LNs draining mucosal sites as compared with DCs (axillary and brachial) draining subcutaneous tissues. To examine whether their state of activation might account for the superior stimulatory capacity of gastric DCs, we examined the surface phenotype of gastric, peripheral, and mesenteric DCs. DCs isolated from mesenteric LNs were included in the analysis because they might resemble gastric LNs in their exposure to chronic stimuli but differ from gastric LNs in their repertoire of tissue-specific self-antigens. As seen in Fig. 2 c, gastric and mesenteric DCs showed higher MHC class II expression than peripheral DCs, whereas similar expression was observed for B7.1/2 molecules and CD40. Nevertheless, only gastric DCs, but not mesenteric or peripheral DCs, were able to induce IFN-γ production by TXA-23 cells in the absence of added antigen.
Gastric DCs Constitutively Process and Present H+/K+-ATPase Antigen In Vivo.
Immature DCs are believed to have a higher capacity for antigen uptake as compared with mature DCs (33–35) but do not form substantial numbers of stable MHC class II–peptide complexes unless they are exposed to inflammatory mediators (8–10, 36). Gastric DCs phenotypically resemble immature/nonactivated DCs (MHC class II+, B71/2lo, and CD40lo) and immunofluorescence analysis of tissue sections revealed the presence of an intracellular pool of serologically recognizable H+/K+-ATPase in some gastric DCs. Therefore, it was important to determine whether the activation of TXA-23 cells by gastric DCs reflected the constitutive in vivo presentation of processed H+/K+-ATPase antigen or whether H+/K+-ATPase protein in intracellular compartments is processed and presented only upon DC activation and/or maturation occurring in culture (37). Fixation of peptide-pulsed gastric DCs with paraformaldehyde before the coculture completely inhibited any TXA-23 activation even using peptide antigen (unpublished data). We therefore analyzed gastric DCs for their capacity to activate TXA-23 T cells in the presence of chloroquine. This drug inhibits acidification and consequently much of the invariant chain and antigen proteolysis that occurs within endocytic vesicles, thereby interfering with the MHC class II–restricted presentation of antigens (38, 39).
TXA-23 cells were stimulated in the presence or absence of chloroquine with either freshly isolated gastric DCs or gastric DCs cultured with H+/K+-ATPase peptide or H+/K+-ATPase protein. Gastric DCs were also exposed to PCs in order to determine whether DCs would also be able to form MHC–peptide complexes from phagocytosed cells, as previously described (31). The concentrations of the H+/K+-ATPase peptide and protein, as well as the gastric DC/PC ratio, were adjusted to generate comparable numbers of IFN-γ+ TXA-23 cells in the absence of drug (Fig. 3 a, top row). Chloroquine failed to inhibit the activation of TXA-23 T cells by gastric DCs used in the absence of any exogenous antigen or when pulsed with peptide antigen, but it effectively inhibited the incremental activation of TXA-23 T cells seen using protein- or PC-pulsed gastric DCs (Fig. 3 a, bottom row).
Figure 3.

Gastric DCs constitutively present H+/K+-ATPase antigen. (a) TXA-23 cells were cultured for 16–18 h in the presence of gastric DCs in the absence (DC alone) or presence of H+/K+-ATPase peptide (DC+peptide; 5 μg/ml), H+/K+-ATPase protein (DC+protein; 50 μg/ml), or a preparation of PCs (DC+PCs; DC/PC ratio = 1:2). Cultures were performed either in the absence (top) or presence (bottom) of chloroquine (final concentration 75 μM). Primary data are shown as dot plots of IFN-γ intracellular staining. One representative experiment out of two is shown. (b) Thymocytes expressing the TXA-23 TCR were labeled with CFSE and transferred into BALB/c mice. After 3 d the transferred transgenic T cells in the gastric and peripheral LN were analyzed for evidence of cell division based on dilution of the CFSE label. One representative experiment out of four is shown.
Because it is difficult to exclude that post-isolation maturation of isolated DCs might contribute to this evidence for processed antigen display even in the presence of chloroquine, we also used an in vivo approach to examine this issue. Naive T cells from a transgenic animal expressing the TXA-23 TCR (16) were labeled with CFSE and transferred into normal BALB/c mice. 3 d later, the gastric and peripheral LNs were removed and the adoptively transferred T cells they contained were analyzed for evidence of cell division based on dilution of the CFSE label. Very few cells showed evidence of such proliferation in the peripheral LN, but extensive division was seen among these transgenic cells in the draining gastric LN (Fig. 3 b). These results are consistent with the staining data showing H+/K+-ATPase only in DCs of the gastric LN and the evidence from in vitro experiments that this protein is actively processed by these gastric DCs in vivo. These findings indicate that H+/K+-ATPase antigen is constitutively processed and presented by gastric DCs in vivo in noninflamed conditions.
CD8αhi and CD8αlo Gastric DCs Are Capable of TXA-23 T Cell Activation.
Distinct populations of DCs can be distinguished according to the expression of surface molecules, including CD8α (40–42). CD8α+ and CD8α− DCs have been suggested to play distinct roles in the induction of tolerance versus immunity, respectively (43, 44). To determine whether this might be due to the selective presentation of self-antigens by one or the other DC subset, we performed a sequential isolation of CD8αhi and CD8αlo subsets from gastric DCs and analyzed both subsets separately for their capacity to induce the activation of TXA-23 T cells. Even though only CD8α− DCs were found to be associated with PCs in the gastric mucosa, both CD8αhi and CD8αlo DC populations from the gastric LN induced similar activation of the TXA-23 clone in the absence of added antigen (Fig. 4 a).
Figure 4.
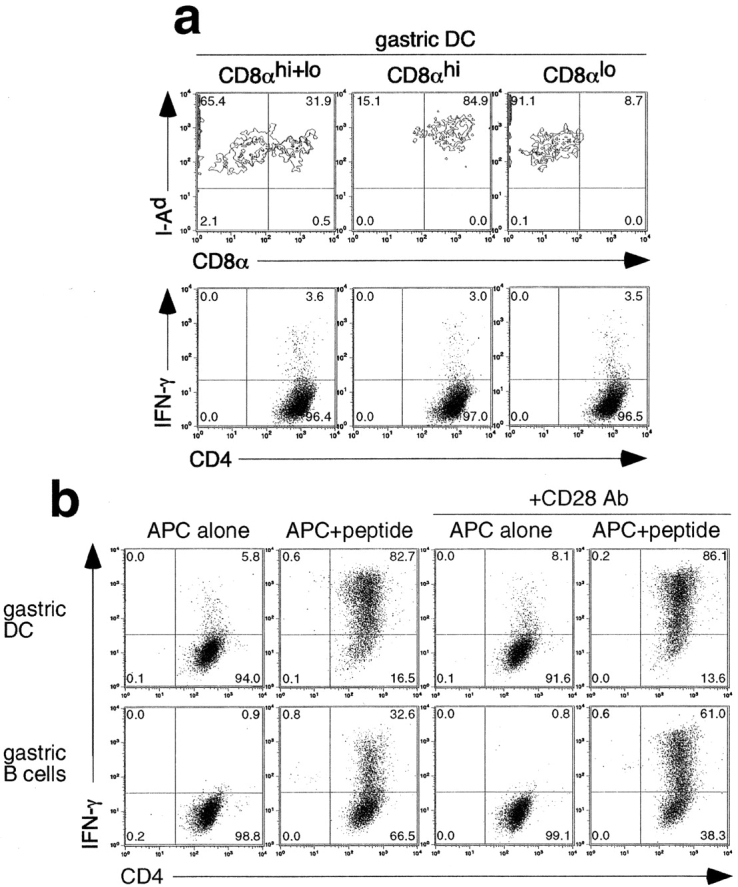
Phenotype and H+/K+-ATPase presenting function of different cell populations. (a) Staining of isolated CD11c+ DCs and of isolated CD11c+ CD8αhi and CD11c+ CD8αlo subpopulations for MHC class II molecule (I-Ad) and CD8α expression (top). TXA-23 cells were cultured for 16–18 h in the presence of these CD11c+ CD8αhi, CD11c+ CD8αlo, or unseparated CD11c+ DCs in the absence of added antigen. The dot plots show intracellular IFN-γ production by TXA-23 cells (bottom). One representative experiment out of three is shown. (b) TXA-23 cells were cultured for 16–18 h in the presence of gastric LN DCs (gastric DC) or gastric LN B cells (gastric B cells) either in the absence (APC alone) or presence (APC+peptide) of specific H+/K+-ATPase peptide (50 μg/ml). The dot plots show intracellular IFN-γ production by TXA-23 cells. Anti-CD28 mAb (final concentration 10 μg/ml) was added to cocultures of TXA-23 cells with either gastric LN DCs (gastric DC) or gastric LN B cells (gastric B) in the absence or presence of H+/K+-ATPase peptide (50 μg/ml). One representative experiment out of three is shown.
DCs Are the only APC in the Gastric LN Able to Activate TXA-23 Cells.
Previous studies have shown that aside from DCs, B cells efficiently present circulating protein in LN draining sites of inflammation (45) and even under noninflammatory conditions, B cells as well as DCs show MHC class II–dependent antigen presentation of determinants derived from soluble proteins present in extracellular fluid (46). Therefore, distinct cell subsets were isolated from the gastric LN and analyzed for their capacity to induce activation of the TXA-23 T cell clone. Only DCs induced IFN-γ production in the absence of added antigen, whereas B cells were unable to do so (Fig. 4 b). Upon the addition of the H+/K+-ATPase peptide, however, gastric B cells were modestly effective in inducing IFN-γ production.
Differential cosignaling, rather than differential TCR ligand display, might account for the failure of gastric B cells to activate the TXA-23 T cell clone in the absence of added antigen (47). Therefore, additional experiments were performed in the absence or presence of anti-CD28 mAb. The relative stimulatory deficiency of the gastric B cells compared with gastric DCs cultured in the presence of H+/K+-ATPase peptide was partially overcome by directly ligating the CD28 molecule on the TXA-23 T cells. However, no IFN-γ production by the TXA-23 clone was seen using B cells without adding specific antigen even with anti-CD28 mAb present (Fig. 4 b), nor did these cells show significant up-regulation of CD69, a sensitive indictor of TCR engagement (unpublished data), suggesting that deficient costimulation by gastric B cells does not obscure the detection of processed tissue-specific antigen.
Increased Number of H+/K+-ATPase–containing DCs in Animals with AIG.
The induction of AIG is associated with the progressive destruction and loss of PCs and chief cells from the gastric mucosa in conjunction with varying degrees of lymphoid infiltration (11). Concomitantly, H+/K+-ATPase–reactive T cells accumulate in the draining gastric LN but not peripheral LN (14, 18). In most autoimmune diseases, severity increases over time and is frequently accompanied by epitope spreading. The latter has been ascribed to increased presentation of tissue antigen as effector-mediated damage makes more cellular material available for processing (for review see references 48 and 49). One implication of these observations is that during T cell–induced AIG there should be an increase in DCs containing, and presumably presenting, a tissue antigen such as H+/K+-ATPase. Therefore, we examined whether changes in the distribution of H+/K+-ATPase Ag, H+/K+-ATPase–containing DCs, or both, occurred after the induction of AIG in BALB/cnu/nu mice.
Stomach sections of untreated BALB/cnu/nu mice closely resembled stomach sections of normal BALB/c mice in terms of morphology and H+/K+-ATPase staining (compare Figs. 1 a and 5 a). 4 wk after adoptive transfer of CD4+ CD25− T cells, lymphoid aggregates at the base of the mucosa and infiltrates of lymphocytes extending from the submucosa into the mucosa were observed as consistent with the induction of AIG (Fig. 5 a, third panel). The extent of H+/K+-ATPase staining in the stomach was considerably reduced (Fig. 5 a, fourth panel), in accordance with the previously described progressive depletion of PCs in AIG (50). In comparison to untreated animals (Fig. 5 b, first panel), there was a substantial increase in the frequency of CD11c+ DCs closely associated with remaining H+/K+-ATPase–expressing PCs (Fig. 5 b, second panel). Additional analysis confirmed the very close juxtaposition of CD11c+ DCs with PC staining for H+/K+-ATPase (Fig. 5 b, third and fourth panels). The loss of H+/K+-ATPase staining in the stomach tissue was paralleled by a striking increase in 2G11 staining localized exclusively to CD11c+ DCs in the draining gastric LN of animals with AIG (Fig. 5 c, second panel), compared with the gastric LN of untreated animals (Fig. 5 c, first panel). Additional analysis by laser scanning confocal microscopy showed an intracellular staining pattern for the H+/K+-ATPase (Fig. 5 d, first panel). A 3D reconstruction of the whole cell was created. By decreasing the opacity of the CD11c staining (red), the intracellular, cytoplasmic localization of the H+/K+-ATPase staining (green) in multiple CD11c+ DCs could be clearly visualized (Fig. 5 d, second and third panels).
Figure 5.
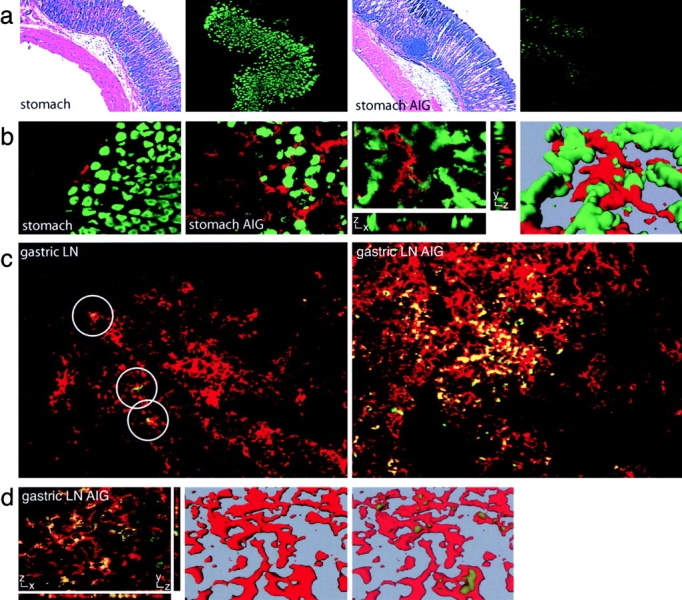
Immunofluorescence staining of H+/K+-ATPase and DCs in the gastric LN and stomach sections of animals with AIG. (a) Stomach sections from untreated BALB/cnu/nu mice (stomach) and BALB/cnu/nu mice with AIG (stomach AIG) were stained with hematoxylin and eosin (first and third panel) or with FITC-2G11 (second and fourth panel). (b) Double staining of stomach sections of untreated BALB/cnu/nu mice (stomach) and BALB/cnu/nu mice with AIG (stomach AIG) for H+/K+-ATPase (green) and anti–CD11c-TXRD (red). Laser scanning confocal microscopy was conducted on a similarly stained stomach section from an animal with AIG. One individual optical section in the z-plane is shown with cross-sectional profiles in the y and x axes (third panel). A 3D reconstruction of the whole cell was produced from these image data, which demonstrates the juxtaposition of H+/K+-ATPase+ PCs (green) and a CD11c+ DC (red; fourth panel). (c) Gastric LN sections of an untreated BALB/cnu/nu mouse (gastric LN) and a BALB/cnu/nu mouse with AIG (gastric LN AIG) were stained with FITC-2G11 (green) and anti–CD11c-TXRD (red). Positive staining with the anti–H+/K+-ATPase mAb is restricted to CD11c+ DCs (circles). (d) Laser scanning confocal microscopy was conducted on a similarly stained gastric LN section from an animal with AIG. One individual optical section in the z-plane is shown with cross-sectional profiles in the y and x axes (left). A 3D reconstruction of the whole cell was produced from these image data (middle) and the intracellular localization of the H+/K+-ATPase staining (green) was visualized by decreasing the opacity of the CD11c staining (red; right).
A quantitative immunohistochemical analysis of H+/K+-ATPase in LN sections of untreated BALB/c mice and animals with AIG was also performed. Positive staining for H+/K+-ATPase was unambiguously identified in gastric, but not peripheral, LN sections of untreated animals (Fig. 6 a). In animals with AIG, a substantial increase in the number of cells staining for H+/K+-ATPase was observed in gastric, but not peripheral, LN sections (Fig. 6 b).
Figure 6.

Quantitative immunohistochemical analysis of H+/K+-ATPase in LN sections of untreated animals and animals with AIG. (a) Anti–H+/K+-ATPase FITC-stained LN sections of untreated BALB/c mice were incubated with secondary anti-FITC HRPO Ab and developed using enhanced diaminobenzidine substrate. Pictures were taken with the bright field setting of the microscope (top) and additionally analyzed using the public domain NIH Image program. Single peaks represent positive staining for H+/K+-ATPase in the threshold histogram plot profile (top) and in the surface plot view (bottom) and can be detected in gastric LN, but not peripheral LN, sections of untreated animals. (b) In animals with AIG, an increase in the staining frequency for H+/K+-ATPase was observed in gastric LN (gastric LN AIG), but not in peripheral LN (peripheral LN AIG), sections.
Increased H+/K+-ATPase–related T Cell Stimulatory Capacity of Gastric DCs During AIG.
Absolute numbers of CD11c+ DCs in the gastric LN were found to be on average 11 ± 7-fold increased in animals with AIG compared with untreated BALB/cnu/nu mice. In contrast, the numbers of CD11c+ DCs in peripheral LNs were similar in untreated mice and animals with AIG (unpublished data). The ability of gastric DCs from untreated BALB/cnu/nu mice to evoke IFN-γ from TXA-23 cells was compared with that of gastric DCs from BALB/cnu/nu mice 4 wk after the induction of AIG. Gastric LN DCs from normal BALB/cnu/nu mice were less active in stimulating the TXA-23 clone than those from normal BALB/c mice, in accordance with previous reports (Fig. 7 , week 0; references 51–53). Strikingly, gastric DCs from BALB/cnu/nu with AIG showed a greater per cell capacity for stimulating IFN-γ production by TXA-23 than DCs from normal BALB/c (14% IFN-γ positive vs. the typical 4 ± 2%; Fig. 7, wk 4). As with cells from the gastric LN of normal mice, both CD8αhi and CD8αlo DCs were active in TXA-23 stimulation (unpublished data). Considering the increased number of CD11c+ DCs, these data suggest a dramatic increase in antigen presentation and/or costimulatory capacity accompanies the development of gastritis in these mice. In line with this notion, preliminary experiments indicate that MHC class II molecule and costimulatory molecule B7.1 expression are increased on gastric DCs from animals with AIG compared with healthy animals (unpublished data).
Figure 7.

Increase of TXA-23 stimulatory capacity of gastric DCs during AIG. The capacity of gastric DCs to induce IFN-γ production of TXA-23 T cells in the absence of added antigen was analyzed in untreated BALB/cnu/nu mice and in BALB/cnu/nu mice 4 wk after the adoptive transfer of CD4+ CD25− T cells to induce AIG. The dot plots show intracellular IFN-γ production by TXA-23 cells upon coculture for 12–16 h with gastric DCs from untreated BALB/cnu/nu mice (week 0) and BALB/cnu/nu with AIG (week 4) in the absence of added antigen. One representative experiment out of three is shown.
Discussion
Here, we show that DCs of the gastric LN, but not of peripheral or mesenteric LN, constitutively acquire, process, and present PC-restricted H+/K+-ATPase in healthy, untreated BALB/c mice. DCs are the only APC type in the gastric LN that show detectable levels of such constitutive antigen acquisition and presentation, with both CD8αhi and CD8αlo gastric DC subsets able to activate H+/K+-ATPase–specific T cells.
Results using both normal animals and transgenic models with artificial “self-antigens” controlled by tissue-specific promoters have suggested that DCs acquire antigen in tissue sites and migrate to draining LN where processed forms of these antigens are presented to T cells (for review see reference 7). In accordance with these prior results, we found MHC class II+ CD11b+ CD8α− CD11c+ DCs residing in stomach tissue in close association with PCs containing H+/K+-ATPase. In rare DCs in the stomach, H+/K+-ATPase or H+/K+-ATPase fragments were present within intracellular vesicles. The number of such antigen-containing DCs visualized by immunofluorescence is likely to be a substantial underestimate of that involved in antigen uptake and transport, given the probability of degradation of the ATPase sufficient to render it undetectable in many cells. H+/K+-ATPase–containing CD8α+ or CD8α− and CD11b+ or CD11b− CD11c+ DEC-205+ DCs were also observed in lymphoid tissue, but only in the gastric LN draining the tissue source of the antigen. Taken together, these data strongly suggest that DCs take up H+/K+-ATPase self-antigen in normal stomach tissues and transport it to the draining gastric LN.
Our ability to detect intracellular H+/K+-ATPase with an Ab suggests that DCs might acquire this antigen in the stomach mucosa by phagocytosis of fragments of PCs or of entire apoptotic bodies. Immature DCs efficiently phagocytose apoptotic cells (31) and the high steady-state turnover rate of gastric PCs (23 d half-life; reference 12) should favor a constant supply of H+/K+-ATPase antigen from dying cells. The number of gastric LN-localized antigen-containing DCs was markedly increased in animals with AIG in concert with an increase in the number of DCs infiltrating the stomach mucosa and with a progressive loss of H+/K+-ATPase–containing PCs. These data in diseased animals are fully consistent with the idea that antigen acquisition involves dead or dying PCs and may also help explain the progressive nature of autoimmune disease as well as the accompanying epitope spreading, which presumably requires increased presentation of what is normally a limiting amount of tissue-specific antigen (13).
DCs in the gastric LN under steady-state conditions not only acquired H+/K+-ATPase self-antigen, but also presented a processed form of this protein in association with MHC class II molecules. Such presentation was not a post-isolation artifact but was shown to represent the display of antigen processed in situ. This is of some importance, as several studies have shown that immature DCs take up antigen efficiently (33–35) but typically do not form substantial numbers of MHC class II–peptide complexes unless exposed to inflammatory mediators (8–10, 36).
Numerous DC subsets have been described based on the expression of various surface proteins such as DEC-205, CD4, and CD8α (for review see reference 54). The role of specific DC subsets in T cell tolerance remains controversial (5, 55–60). The capacity of both CD8αhigh and CD8αlow DCs to present H+/K+-ATPase antigen in our study may reflect the detection of both resident and migratory DCs, respectively, which, due to activating signals, are both able to process some of the acquired antigen. Alternatively, CD8α+ DCs have recently been reported to derive from CD8α− DCs by a maturation process (61–63). Both the maturing CD8αlow and more mature CD8αhigh DCs might be capable of H+/K+-ATPase antigen presentation in the present model.
Constitutive presentation of H+/K+-ATPase in normal mice does not result in the induction of AIG nor, it seems, in the total inactivation and/or deletion of H+/K+-ATPase–specific T cells, as documented by the numerous examples of AIG induction after the transfer of CD4+ CD25− T cells from normal donors into T cell–deficient hosts (19, 64) including those reported here. The presence of functional autoreactive cells in animals in which self-antigen presentation by DCs is apparent stands in contrast to a variety of reports in transgenic systems (for review see references 7 and 65) and with the clear evidence for antigen-specific T cell deletion mediated by DCs under “steady-state” noninflammatory conditions (5). Many of these deletion models involve just one TCR borne on transgenic T cells and it is possible that only those T cells with TCR that are of the highest affinity for the presented self-antigen undergo deletion by DC-borne determinants. In addition, the deletion of low affinity T cells by a transgenic self-antigen can take several weeks (66). Because normal animals maintain a substantial thymic output, it is possible that steady-state antigen presentation by DCs continually removes mature antigen-specific cells from the repertoire, but that at any given moment the recently exported cells not yet eliminated account for the capacity of transferred CD4+ CD25− T cells to induce disease. Finally, the high level of H+/K+-ATPase acquired by DCs might result in the presentation of this antigen by both tolerogenic and stimulatory subsets. Interaction of the latter with T cells may impose a need for control of the activation by CD25+ regulatory T cells (12). Thus, these findings support the emerging view that both DC-mediated deletion and immunoregulatory processes contribute to the maintenance of an effective state of peripheral tolerance in the normal individual.
Acknowledgments
We thank Owen Schwartz (NIAID Imaging Facility) for confocal microscopy expertise. We also thank members of the Editorial Board of the Journal for help in making this report more focused and accessible to the reader.
This work was supported by the Austrian Science Fund, grants J1863-MED and J2059-PAT.
The online version of this article contains supplemental material.
Footnotes
Abbreviations used in this paper: aa, amino acid; AIG, autoimmune gastritis; CFSE, carboxyfluorescein succinimidyl ester; DC, dendritic cell; HRPO, horseradish peroxidase; PC, parietal cell; TXRD, Texas red.
References
- 1.von Boehmer, H. 1988. The developmental biology of T lymphocytes. Annu. Rev. Immunol. 6:309–326. [DOI] [PubMed] [Google Scholar]
- 2.Robey, E., and B.J. Fowlkes. 1994. Selective events in T cell development. Annu. Rev. Immunol. 12:675–705. [DOI] [PubMed] [Google Scholar]
- 3.Schwartz, R.H. 1989. Acquisition of immunologic self-tolerance. Cell. 57:1073–1081. [DOI] [PubMed] [Google Scholar]
- 4.Schonrich, G., F. Momburg, M. Malissen, A.M. Schmitt-Verhulst, B. Malissen, G.J. Hämmerling, and B. Arnold. 1992. Distinct mechanisms of extrathymic T-cell tolerance due to differential expression of self antigen. Int. Immunol. 4:581–590. [DOI] [PubMed] [Google Scholar]
- 5.Hawiger, D., K. Inaba, Y. Dorsett, M. Guo, K. Mahnke, M. Rivera, J.V. Ravetch, R.M. Steinman, and M.C. Nussenzweig. 2001. Dendritic cells induce peripheral T cell unresponsiveness under steady state conditions in vivo. J. Exp. Med. 194:769–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Banchereau, J., and R.M. Steinman. 1998. Dendritic cells and the control of immunity. Nature. 392:245–252. [DOI] [PubMed] [Google Scholar]
- 7.Steinman, R.M., and M.C. Nussenzweig. 2002. Avoiding horror autotoxicus: the importance of dendritic cells in peripheral T cell tolerance. Proc. Natl. Acad. Sci. USA. 99:351–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reis e Sousa, C., and R.N. Germain. 1999. Analysis of adjuvant function by direct visualization of antigen presentation in vivo: endotoxin promotes accumulation of antigen-bearing dendritic cells in the T cell areas of lymphoid tissue. J. Immunol. 162:6552–6561. [PubMed] [Google Scholar]
- 9.Inaba, K., S. Turley, T. Iyoda, F. Yamaide, S. Shimoyama, C. Reis e Sousa, R.N. Germain, I. Mellman, and R.M. Steinman. 2000. The formation of immunogenic major histocompatibility complex class II–peptide ligands in lysosomal compartments of dendritic cells is regulated by inflammatory stimuli. J. Exp. Med. 191:927–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Turley, S.J., K. Inaba, W.S. Garrett, M. Ebersold, J. Unternaehrer, R.M. Steinman, and I. Mellman. 2000. Transport of peptide-MHC class II complexes in developing dendritic cells. Science. 288:522–527. [DOI] [PubMed] [Google Scholar]
- 11.Toh, B.H., J.W. Sentry, and F. Alderuccio. 2000. The causative H+/K+ ATPase antigen in the pathogenesis of autoimmune gastritis. Immunol. Today. 21:348–354. [DOI] [PubMed] [Google Scholar]
- 12.Shevach, E.M. 2000. Suppressor T cells: rebirth, function and homeostasis. Curr. Biol. 10:R572–R575. [DOI] [PubMed] [Google Scholar]
- 13.Sercarz, E.E., P.V. Lehmann, A. Ametani, G. Benichou, A. Miller, and K. Moudgil. 1993. Dominance and crypticity of T cell antigenic determinants. Annu. Rev. Immunol. 11:729–766. [DOI] [PubMed] [Google Scholar]
- 14.Suri-Payer, E., A.Z. Amar, R. McHugh, K. Natarajan, D.H. Margulies, and E.M. Shevach. 1999. Post-thymectomy autoimmune gastritis: fine specificity and pathogenicity of anti-H/K ATPase-reactive T cells. Eur. J. Immunol. 29:669–677. [DOI] [PubMed] [Google Scholar]
- 15.Murphy, K.M., A.B. Heimberger, and D.Y. Loh. 1990. Induction by antigen of intrathymic apoptosis of CD4+ CD8+TCRlo thymocytes in vivo. Science. 250:1720–1723. [DOI] [PubMed] [Google Scholar]
- 16.McHugh, R.S., E.M. Shevach, D.H. Margulies, and K. Natarajan. 2001. A T cell receptor transgenic model of severe, spontaneous organ-specific autoimmunity. Eur. J. Immunol. 31:2094–2103. [DOI] [PubMed] [Google Scholar]
- 17.Metlay, J.P., M.D. Witmer-Pack, R. Agger, M.T. Crowley, D. Lawless, and R.M. Steinman. 1990. The distinct leukocyte integrins of mouse spleen dendritic cells as identified with new hamster monoclonal antibodies. J. Exp. Med. 171:1753–1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Suri-Payer, E., P.J. Kehn, A.W. Cheever, and E.M. Shevach. 1996. Pathogenesis of post-thymectomy autoimmune gastritis. Identification of anti-H/K adenosine triphosphatase-reactive T cells. J. Immunol. 157:1799–1805. [PubMed] [Google Scholar]
- 19.Sakaguchi, S., N. Sakaguchi, M. Asano, M. Itoh, and M. Toda. 1995. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor α-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J. Immunol. 155:1151–1164. [PubMed] [Google Scholar]
- 20.Guery, J.C., and L. Adorini. 1995. Dendritic cells are the most efficient in presenting endogenous naturally processed self-epitopes to class II-restricted T cells. J. Immunol. 154:536–544. [PubMed] [Google Scholar]
- 21.Dittel, B.N., I. Visintin, R.M. Merchant, and C.A. Janeway, Jr. 1999. Presentation of the self antigen myelin basic protein by dendritic cells leads to experimental autoimmune encephalomyelitis. J. Immunol. 163:32–39. [PubMed] [Google Scholar]
- 22.Albert, M.L., B. Sauter, and N. Bhardwaj. 1998. Dendritic cells acquire antigen from apoptotic cells and induce class I-restricted CTLs. Nature. 392:86–89. [DOI] [PubMed] [Google Scholar]
- 23.Kurts, C., M. Cannarile, I. Klebba, and T. Brocker. 2001. Dendritic cells are sufficient to cross-present self-antigens to CD8 T cells in vivo. J. Immunol. 166:1439–1442. [DOI] [PubMed] [Google Scholar]
- 24.Aharoni, R., D. Teitelbaum, R. Arnon, and J. Puri. 1991. Immunomodulation of experimental allergic encephalomyelitis by antibodies to the antigen-Ia complex. Nature. 351:147–150. [DOI] [PubMed] [Google Scholar]
- 25.Murphy, D.B., S. Rath, E. Pizzo, A.Y. Rudensky, A. George, J.K. Larson, and C.A. Janeway, Jr. 1992. Monoclonal antibody detection of a major self peptide-MHC class-II complex. J. Immunol. 148:3483–3491. [PubMed] [Google Scholar]
- 26.Puri, J., R. Arnon, E. Gurevich, and D. Teitelbaum. 1997. Modulation of the immune response in multiple sclerosis: production of monoclonal antibodies specific to HLA/myelin basic protein. J. Immunol. 158:2471–2476. [PubMed] [Google Scholar]
- 27.Zhong, G., C. Reis e Sousa, and R.N. Germain. 1997. Production, specificity, and functionality of monoclonal antibodies to specific peptide: MHC class II complexes formed by active processing of exogenous protein. Proc. Natl. Acad. Sci. USA. 94:13856–13861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dadaglio, G., C.A. Nelson, M.B. Deck, S.J. Petzold, and E.R. Unanue. 1997. Characterization and quantitation of peptide-MHC complexes produced from hen egg lysozyme using a monoclonal antibody. Immunity. 6:727–738. [DOI] [PubMed] [Google Scholar]
- 29.Krogsgaard, M., K.W. Wucherpfennig, B. Canella, B.E. Hansen, A. Svejgaard, J. Pyrdol, H. Ditzel, C. Raine, J. Engberg, and L. Fugger. 2000. Visualization of myelin basic protein (MBP) T cell epitopes in multiple sclerosis lesions using a monoclonal antibody specific for the human histocompatibility leukocyte antigen (HLA)-DR2–MBP 85–99 complex. J. Exp. Med. 191:1395–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chow, D.C., and J.G. Forte. 1993. Characterization of the β-subunit of the H(+)-K(+)-ATPase using an inhibitory monoclonal antibody. Am. J. Physiol. 265:C1562–C1570. [DOI] [PubMed] [Google Scholar]
- 31.Inaba, K., S. Turley, F. Yamaide, T. Iyoda, K. Mahnke, M. Inaba, M. Pack, M. Subklewe, B. Sauter, D. Sheff, et al. 1998. Efficient presentation of phagocytosed cellular fragments on the major histocompatibility complex class II products of dendritic cells. J. Exp. Med. 188:2163–2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Crowley, M., K. Inaba, and R.M. Steinman. 1990. Dendritic cells are the principal cells in mouse spleen bearing immunogenic fragments of foreign proteins. J. Exp. Med. 172:383–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Romani, N., S. Koide, M. Crowley, M. Witmer-Pack, A.M. Livingstone, C.G. Fathman, K. Inaba, and R.M. Steinman. 1989. Presentation of exogenous protein antigens by dendritic cells to T cell clones. Intact protein is presented best by immature, epidermal Langerhans cells. J. Exp. Med. 169:1169–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pure, E., K. Inaba, M.T. Crowley, L. Tardelli, M.D. Witmer-Pack, G. Ruberti, G. Fathman, and R.M. Steinman. 1990. Antigen processing by epidermal Langerhans cells correlates with the level of biosynthesis of major histocompatibility complex class II molecules and expression of invariant chain. J. Exp. Med. 172:1459–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Reis e Sousa, C., P.D. Stahl, and J.M. Austyn. 1993. Phagocytosis of antigens by Langerhans cells in vitro. J. Exp. Med. 178:509–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sallusto, F., and A. Lanzavecchia. 1994. Efficient presentation of soluble antigen by cultured human dendritic cells is maintained by granulocyte/macrophage colony-stimulating factor plus interleukin 4 and downregulated by tumor necrosis factor alpha. J. Exp. Med. 179:1109–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gallucci, S., M. Lolkema, and P. Matzinger. 1999. Natural adjuvants: endogenous activators of dendritic cells. Nat. Med. 5:1249–1255. [DOI] [PubMed] [Google Scholar]
- 38.McCoy, K.L., J. Miller, M. Jenkins, F. Ronchese, R.N. Germain, and R.H. Schwartz. 1989. Diminished antigen processing by endosomal acidification mutant antigen-presenting cells. J. Immunol. 143:29–38. [PubMed] [Google Scholar]
- 39.Stockinger, B., U. Pessara, R.H. Lin, J. Habicht, M. Grez, and N. Koch. 1989. A role of Ia-associated invariant chains in antigen processing and presentation. Cell. 56:683–689. [DOI] [PubMed] [Google Scholar]
- 40.Ardavin, C., L. Wu, C.L. Li, and K. Shortman. 1993. Thymic dendritic cells and T cells develop simultaneously in the thymus from a common precursor population. Nature. 362:761–763. [DOI] [PubMed] [Google Scholar]
- 41.Wu, L., C.L. Li, and K. Shortman. 1996. Thymic dendritic cell precursors: relationship to the T lymphocyte lineage and phenotype of the dendritic cell progeny. J. Exp. Med. 184:903–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vremec, D., and K. Shortman. 1997. Dendritic cell subtypes in mouse lymphoid organs: cross-correlation of surface markers, changes with incubation, and differences among thymus, spleen, and lymph nodes. J. Immunol. 159:565–573. [PubMed] [Google Scholar]
- 43.de St Groth, B.F. 1998. The evolution of self-tolerance: a new cell arises to meet the challenge of self-reactivity. Immunol. Today. 19:448–454. [DOI] [PubMed] [Google Scholar]
- 44.de St Groth, B.F. 2001. DCs and peripheral T cell tolerance. Semin. Immunol. 13:311–322. [DOI] [PubMed] [Google Scholar]
- 45.Guery, J.C., F. Ria, F. Galbiati, S. Smiroldo, and L. Adorini. 1997. The mode of protein antigen administration determines preferential presentation of peptide-class II complexes by lymph node dendritic or B cells. Int. Immunol. 9:9–15. [DOI] [PubMed] [Google Scholar]
- 46.Zhong, G., C. Reis e Sousa, and R.N. Germain. 1997. Antigen-unspecific B cells and lymphoid dendritic cells both show extensive surface expression of processed antigen-major histocompatibility complex class II complexes after soluble protein exposure in vivo or in vitro. J. Exp. Med. 186:673–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cassell, D.J., and R.H. Schwartz. 1994. A quantitative analysis of antigen-presenting cell function: activated B cells stimulate naive CD4 T cells but are inferior to dendritic cells in providing costimulation. J. Exp. Med. 180:1829–1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Miller, S.D., B.L. McRae, C.L. Vanderlugt, K.M. Nikcevich, J.G. Pope, L. Pope, and W.J. Karpus. 1995. Evolution of the T-cell repertoire during the course of experimental immune-mediated demyelinating diseases. Immunol. Rev. 144:225–244. [DOI] [PubMed] [Google Scholar]
- 49.Mamula, M.J. 1998. Epitope spreading: the role of self peptides and autoantigen processing by B lymphocytes. Immunol. Rev. 164:231–239. [DOI] [PubMed] [Google Scholar]
- 50.Judd, L.M., P.A. Gleeson, B.H. Toh, and I.R. van Driel. 1999. Autoimmune gastritis results in disruption of gastric epithelial cell development. Am. J. Physiol. 277:G209–G218. [DOI] [PubMed] [Google Scholar]
- 51.Grabbe, S., R.L. Gallo, A. Lindgren, and R.D. Granstein. 1993. Deficient antigen presentation by Langerhans cells from athymic (nu/nu) mice. Restoration with thymic transplantation or administration of cytokines. J. Immunol. 151:3430–3439. [PubMed] [Google Scholar]
- 52.Burkly, L., C. Hession, L. Ogata, C. Reilly, L.A. Marconi, D. Olson, R. Tizard, R. Cate, and D. Lo. 1995. Expression of relB is required for the development of thymic medulla and dendritic cells. Nature. 373:531–536. [DOI] [PubMed] [Google Scholar]
- 53.Shreedhar, V., A.M. Moodycliffe, S.E. Ullrich, C. Bucana, M.L. Kripke, and L. Flores-Romo. 1999. Dendritic cells require T cells for functional maturation in vivo. Immunity. 11:625–636. [DOI] [PubMed] [Google Scholar]
- 54.Shortman, K., and Y.J. Liu. 2002. Mouse and human dendritic cell subtypes. Nat. Rev. Immunol. 2:151–161. [DOI] [PubMed] [Google Scholar]
- 55.Suss, G., and K. Shortman. 1996. A subclass of dendritic cells kills CD4 T cells via Fas/Fas-ligand–induced apoptosis. J. Exp. Med. 183:1789–1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kronin, V., K. Winkel, G. Suss, A. Kelso, W. Heath, J. Kirberg, H. von Boehmer, and K. Shortman. 1996. A subclass of dendritic cells regulates the response of naive CD8 T cells by limiting their IL-2 production. J. Immunol. 157:3819–3827. [PubMed] [Google Scholar]
- 57.Inaba, K., M. Pack, M. Inaba, H. Sakuta, F. Isdell, and R.M. Steinman. 1997. High levels of a major histocompatibility complex II–self peptide complex on dendritic cells from the T cell areas of lymph nodes. J. Exp. Med. 186:665–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kronin, V., C.J. Fitzmaurice, I. Caminschi, K. Shortman, D.C. Jackson, and L.E. Brown. 2001. Differential effect of CD8(+) and CD8(−) dendritic cells in the stimulation of secondary CD4(+) T cells. Int. Immunol. 13:465–473. [DOI] [PubMed] [Google Scholar]
- 59.Hugues, S., E. Mougneau, W. Ferlin, D. Jeske, P. Hofman, D. Homann, L. Beaun, C. Schrike, M. Von Herrath, A. Lehuen, et al. 2002. Tolerance to islet antigens and prevention from diabetes induced by limited apoptosis of pancreatic beta cells. Immunity. 16:169–181. [DOI] [PubMed] [Google Scholar]
- 60.Iyoda, T., S. Shimoyama, K. Liu, Y. Omatsu, Y. Akiyama, Y. Maeda, K. Takahara, R.M. Steinman, and K. Inaba. 2002. The CD8+ dendritic cell subset selectively endocytoses dying cells in culture and in vivo. J. Exp. Med. 195:1289–1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Merad, M., L. Fong, J. Bogenberger, and E.G. Engleman. 2000. Differentiation of myeloid dendritic cells into CD8α-positive dendritic cells in vivo. Blood. 96:1865–1872. [PubMed] [Google Scholar]
- 62.Traver, D., K. Akashi, M. Manz, M. Merad, T. Miyamoto, E.G. Engleman, and I.L. Weissman. 2000. Development of CD8α-positive dendritic cells from a common myeloid progenitor. Science. 290:2152–2154. [DOI] [PubMed] [Google Scholar]
- 63.del Hoyo, G.M., P. Martin, C.F. Arias, A.R. Marin, and C. Ardavin. 2002. CD8 α (+) dendritic cells originate from the CD8 α (−) dendritic cell subset by a maturation process involving CD8 α, DEC-205, and CD24 up-regulation. Blood. 99:999–1004. [DOI] [PubMed] [Google Scholar]
- 64.Asano, M., M. Toda, N. Sakaguchi, and S. Sakaguchi. 1996. Autoimmune disease as a consequence of developmental abnormality of a T cell subpopulation. J. Exp. Med. 184:387–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Stockinger, B. 1999. T lymphocyte tolerance: from thymic deletion to peripheral control mechanisms. Adv. Immunol. 71:229–265. [DOI] [PubMed] [Google Scholar]
- 66.Morgan, D.J., H.T. Kreuwel, and L.A. Sherman. 1999. Antigen concentration and precursor frequency determine the rate of CD8+ T cell tolerance to peripherally expressed antigens. J. Immunol. 163:723–727. [PubMed] [Google Scholar]