

Abstract
A persistent immune response to hepatitis viruses is a well-recognized risk factor for hepatocellular carcinoma. However, the molecular and cellular basis for the procarcinogenic potential of the immune response is not well defined. Here, using a unique animal model of chronic hepatitis that induces hepatocellular carcinogenesis, we demonstrate that neutralization of the activity of Fas ligand prevented hepatocyte apoptosis, proliferation, liver inflammation, and the eventual development of hepatocellular carcinoma. The results indicate that Fas ligand is involved not only in direct hepatocyte killing but also in the process of inflammation and hepatocellular carcinogenesis in chronic hepatitis. This is the first demonstration that amelioration of chronic inflammation by some treatment actually caused reduction of cancer development.
Keywords: disease model, apoptosis, inflammation, cytotoxic T lymphocytes, cancer
Introduction
Hepatitis B virus (HBV) is one of the most common pathogens; more than 350 million people are estimated to be chronically infected with it, worldwide. Hepatitis C virus is also a widespread pathogen, which has a worldwide seroprevalence of ∼1%. These two pathogens are the major cause of chronic liver inflammation, leading to hepatocellular carcinoma (HCC).
CTLs have been implicated in both the eradication of viruses and liver injury in viral hepatitis patients (1, 2). It has been previously demonstrated that the transfusion of a highly active CD8+ CTL clone that is specific for the hepatitis B surface antigen (HBsAg) induces lethal fulminant hepatitis in transgenic mice that express the HBsAg specifically in the liver (3). We further demonstrated that Fas ligand (FasL), one of the major cytocidal molecules produced by CTLs (4, 5) plays an important role in the pathogenesis of this disease, and that the administration of soluble Fas (Fas–Fc fusion protein that can neutralize FasL) rescues mice from the fatal disease (6, 7). On the other hand, it is generally believed that apoptosis is a mechanism to prevent carcinogenesis. Therefore, it was possible that treatment of hepatitis patients with a FasL-neutralizing agent might increase the risk of hepatic cancer. However, we have not been able to investigate this possibility, because the CTL clone induces neither chronic liver diseases nor HCC.
HCC occurs after many years of chronic hepatitis. The cycles of liver cell destruction and regeneration by repetitive inflammation are thought to set up the mitogenic and mutagenic environment leading to HCC development (8–11). In an effort to clarify the carcinogenic potential of persistent inflammation, one of us (Y. Nakamoto) recently developed a unique animal model of chronic hepatitis that leads to HCC (12). In this model, an investigator transfuses HBsAg-primed splenocytes from wild-type mice into the aforementioned HBsAg transgenic mice. Immune responses against HBsAg are essentially involved in the development of liver diseases including HCC, because the transgenic mice are healthy unless primed splenocytes are transfused. Using this model, we have investigated how treatment by an anti-FasL neutralizing antibody affects in the progression of chronic hepatitis and the development of HCC.
Materials and Methods
HBV Transgenic Mice.
HBsAg transgenic mouse lineage 107–5D (official designation Tg[Alb-1,HBV]Bri66; inbred B10D2, H-2d) was provided by Dr. F.V. Chisari (The Scripps Research Institute, La Jolla, CA; reference 13). Lineage 107–5D contains the entire HBV envelope-coding region (subtype ayw) under the constitutive transcriptional control of the mouse albumin promoter (13). These mice express the HBV small, middle, and large envelope proteins in their hepatocytes (13). They are immunologically tolerant to HBsAg at the T cell level (14) and they display no evidence of liver disease during their lifetime, without the adoptive transfer of HBsAg-specific CTLs (13, 15). There is no X-RNA or X-protein expression detectable in the livers of these animals (unpublished data).
Disease Model.
The animal model of chronic hepatitis was generated as described previously (12). Briefly, male HBsAg transgenic mice were thymectomized, irradiated (900 cGy), and their hemopoietic system was reconstituted with bone marrow cells from syngeneic nontransgenic B10D2 (H-2d) mice. 1 wk after the bone marrow transfer, the animals received the indicated numbers of splenocytes from nontransgenic B10D2 (H-2d) mice that were infected intraperitoneally with a recombinant vaccinia virus expressing HBsAg (HBs-vac) 3 wk before the splenocyte transfer (15).
Preparation and Administration of Anti-FasL mAb.
The anti–mouse FasL mAb (FLIM58) was produced and purified using a protein A column as described previously (16). 2 ng of anti-FasL mAb used in this study completely neutralized the cytotoxicity of 5 units of recombinant mouse FasL. 500 μg of the anti-mouse FasL mAb or control hamster IgG (ICN Biomedicals), or PBS alone was administered intraperitoneally or subcutaneously daily for seven consecutive days (day 0 to 6), and 200 μg anti–mouse FasL mAb, control IgG, or PBS alone was injected every other day between the second and fourth week (day 7 to 28) after adoptive transfer of HBsAg-primed nontransgenic mouse splenocytes.
Measurement of Serum Alanine Aminotransferase Activity and IL-18.
Serum alanine aminotransferase (ALT) activity was determined as described previously (11). Serum IL-18 was measured by ELISA kits provided by Hayashibara (Okayama, Japan), as reported previously (17).
Immunohistochemical Analysis.
Tissue samples were fixed in buffered zinc formalin (Anatech Ltd.), embedded in paraffin, sectioned (at 3 μm), and stained with hematoxylin and eosin as described previously (12). Some of the paraffin sections were treated with anti-proliferating cell nuclear antigen (PCNA) primary solution (Dako) at a 1:10 dilution, followed by biotin-conjugated secondary antibody (Vector Laboratories). PCNA+ cells were then visualized using a VECTASTAIN ABC Standard Kit (Vector Laboratories), and the tissue sections were counterstained with hematoxylin before mounting. Liver tissues were also embedded in OCT compound (Sakura Finetek) and snap-frozen in liquid nitrogen. Cryostat sections of frozen tissues were fixed in 4% paraformaldehyde overnight at 4°C. After blocking biotin, the tissue sections were incubated with rabbit anti–mouse active caspase-3 antibodies (18) at a 1:400 dilution for 30 min at room temperature, followed by biotin-conjugated goat anti–rabbit IgG secondary antibodies (Vector Laboratories). The reaction was visualized in the same way as the PCNA staining described above. The TdT-mediated digoxigenin-dUTP nick-end labeling (TUNEL) analysis was performed on serial liver sections according to the manufacturer's instructions (Roche).
Detection of HBV-specific CD8+ T Lymphocytes.
Intrahepatic lymphocytes (IHLs) were stained with Cy-Chrome–conjugated anti–mouse CD8 mAb (53–6.7; BD Biosciences) in round-bottom 96-well plates. Otherwise, IHLs were cultured for 5 h at 37°C in complete RPMI medium containing 10% FBS in the presence or absence of 10−7 M of the peptide representing residues 28–39 of HBsAg (IPQSLDSWWTSL) or the lymphocytic choriomeningitis virus nucleoprotein (LCMV NP) peptide (PQASGVYML). Brefeldin A (Sigma-Aldrich), which inhibits exocytosis of the cytokines, was added at a final concentration 2 μg/ml. Subsequently, the cells were stained with Cy-Chrome-anti–mouse CD8 mAb. They were then permeabilized using the Cytofix/Cytoperm kit (BD Biosciences) and stained with allophycocyanin (APC)-conjugated rat mAb specific for mouse IFN-γ (XMG1.2) or its isotype control Ab (rat IgG1; BD Biosciences). Cells were resuspended in PBS containing 2% formaldehyde, and analyzed on a FACSCalibur™ flow cytometer (50,000–300,000 gated events acquired per sample) using CELLQuest™ software (Becton Dickinson).
Results
Anti-FasL mAb Diminished Not Only Hepatocyte Apoptosis but Also Inflammation and Hepatocyte Proliferation.
As we reported previously (12), transplantation of total splenocytes from HBsAg-primed mice into HBsAg-transgenic mice induced relatively slow and prolonged acute-phase liver injury, compared with CTL clone-induced hepatitis (3) or other animal models of acute hepatitis. The liver injury peaked 1 wk after the transplantation and gradually resolved thereafter, as revealed by the change in the serum ALT level (Fig. 1 A). Consistent with this, on day 7 we observed strong activation of caspase-3 and DNA degradation (shown by TUNEL), indicating massive hepatocyte apoptosis that occurred along the edge of inflammatory infiltration (Fig. 1 B). Interestingly, the number of active caspase-3+ cells peaked in the second week and was much greater than the number of TUNEL+ cells at the same time point (Fig. 1 C). This observation may suggest that some hepatocytes became relatively resistant to caspase-3 activation after prolonged inflammation.
Figure 1.



Anti-FasL mAb reduced hepatocellular apoptosis, inflammation, and regeneration in the prolonged acute-phase liver injury. (A) Anti–mouse FasL mAb or PBS was administered to HBsAg transgenic mice intraperitoneally or subcutaneously after the transfer of 5 × 107 HBsAg-primed nontransgenic splenocytes. Liver injury was evaluated by monitoring serum ALT activity. Vertical lines indicate standard errors. Each group represents five animals. Data shown represent three similar experiments. (B) Immunohistochemical analyses for active caspase-3, TUNEL, and PCNA were performed on liver sections from HBsAg transgenic mice that were treated with anti-FasL mAb or control hamster IgG, 14 d after the splenocyte transfer. TUNEL+ and PCNA+ hepatocyte nuclei are indicated with arrows. The bar represents 40 μm. (C) The proportions (%) of active caspase-3+, TUNEL+, or PCNA+ hepatocytes were quantified in the transgenic mice that were treated with anti-FasL mAb (white bars) or control IgG (black bars) and killed at the indicated time points after the splenocyte transfer. Each group represents three animals. (D) The serum IL-18 levels before or 7 d after the splenocyte transfer were determined by ELISA.
Because we previously demonstrated that the Fas–Fc fusion protein that can neutralize FasL has therapeutic potential against the fulminant hepatitis induced by a CD8+ CTL clone (6), we thought that treatment with an anti-FasL neutralizing antibody might suppress the activity of hepatitis in this model. As we expected, administration of anti-FasL mAb intraperitoneally and subcutaneously decreased the mean ALT level at day 7 to ∼30% and 60%, respectively, of that in the mice that received PBS (Fig. 1 A). Therefore, in the following experiments, we administered mAb intraperitoneally. In accordance with the reduction in serum ALT levels, both caspase-3 activation and DNA degradation in hepatocytes 1 to 4 wk after the splenocyte transfer were greatly diminished by the anti-FasL mAb treatment (Fig. 1, B and C). The anti-FasL mAb treatment not only inhibited hepatocyte apoptosis but also reduced the size and number of inflammatory foci in the liver (Fig. 1 B, and data not depicted). We investigated serum IL-18 levels, because we previously discovered that FasL induces the activation of IL-1β and IL-18 in neutrophils and macrophages, respectively, and that IL-18 is at least partly involved in the FasL-induced liver injury (19, 20). Serum IL-18 increased after the splenocyte transfer and anti-FasL mAb significantly reduced it (Fig. 1 D). These results indicate that anti-FasL mAb treatment inhibited inflammatory responses in this model. The anti-FasL mAb treatment also reduced the number of PCNA+ hepatocytes, indicating suppression of the regenerative proliferation of hepatocytes (Fig. 1, B and C).
Anti-FasL mAb Did Not Deplete CTLs.
CD8 CTL plays a primary role in the acute-phase liver injury in this animal model (reference 12, and unpublished data). Therefore, the reason that anti-FasL mAb protected the liver could be because it eliminated CTLs. To test this possibility, we compared the proportion of intrahepatic CD8+ cells in control and anti-FasL mAb-treated mice at day 7. Approximately 15% of the IHLs were CD8+ in transgenic mice that had been transfused with primed splenocytes (Table I). The anti-FasL mAb treatment did not reduce the proportion of CD8+ cells. Interestingly, we reproducibly detected low but significant numbers of CD8+ IHLs that produced IFN-γ in response to stimulation by the peptide representing residues 28–39 of HBsAg in transgenic mice transfused with primed splenocytes. This peptide is the major epitope of the HBsAg-specific CTLs detected in this model (12). Therefore, these IFN-γ–producing CD8+ cells are likely to be the HBsAg-specific CTLs. The proportion of the IFN-γ–producing cells in the total CD8+ cells was ∼0.18%. This percentage is similar to the percentage of CD8+ cells that stained with the phycoerythrin-labeled tetramer consisting of MHC class I molecule (H-2Ld) and the peptide epitope of HBsAg (data not depicted). In contrast, the percentage of IFN-γ–producing CD8+ cells specific for LCMV NP peptide was less than 0.02%. The percentage of HBsAg peptide-induced IFN-γ–producing CD8+ cells in unmanipulated transgenic mice was also less than 0.02%. Importantly, the anti-FasL mAb treatment did not significantly affect the percentage of IFN-γ–producing CD8+ cells. Therefore, the liver protective effect of anti-FasL mAb was not a result of CTL depletion, and thus it was likely due to its neutralizing effect against FasL.
Table I.
Anti-FasL mAb Did Not Deplete HBsAg-specific CD8+ T Lymphocytes
| CD8+IFN-γ+
stimulated with: |
|||
|---|---|---|---|
| Treatment | CD8+ | HBV S28–39 | LCMV NP |
| Control IgG | 15.4 ± 0.5 | 0.18 ± 0.02 | <0.02 |
| Anti-FasL | 16.1 ± 0.8 | 0.13 ± 0.02 | <0.02 |
| Unmanipulated | 9.1 ± 1.1 | <0.02 | <0.02 |
IHLs were isolated from control IgG or anti-FasL mAb treated transgenic mice 7 d after the splenocyte transfer, or from unmanipulated transgenic mice. The numbers give the proportion of CD8+ cells ([%] = [number of CD8+ cells]/[number of total cells] × 100) or CD8+IFN-γ+ cells ([%] = [number of CD8+IFN-γ+ cells]/[number of CD8+ cells] × 100) ± standard deviation.
Prevention of Chronic Liver Disease Including Hepatocarcinogenesis by the Administration of Anti-FasL mAb.
To evaluate the effect of anti-FasL mAb treatment on the development of chronic liver disease, including HCC, we histopathologically examined the livers of mice that were treated with a control substance or with anti-FasL mAb and assessed tumor development by autopsy. 9 mo after splenocyte transfer, the control mice, which received PBS, displayed portal lymphocytic infiltrates, lobular disarray, and marked variation in the size and shape of hepatocytes, reflecting long-term, persistent hepatitis (Fig. 2 A, left). In contrast, the mice treated with anti-FasL mAb showed minimal inflammatory infiltrates and no dysplastic or preneoplastic changes in the liver (Fig. 2 A, right). After one year or later, all the control animals displayed marked hepatic atrophy and developed multiple liver tumors, most of which were larger than 4 mm in diameter with the largest ranging up to 20 mm in diameter (Fig. 2 B, and Table II). The tumor specimens illustrated the classical histological features of HCC, consisting of relatively poorly differentiated hepatoma cells (Fig. 2 C, left). The surrounding hepatic parenchyma displayed focal lobular inflammatory infiltrates associated with degenerating hepatocytes and marked lobular disarray. In contrast, most of the anti-FasL mAb-treated livers had an almost normal appearance both macro- and microscopically (Fig. 2, B and C, right). Only two of the 15 animals developed solitary liver tumors, one of which was histologically classified as HCC (Table II). Collectively, these results demonstrated that the hepatocarcinogenesis was greatly suppressed by the administration of anti-FasL mAb in this chronic hepatitis model, suggesting that FasL expressed on liver-infiltrating CD8+ T cells activated the caspase cascade in the hepatocytes and induced hepatocellular apoptosis, liver inflammation, regeneration, and the eventual development of HCC.
Figure 2.
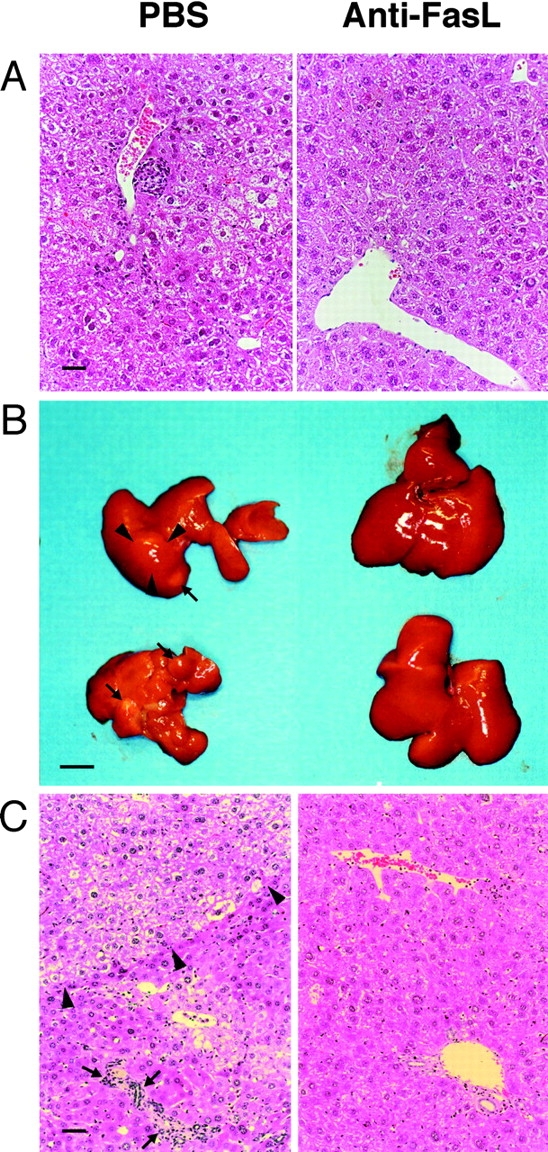
Prevention of progressive liver dysplasia and HCC development by anti-FasL mAb treatment. The transgenic mice described in the legend to Fig. 1 were killed 9 (A) and 15 (B and C) mo after the splenocyte transfer. (A) A 9-mo liver specimen with PBS (left) or anti-FasL mAb treatment (right). (B and C) 15 mo after the splenocyte transfer, livers from PBS-injected animals displayed marked atrophy and multiple liver tumors (arrows) up to 11 mm in diameter (arrowheads; B, left). A representative specimen illustrates the classical histological features of HCC (arrowheads), and the surrounding hepatic parenchyma displays focal lobular inflammatory infiltrates associated with degenerating hepatocytes (arrows; C, left). Most of livers from anti-FasL mAb-injected animals did not show apparent atrophy or liver tumors (B, right). A representative specimen demonstrates minimal portal infiltrates and very mild lobular disarray (C, right). Liver sections were stained with hematoxylin and eosin. The bars represent 40 μm (A and C), and 10 mm (B).
Table II.
Prevention of Hepatocarcinogenesis by Anti-FasL mAb Treatment
| Mouse ID | Mo after spl. transfer |
Age (mo) at killing |
No. of tumors |
Largest tumor (mm)a |
Tumor histology |
|---|---|---|---|---|---|
| Treated with PBS or control hamster IgG (intraperitoneally) | |||||
| 110 | 9 | 16 | 1 | 2 | Adenoma |
| 113 | 8 | 15 | 2 | 2 | Adenoma |
| 189 | 9 | 15 | 1 | 4 | Adenoma |
| 266 | 13 | 17 | 1 | 11 | HCC |
| 276 | 15 | 18 | 1 | 5 | HCC |
| 323 | 15 | 18 | 2 | 11 | HCC |
| 341 | 15 | 18 | 4 | 3 | HCC |
| 343 | 14 | 17 | 11 | 3 | HCC |
| 391 | 15 | 17 | 3 | 7 | HCC |
| 6 | 17 | 19 | 1 | 10 | HCC |
| 55 | 17 | 19 | 3 | 10 | HCC |
| 59 | 20 | 22 | 2 | 20 | HCC |
| Treated with anti-FasL mAb (intraperitoneally) | |||||
| 33 | 7 | 17 | 0 | 0 | |
| 93 | 7 | 12 | 0 | 0 | |
| 96 | 7 | 12 | 0 | 0 | |
| 135 | 9 | 16 | 0 | 0 | |
| 136 | 13 | 20 | 1 | 3 | Adenoma |
| 147 | 13 | 20 | 0 | 0 | |
| 179 | 12 | 16 | 0 | 0 | |
| 287 | 18 | 20 | 0 | 0 | |
| 243 | 17 | 23 | 1 | 12 | HCC |
| 258 | 18 | 21 | 0 | 0 | |
| 283 | 17 | 19 | 0 | 0 | |
| 349 | 12 | 14 | 0 | 0 | |
| 351 | 15 | 17 | 0 | 0 | |
| 360 | 15 | 17 | 0 | 0 | |
| 78 | 23 | 25 | 0 | 0 | |
spl., splenocyte.
Diameter in the major axis.
Discussion
In this model, the major mechanism of hepatocyte injury seemed to be apoptosis, because we observed massive apoptotic hepatocytes associated with an elevation of the serum ALT level. The anti-FasL mAb treatment markedly attenuated the hepatocyte apoptosis. However, because the proportion of HBsAg-reactive CD8+ T cells to the total population of infiltrates in the liver was so low (Table I), it is unlikely that FasL expressed on the surface of CTLs induced all the apoptosis. Recent studies have established that FasL has a proinflammatory activity (21–23). We have previously demonstrated that FasL induces the release of the activated form of proinflammatory cytokines such as IL-1β and IL-18 from neutrophils and/or macrophages (19, 20). Consistent with this, we found strong inflammatory infiltration in the liver and an elevated serum IL-18 level in this model, and anti-FasL mAb treatment reduced them (Fig. 1, B and D). Thus, it is more likely that the massive apoptosis was induced by inflammation that was exaggerated by FasL.
It is widely believed that apoptosis is a mechanism for preventing oncogenesis. Therefore, one might be concerned that inhibition of FasL, an apoptosis-inducing factor, may increase the risk of cancer. However, our observations indicate that anti-FasL mAb prevented both apoptosis and HCC development. The precise mechanism for the HCC prevention by anti-FasL mAb is not perfectly clear at this moment. However, it is likely that inhibition of persistent inflammation is the major mechanism for it, because it has been suggested that persistent inflammation is a strong risk factor for virus-induced hepatocarcinogenesis (8–11), and HCC development in the animal model used here is strictly dependent on an immune response to HBsAg (12). Additional experiments involving other antiinflammatory treatment are necessary to confirm this notion. In addition, inhibition of hepatocyte proliferation by anti-FasL mAb treatment may be an important factor, because hepatocyte proliferation may enhance the chance of oncogenesis. We found many PCNA+ hepatocytes after the acute-phase injury, and anti-FasL antibody treatment reduced it. It is likely that massive cell loss by inflammation induced the regenerative proliferation of hepatocytes, and FasL might indirectly cause the latter by inducing the former. Alternatively, FasL may be directly involved in the inflammation-induced hepatocyte proliferation, because it was recently reported that FasL is involved in hepatocyte proliferation in the regenerating liver after partial hepatectomy (24).
The model used in this study is of obvious value in terms of the similarity of the disease process to human viral hepatitis. Meanwhile, there may be a need to tone down the view that the model is a faithful representation of the natural disease because these mice do not carry full HBV genome. However, HBV transgenic mouse lineages that contain full viral genome develop neither inflammation nor tumors in the liver spontaneously (25). Additional studies may be of great interest to examine whether adoptive transfer of splenocytes primed with HBsAg or other viral antigens induces persistent liver inflammation and reproduces the process of hepatocarcinogenesis in these lineages.
In any case, we have demonstrated here that the inhibition of FasL activity not only ameliorated acute liver injury but also chronic liver dysplasia and HCC development. These results provide a rationale for developing a therapy for hepatitis using anti-FasL antibody or inhibitors for the Fas signal transduction pathway.
Acknowledgments
We thank Dr. F.V. Chisari for kindly providing us the HBsAg transgenic mice and for his helpful comments on the manuscript. We also thank Ms. I. Hashitani for secretarial and technical assistance, and Ms. A. Nakano, Ms. Y. Hashizume, and Ms. Y. Hashimoto for technical assistance.
This study was supported in part by Special Coordination Funds for Promoting Science and Technology, and Grants-in-Aid for Scientific Research on Priority Areas (Diagnosis and Treatment of Cancer) from the Ministry of Education, Culture, Sports, Science and Technology, the Japanese Government.
References
- 1.Chisari, F.V., and C. Ferrari. 1995. Hepatitis B virus immunopathology. Springer Semin. Immunopathol. 17:261–281. [DOI] [PubMed] [Google Scholar]
- 2.Bertoletti, A., and M.K. Maini. 2000. Protection or damage: a dual role for the virus-specific cytotoxic T lymphocyte response in hepatitis B and C infection? Curr. Opin. Microbiol. 3:387–392. [DOI] [PubMed] [Google Scholar]
- 3.Ando, K., T. Moriyama, L.G. Guidotti, S. Wirth, R.D. Schreiber, H.J. Schlicht, S. Huang, and F.V. Chisari. 1993. Mechanisms of class I restricted immunopathology. A transgenic mouse model of fulminant hepatitis. J. Exp. Med. 178:1541–1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Suda, T., T. Okazaki, Y. Naito, T. Yokota, N. Arai, S. Ozaki, K. Nakao, and S. Nagata. 1995. Expression of the Fas ligand in cells of T cell lineage. J. Immunol. 154:3806–3813. [PubMed] [Google Scholar]
- 5.Lowin, B., M. Hahne, C. Mattmann, and J. Tschopp. 1994. Cytolytic T-cell cytotoxicity is mediated through perforin and Fas lytic pathways. Nature. 370:650–652. [DOI] [PubMed] [Google Scholar]
- 6.Kondo, T., T. Suda, H. Fukuyama, M. Adachi, and S. Nagata. 1997. Essential roles of the Fas ligand in the development of hepatitis. Nat. Med. 3:409–413. [DOI] [PubMed] [Google Scholar]
- 7.Nakamoto, Y., L.G. Guidotti, V. Pasquetto, R.D. Schreiber, and F.V. Chisari. 1997. Differential target cell sensitivity to CTL-activated death pathways in hepatitis B virus transgenic mice. J. Immunol. 158:5692–5697. [PubMed] [Google Scholar]
- 8.Robinson, W.S., L. Klote, and N. Aoki. 1990. Hepadnaviruses in cirrhotic liver and hepatocellular carcinoma. J. Med. Virol. 31:18–32. [DOI] [PubMed] [Google Scholar]
- 9.Colombo, M., R. de Franchis, E. Del Ninno, A. Sangiovanni, C. De Fazio, M. Tommasini, M.F. Donato, A. Piva, V. Di Carlo, and N. Dioguardi. 1991. Hepatocellular carcinoma in Italian patients with cirrhosis. N. Engl. J. Med. 325:675–680. [DOI] [PubMed] [Google Scholar]
- 10.Tsukuma, H., T. Hiyama, S. Tanaka, M. Nakao, T. Yabuuchi, T. Kitamura, K. Nakanishi, I. Fujimoto, A. Inoue, H. Yamazaki, et al. 1993. Risk factors for hepatocellular carcinoma among patients with chronic liver disease. N. Engl. J. Med. 328:1797–1801. [DOI] [PubMed] [Google Scholar]
- 11.Chisari, F.V., K. Klopchin, T. Moriyama, C. Pasquinelli, H.A. Dunsford, S. Sell, C.A. Pinkert, R.L. Brinster, and R.D. Palmiter. 1989. Molecular pathogenesis of hepatocellular carcinoma in hepatitis B virus transgenic mice. Cell. 59:1145–1156. [DOI] [PubMed] [Google Scholar]
- 12.Nakamoto, Y., L.G. Guidotti, C.V. Kuhlen, P. Fowler, and F.V. Chisari. 1998. Immune pathogenesis of hepatocellular carcinoma. J. Exp. Med. 188:341–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chisari, F.V., P. Filippi, A. McLachlan, D.R. Milich, M. Riggs, S. Lee, R.D. Palmiter, C.A. Pinkert, and R.L. Brinster. 1986. Expression of hepatitis B virus large envelope polypeptide inhibits hepatitis B surface antigen secretion in transgenic mice. J. Virol. 60:880–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wirth, S., L.G. Guidotti, K. Ando, H.J. Schlicht, and F.V. Chisari. 1995. Breaking tolerance leads to autoantibody production but not autoimmune liver disease in hepatitis B virus envelope transgenic mice. J. Immunol. 154:2504–2515. [PubMed] [Google Scholar]
- 15.Moriyama, T., S. Guilhot, K. Klopchin, B. Moss, C.A. Pinkert, R.D. Palmiter, R.L. Brinster, O. Kanagawa, and F.V. Chisari. 1990. Immunobiology and pathogenesis of hepatocellular injury in hepatitis B virus transgenic mice. Science. 248:361–364. [DOI] [PubMed] [Google Scholar]
- 16.Miwa, K., H. Hashimoto, T. Yatomi, N. Nakamura, S. Nagata, and T. Suda. 1999. Therapeutic effect of an anti-Fas ligand mAb on lethal graft-versus-host disease. Int. Immunol. 11:925–931. [DOI] [PubMed] [Google Scholar]
- 17.Matsui, K., T. Yoshimoto, H. Tsutsui, Y. Hyodo, N. Hayashi, K. Hiroishi, N. Kawada, H. Okamura, K. Nakanishi, and K. Higashino. 1997. Propionibacterium acnes treatment diminishes CD4+ NK1.1+ T cells but induces type I T cells in the liver by induction of IL-12 and IL-18 production from Kupffer cells. J. Immunol. 159:97–106. [PubMed] [Google Scholar]
- 18.Urase, K., E. Fujita, Y. Miho, Y. Kouroku, T. Mukasa, Y. Yagi, M.Y. Momoi, and T. Momoi. 1998. Detection of activated caspase-3 (CPP32) in the vertebrate nervous system during development by a cleavage site-directed antiserum. Brain Res. Dev. Brain Res. 111:77–87. [DOI] [PubMed] [Google Scholar]
- 19.Miwa, K., M. Asano, R. Horai, Y. Iwakura, S. Nagata, and T. Suda. 1998. Caspase 1-independent IL-1beta release and inflammation induced by the apoptosis inducer Fas ligand. Nat. Med. 4:1287–1292. [DOI] [PubMed] [Google Scholar]
- 20.Tsutsui, H., N. Kayagaki, K. Kuida, H. Nakano, N. Hayashi, K. Takeda, K. Matsui, S. Kashiwamura, T. Hada, S. Akira, et al. 1999. Caspase-1-independent, Fas/Fas ligand-mediated IL-18 secretion from macrophages causes acute liver injury in mice. Immunity. 11:359–367. [DOI] [PubMed] [Google Scholar]
- 21.Seino, K., N. Kayagaki, K. Okumura, and H. Yagita. 1997. Antitumor effect of locally produced CD95 ligand. Nat. Med. 3:165–170. [DOI] [PubMed] [Google Scholar]
- 22.Kang, S.M., D.B. Schneider, Z. Lin, D. Hanahan, D.A. Dichek, P.G. Stock, and S. Baekkeskov. 1997. Fas ligand expression in islets of Langerhans does not confer immune privilege and instead targets them for rapid destruction. Nat. Med. 3:738–743. [DOI] [PubMed] [Google Scholar]
- 23.Allison, J., H.M. Georgiou, A. Strasser, and D.L. Vaux. 1997. Transgenic expression of CD95 ligand on islet beta cells induces a granulocytic infiltration but does not confer immune privilege upon islet allografts. Proc. Natl. Acad. Sci. USA. 94:3943–3947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Desbarats, J., and M.K. Newell. 2000. Fas engagement accelerates liver regeneration after partial hepatectomy. Nat. Med. 6:920–923. [DOI] [PubMed] [Google Scholar]
- 25.Guidotti, L.G., B. Matzke, H. Schaller, and F.V. Chisari. 1995. High-level hepatitis B virus replication in transgenic mice. J. Virol. 69:6158–6169. [DOI] [PMC free article] [PubMed] [Google Scholar]