

Abstract
The guanine nucleotide exchange factor Vav1 regulates actin polymerization and contributes to cytotoxicity by natural killer (NK) cells. An open question is how Vav1 becomes activated and what receptor can signal upstream of actin cytoskeleton rearrangement upon NK cell contact with target cells. Using transfected insect cells that express ligands of human NK cell receptors, we show that engagement of the β2 integrin LFA-1 on NK cells by intercellular adhesion molecule (ICAM)-1 led to a tyrosine phosphorylation of Vav1 that was not sensitive to cholesterol depletion and to inhibition of actin polymerization. Vav1 phosphorylation was blocked by an inhibitor of Src-family kinases, and correlated with activation of its downstream effector PAK. Binding of activation receptor 2B4 to its ligand CD48 was not sufficient for Vav1 phosphorylation. However, coengagement of 2B4 with LFA-1 resulted in an enhancement of Vav1 phosphorylation that was sensitive to cholesterol depletion and to inhibition of actin polymerization. Vav1 was recruited to a detergent-resistant membrane (DRM) fraction only when 2B4 and LFA-1 were coengaged, but not after LFA-1 engagement. Therefore, binding of LFA-1 to ICAM-1 on target cells may initiate an early signaling cascade in NK cells through activation of Vav1, leading to cytoskeleton reorganization and amplification of signals from other activation receptors.
Keywords: natural killer cell, lipid raft, LFA-1, Vav1, 2B4
Introduction
Target cell lysis by NK cells is activated by a number of receptors that bind to ligands expressed on tumor cells and virus-infected cells. Several NK cell receptors signal for cytotoxicity through their association with partner chains that carry immunoreceptor tyrosine-based activation motifs (ITAM) in their cytoplasmic tails, such as DAP12 and the Fc receptor γ chain (1). The activation receptor NKG2D signals through its association with DAP10, which carries a YxxM cytoplasmic tyrosine motif, and, at least in the mouse, with the ITAM-containing DAP12 (2–4). However, neither ITAM-mediated signals nor NKG2D are necessary to induce natural cytotoxicity, as demonstrated with mouse NK cells deficient in both Syk and ZAP70 tyrosine kinases, and with target cells lacking NKG2D ligands (5). Other receptors that contribute to natural cytotoxicity include the β2 integrin LFA-1 (6), which is essential for adhesion to target cells, and a number of costimulatory receptors, such as CD2, 2B4 (CD244), and CD44 (7, 8).
In T cells, the initial signals that lead to enhanced adhesion through LFA-1, actin cytoskeleton rearrangement, and receptor clustering at the cellular interface upon contact with an APC are delivered by TCR engagement (9). The guanine nucleotide exchange factor (GEF) domain-containing Vav1, through activation of the small GTPases Rac1 and Cdc42, is an essential mediator of the TCR-induced cytoskeleton reorganization and polarization of cholesterol-rich lipid rafts (10–12). The GEF activity of Vav1 is enhanced by phosphorylation of several tyrosines in Vav1 (13). Vav1 and Rac1 both contribute to target cell lysis by NK cells (14–17), but the receptors and early signals that activate Vav1 are not known. NK cells rapidly form tight conjugates with cells expressing ICAM-1, through engagement of the β2 integrin LFA-1 (18). To test for the outcome of LFA-1 engagement by its natural ligand, and to avoid the multiple receptor–ligand interactions that occur between NK cells and mammalian target cells, we have transfected insect cells with ICAM-1, in combination with other ligands of human NK cell receptors (18). Transfected insect cells have been used successfully to define the minimal requirements for T cell activation (19). Here we show that contact of NK cells with insect cells expressing ICAM-1 alone resulted in a tyrosine phosphorylation of Vav1 that was not dependent on actin polymerization and on lipid rafts. In contrast, Vav1 phosphorylation induced by coengagement of 2B4 with LFA-1 was dependent on actin polymerization and on lipid rafts. Therefore, LFA-1 has the properties required of an early activation receptor on NK cells.
Materials and Methods
Antibodies and Cells.
For Western blot analysis, the following Abs were used: biotin-conjugated anti-phosphotyrosine (clone PY-20; Transduction Laboratories), mouse anti-Vav1 (Upstate), and anti-CD45 (Transduction Laboratories). For blocking experiments, anti-CD11a (clone TS1/22; Endogen) and anti-CD11b Abs (clone LM2/1; Biosource International) were used. The rabbit polyclonal antibodies H-211 to Vav1 and C-19 to PAK (Santa Cruz Biotechnology, Inc.) were used for immunoprecipitations. For cross-linking experiments we used the anti CD11a (BD Biosciences) and F(ab′)2 goat anti–mouse IgG (Jackson ImmunoResearch Laboratories). Human polyclonal NK cells were isolated from PBL using the MACS NK cell isolation kit (Miltenyi Biotech). NK cells and transfected Drosophila Schneider 2 (SC2) were obtained and maintained in culture as described (18).
Receptor Cross-Linking.
For antibody-mediated cross-linking of CD11a, 3 × 107 polyclonal NK cells were incubated with 5 μg of anti CD11a Ab for 15 min at 4°C. Cells were washed once with Iscove's medium and 50 μg/ml of F(ab′)2 goat anti–mouse were added. Cells were washed again and cross-linking was performed for the indicated times at 37°C. Cells were centrifuged and lysed before Western blot analysis.
Cell Mixing.
NK and target cells were mixed at an effector to target ratio of 6:1, briefly centrifuged, and allowed to recover for 20 min at room temperature before incubation at 37°C for 20 min. Cells were then lysed in ice-cold lysis buffer (1% Triton X-100, 50 mM NaCl, 10 mM EDTA, 50 mM NaFluoride, 10 mM NaPhosphate, 1 mM NaVO4, PMSF). For detergent-resistant membrane (DRM) experiments, cell mixing of 3 × 108 polyclonal human NK cells were mixed with 0.5 × 108 SC2 transfectants, as described above.
In Vitro Kinase Assay.
NK and target cells were mixed at a ratio of 6:1 and allowed to recover for 10 min at room temperature before incubation at 37°C for 5 min. Cells were then lysed in 1% NP-40, 140 mM NaCl, 1 mM EDTA, 10 mM Tris-HCl, pH 8, 1 mM NaVO4, and PMSF. Lysates were centrifuged at 14,000 g for 20 min and precleared with a control Ab for 1 h, followed by immunoprecipitation with polyclonal anti-PAK. Immunoprecipitated proteins were washed 3 times in lysis buffer and incubated with 10 μCi of [γ-32P] ATP in 30 mM Tris, 20 mM MgCl2, 2 mM MnCl2, 10 μM ATP, and 5 μg myelin basic protein (MBP; Sigma-Aldrich) for 30 min at 30°C. Sample buffer was added and the mixture was electrophoresed on a 14% SDS gel. After drying, 32P-labeled MBP was detected by autoradiography.
Blocking Experiments.
For antibody blocking experiments, NK cells were preincubated with 10 μg/ml of purified CD11a or CD11b mAb for 30 min at 4°C, and washed once before cell mixing. For inhibition of tyrosine kinases and of actin polymerization, NK cells were preincubated for 40 min at 37°C with 30 μM PP1, 12 μM Piceatannol, 20 μM cytochalasin D (BIOMOL Research Laboratories, Inc.), or 20 μM Latrunculin A (Molecular Probes) and used in mixing experiments in the presence of the inhibitors. For cholesterol depletion, NK cells were washed in PBS, incubated 30 min at 37°C with 12.5 mM methyl-β-Cyclodextrin (MCD; Sigma-Aldrich), and washed again before cell mixing experiments.
Immunoprecipitation and Western Blot Analysis.
Cells were lysed in ice-cold lysis buffer. Lysates were centrifuged at 12,000 g for 20 min and supernatants were precleared with protein G for 1 h. Specific immunoprecipitation was performed overnight at 4°C. Total proteins or immunoprecipitated proteins were separated by SDS-PAGE. The gels were blotted onto PVDF membranes (Immobilon P; Millipore). Membranes were then blocked 30 min with 5% BSA in PBS 0.1% Tween-20 and incubated overnight with the specific Ab at 4°C. After washing, the membranes were incubated with streptavidin-horseradish peroxidase-conjugate (Amersham Biosciences) or goat anti–mouse IgG peroxidase (Jackson ImmunoResearch Laboratories), washed, stained with enhanced chemiluminescence reagent (Pierce Chemical Co.), and exposed to X-ray film.
Isolation of DRM.
DRMs were isolated by ultracentrifugation in a sucrose gradient as described (20) except that cells were lysed in a buffer containing 10 mM Tris-HCl, pH 7.4, 150 mM NaCl, 5 mM EDTA, and 0.5% Triton X-100. The fractions containing DRM were identified by Western blotting with peroxidase-conjugated Cholera Toxin B-subunit (Sigma-Aldrich).
Results and Discussion
Engagement of LFA-1 on Human NK Cells Induces Tyrosine Phosphorylation of Vav1.
To investigate the signals delivered by LFA-1 alone, a mAb specific for CD11a was used to cross-link LFA-1 on polyclonal NK cells. Western blotting of cell lysates with the anti-phosphotyrosine mAb PY-20 revealed tyrosine-phosphorylated proteins specifically induced by LFA-1 cross-linking for 1, 5, and 10 min (Fig. 1 A). No tyrosine phosphorylation occurred by cross-linking CD56 on NK cells (unpublished data). One of the prominent tyrosine phosphorylated proteins was similar in size to Vav1, as shown by reprobing the membrane with an anti-Vav1 Ab (Fig. 1 A). Immunoprecipitations with a rabbit anti-Vav1 Ab and blotting for tyrosine phosphorylation showed that Vav1 was phosphorylated after 5 and 10 min of CD11a cross-linking (Fig. 1 B). No phosphorylated Vav1 was detected after CD56 cross-linking (unpublished data). These results are similar to the Vav1 phosphorylation detected in neutrophils after cross-linking β2 integrin with mAb (21), as well as Vav1 phosphorylation in NK cells after cross-linking β1 integrin with mAb (22). Secretion of TNFα by NK cells upon cross-linking of LFA-1 by mAb, even in the absence of cell aggregation, suggested also a signaling function of LFA-1 (23).
Figure 1.

Tyrosine phosphorylation of Vav1 after antibody-induced engagement of LFA-1 in NK cells. LFA-1 on NK cells was cross-linked with anti-CD11a and a F(ab′)2 goat anti–mouse antibody for the indicated times at 37°C. (A) Cell lysates were resolved by 4–20% SDS-PAGE and immunoblotted with biotinylated anti phosphotyrosine Ab, PY-20 (p-Tyr, left). (B) Vav1 was immunoprecipitated from cell lysates, separated on 6% SDS-PAGE, and blotted with PY-20 mAb (p-Tyr, left). The same blots were reprobed with an anti-Vav1 Ab (Vav1, right).
To test the outcome of LFA-1 engagement in a more physiological context, NK cells were mixed with the Drosophila SC2 cell line expressing human ICAM-1 (SC2-ICAM). Phospho-Vav1 was detected in NK cells mixed with SC2-ICAM, but not with SC2 cells, after immunoprecipitation of Vav1 and blotting with anti-phosphotyrosine mAb PY-20 (Fig. 2 A). Experiments with unmixed SC2 and SC2-ICAM cells confirmed that anti-Vav1 antibody did not precipitate nonspecific bands from SC2 cells (unpublished data). NK cells express the two β2 integrins LFA-1 and Mac-1. Blocking experiments with Abs to CD11a (LFA-1) and CD11b (Mac-1) showed that Vav1 phosphorylation was induced mainly by LFA-1 (Fig. 2 A). We therefore conclude that ICAM-1, expressed on SC2 cells, induces Vav1 phosphorylation in NK cells mostly through interaction with LFA-1.
Figure 2.
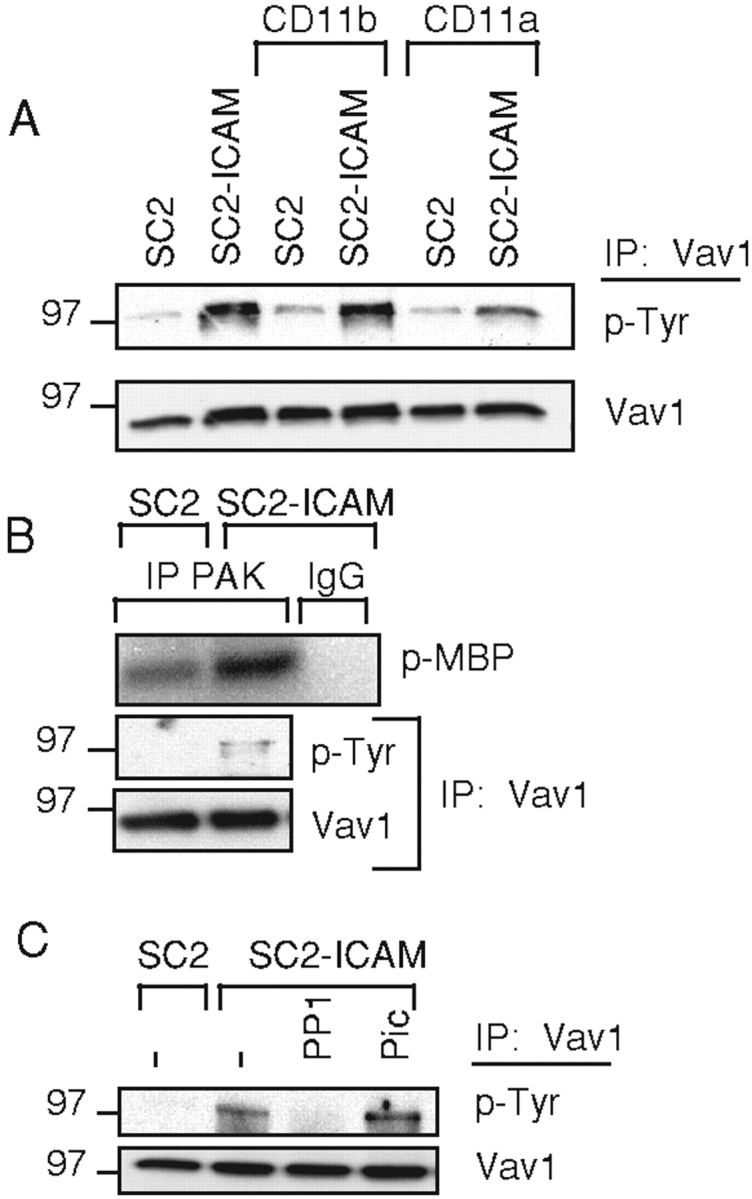
Phosphorylation of Vav1 induced by engagement of LFA-1 on NK cells. (A) NK cells, pretreated with anti-CD11b or anti-CD11a, were mixed with SC2 or SC2-ICAM-1 target cells. Vav1 was immunoprecipitated from cell lysates, resolved on a 6% SDS-PAGE, and blotted with biotinylated anti phosphotyrosine Ab PY-20 (p-Tyr). The same blot was reblotted with an anti-Vav1 Ab (Vav1). (B) PAK kinase activity in NK cells mixed with SC2 and SC2-ICAM cells. PAK or control (rabbit IgG) immunoprecipitates were subjected to an in vitro kinase assay. Phosphorylation of the MBP substrate (p-MBP) was detected by autoradiography. Vav1 was immunoprecipitated from the supernatant of the PAK immunoprecipitation, and tested for tyrosine phosphorylation as in (A). (C) Inhibition of Vav1 tyrosine phosphorylation. Vav1 phosphorylation was tested in NK cells either untreated (−), treated with PP1, or treated with Piceatannol (Pic), after mixing with SC2 and SC2-ICAM cells, as indicated.
To test if the phosphorylation of Vav1 was accompanied by enhanced GEF activity, the kinase activity of PAK, a downstream effector of Vav1-Rac1 signals, was measured after mixing NK cells with SC2 cells. Increased PAK activity in NK cells was detected after mixing with SC2-ICAM cells for 5 min (Fig. 2 B). Vav1 phosphorylation in the same cells was also detected (Fig. 2 B). Whereas Vav1 phosphorylation increased between 5 and 20 min, PAK activity decreased (unpublished data), indicating a complex regulation of this signaling pathway. The GEF activity of Vav1 is enhanced by phosphorylation through Src-family kinases (13), and Vav1 phosphorylation during antigen-specific activation of T cells requires Fyn (24). On the other hand, ligation of β2 integrin on neutrophils activates the tyrosine kinase Syk, which is required for neutrophil activation (25), and attachment to fibrinogen of CHO cells expressing β3 integrin results in a Syk-dependent Vav1 phosphorylation (26). We therefore tested if Vav1 phosphorylation induced by LFA-1 in NK cells required a Src-family kinase, a Syk/ZAP70 kinase, or both. Treatment of NK cells with PP1, an inhibitor of src-family kinases, but not with Piceatannol, an inhibitor of the Syk/ZAP70 kinases, blocked the Vav1 phosphorylation induced by SC2-ICAM cells (Fig. 2 C). In parallel, phosphorylation of receptor 2B4 was monitored after incubation of the same NK cells with a human target cell expressing the 2B4 ligand CD48. As reported previously (27), phosphorylation of 2B4 was completely inhibited by PP1 and was greatly reduced by Piceatannol (unpublished data). These results indicate that Src-family kinase activity but not Syk or ZAP70 is required for the LFA-1–induced phosphorylation of Vav1.
Vav1 phosphorylation was measured after incubation of NK cells with SC2 cells expressing ICAM-1, CD48 (SC2-CD48), or coexpressing both (SC2-ICAM-CD48). Cell sorting was used to obtain matched levels of ICAM-1 expressed on SC2-ICAM and SC2-ICAM-CD48 cells, and of CD48 on SC2-CD48 and SC2-ICAM-CD48 cells (18). Vav1 phosphorylation was not detectable after mixing with SC2-CD48 cells (Fig. 3 A). This result is not simply due to a lack of 2B4 signaling because strong tyrosine phosphorylation of 2B4 occurs after mixing with SC2-CD48 cells (unpublished data). On the other hand, coengagement of LFA-1 and 2B4 led to an increase in Vav1 phosphorylation, as compared with that obtained by engagement of LFA-1 alone (Fig. 3 A). Therefore, signals through LFA-1 but not 2B4 lead to a phosphorylation of Vav1 which can be enhanced by 2B4 signals. The possibility that another receptor on NK cells binds to something on insect cells cannot be excluded. However, if such a receptor were to exist it is not sufficient to induce Vav1 phosphorylation, nor can it enhance the LFA-1–induced Vav1 phosphorylation as well as 2B4 does.
Figure 3.
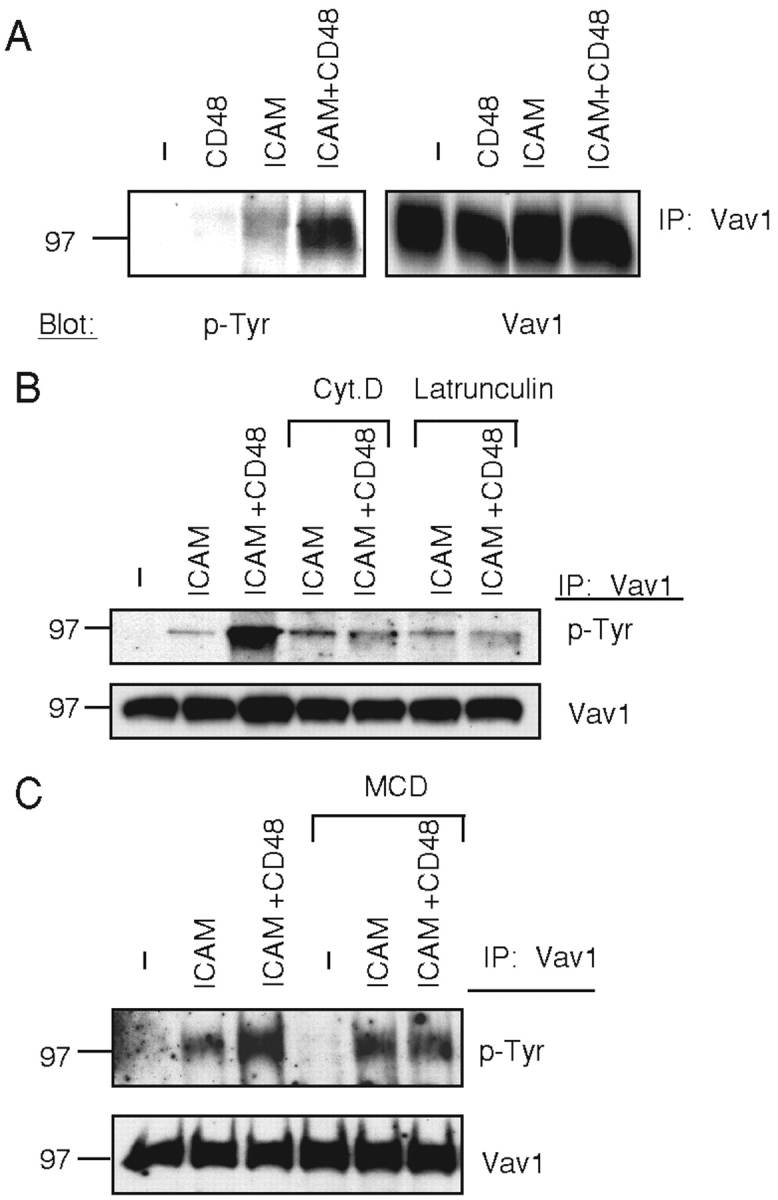
Phosphorylation of Vav1 induced by engagement of LFA-1 and of 2B4 on NK cells. (A) NK cells were mixed with SC2 (−), SC2-CD48 (CD48), SC2-ICAM-1 (ICAM), or SC2-ICAM-CD48 (ICAM+CD48) target cells. Vav1 was immunoprecipitated, resolved on a 6% SDS-PAGE, and blotted with biotinylated-PY-20 (p-Tyr, left). The same blot was reblotted with an anti-Vav1 Ab (Vav1). (B and C) Vav1 phosphorylation induced by LFA-1 is independent of actin polymerization and of lipid rafts. NK cells treated with cytochalasin D (Cyt.D) and Latrunculin A (B), or with MCD (C) were mixed with SC2 (−), SC2-ICAM (ICAM), or SC2-ICAM-CD48 (ICAM+CD48) cells. Vav1 phosphorylation was detected as in (A).
Role of Actin Cytoskeleton and of Lipid Rafts in Vav1 Phosphorylation.
Work in T cells has demonstrated that actin cytoskeleton rearrangement and lipid raft polarization induced by contact with an APC are dependent on Vav1 activity (10–12). Therefore, early signals upstream of actin polymerization that induce Vav1 phosphorylation must exist. Activation receptor 2B4 on NK cells is not likely to provide such early signals because 2B4 phosphorylation is itself dependent on actin polymerization (20). To test if LFA-1 engagement on NK cells can activate Vav1 upstream of cytoskeleton rearrangements, actin polymerization was blocked by treatment with cytochalasin D and Latrunculin A. As shown in Fig. 3 B, these inhibitors did not block the Vav1 phosphorylation induced by SC2-ICAM cells. In contrast, and consistent with the actin polymerization-dependent phosphorylation of 2B4 (20), the enhancement of Vav1 phosphorylation due to coengagement of 2B4 with LFA-1 was blocked by cytochalasin D and Latrunculin A (Fig. 3 B). These results show that these inhibitors were effective in blocking the 2B4 signal leading to enhanced Vav1 phosphorylation, but not the LFA-1 signal.
Cholesterol-rich membrane domains, referred to as lipid rafts or DRM domains, contribute to cytotoxicity of NK cells, and lipid raft clustering at the NK-target cell contact correlates with target cell lysis (28). Furthermore, phosphorylation of 2B4 and 2B4-mediated NK cell activation are sensitive to cholesterol depletion, and phosphorylated 2B4 is found exclusively in DRM domains (20). Cholesterol was depleted from NK cell membranes by treatment with MCD in order to test whether Vav1 phosphorylation induced by LFA-1 required lipid rafts. Cholesterol depletion did not block Vav1 phosphorylation induced by LFA-1, but did prevent the 2B4-induced enhancement in Vav1 phosphorylation (Fig. 3 C). These results show that engagement of LFA-1 by its natural ligand on target cells leads to Vav1 tyrosine phosphorylation without a requirement for actin polymerization or lipid rafts. Therefore, signals upstream of actin polymerization and lipid raft clustering induce Vav1 phosphorylation, which is then enhanced by downstream signals. Similarly, in T cells activated by TCR engagement a first wave of Vav1 tyrosine phosphorylation independent of lipid rafts was detected, whereas a second wave of Vav1 phosphorylation occurred after raft coalescence and F-actin reorganization (29). In addition, once it is activated, LFA-1 on T cells induces actin polymerization upon binding to ICAM-1 (30).
The recruitment of Vav1 into DRM domains was evaluated. The separation of DRM from soluble membrane fractions by sucrose gradient ultracentrifugation was monitored by blotting gradient fractions with Cholera Toxin subunit B, which binds to the DRM-resident ganglioside GM1, and for CD45, which resides in the soluble fraction (Fig. 4 A). In nonstimulated NK cells, Vav1 was present exclusively in the detergent-soluble fractions (Fig. 4 A). DRM and soluble membrane domains were isolated from NK cells mixed with SC2, SC2-ICAM, and SC2-ICAM-CD48 cells (Fig. 4 B). Immunoprecipitated Vav1 was probed by Western blot with an anti-phosphotyrosine mAb and with an anti-Vav1 mAb. The results with NK cells mixed with SC2-ICAM cells confirmed the actin polymerization-independent phosphorylation of Vav1 and showed that phospho-Vav1 remained in the soluble fraction (Fig. 4 B). The actin polymerization-dependent enhancement of Vav1 phosphorylation in NK cells mixed with SC2-ICAM-CD48 cells was confirmed (Fig. 4 B). In addition, Vav1 translocation into DRM was detected, and DRM-associated Vav1 was phosphorylated. Movement of Vav1 to DRM was sensitive to cytochalasin D (Fig. 4 B). Therefore, recruitment of Vav1 to lipid rafts requires signals that are downstream of actin polymerization.
Figure 4.

Vav1 phosphorylation and translocation into DRM fractions. (A) DRM domains of polyclonal NK cells were characterized by Western blot analysis. Blots were probed with cholera toxin B subunit (CtxB) and anti-CD45 Ab (CD45). Vav1 immunoprecipitated from gradient fractions 2–12 was Western blotted with anti-Vav1 Ab (Vav1). (B) NK cells that were pretreated with (+) or without (−) cytochalasin D (Cyt D), were mixed with SC2 (−), SC2-ICAM (ICAM), or SC2- ICAM-CD48 (ICAM+CD48) cells. Cells were lysed, and DRM isolated using sucrose gradient ultracentrifugation. Fractions 3, 4, and 5 were pooled and correspond to the DRM fractions (DRM), whereas pooled fractions 10, 11, and 12 correspond to the soluble fraction (soluble). Lysates of each pool were analyzed by Western blot with cholera toxin B subunit (CtxB) and anti-CD45 (CD45) Ab. Immunoprecipitated Vav1 was detected by Western blot with anti Vav1 Ab (Vav1). Phospho-Vav1 was detected by blotting the same membrane with an anti-phosphotyrosine Ab, PY-20 (p-Tyr).
Conclusions.
Our results show that engagement of LFA-1 by ICAM-1 on target cells leads to a tyrosine phosphorylation of Vav1 that occurs upstream of actin polymerization and of lipid raft clustering. Thus, LFA-1 may provide an essential early signal to NK cells upon contact with ICAM-expressing target cells, which leads to downstream signals via Vav1 that are required for further receptor clustering. The LFA-1 signal is most likely relevant to NK cell effector function, as NK cells lysed insect cells expressing ICAM-1 (unpublished data). We propose the following model for the regulation of NK cell cytotoxicity. Upon contact with ICAM-expressing target cells, NK cells adhere rapidly through engagement of LFA-1 (18). In addition to its role in adhesion, LFA-1 delivers activation signals that result in tyrosine phosphorylation of Vav1 by a Src-family kinase, and Rac1 activation. Cytoskeletal rearrangements induce clustering and phosphorylation of activation receptors, such as 2B4, in lipid rafts (20). In turn, 2B4 contributes additional signals for the activation of cytotoxicity, including Vav1 translocation into lipid rafts, and enhanced Vav1 phosphorylation. The actin polymerization- and lipid raft-dependent 2B4 signaling provides a point at which NK cell cytotoxicity can be controlled. Indeed, coengagement of MHC class I–specific inhibitory receptors on NK cells blocks the recruitment of 2B4 into lipid rafts, thereby preventing NK cell activation at an early step (20).
Binding of LFA-1 to ICAM-1 on insect cells induces signals that lead to cytotoxicity by NK cells. β2 integrin binding to ICAM-1 induces degranulation by activated neutrophils (25). In contrast to T cells, which rely on the TCR to initiate activation signals and up-regulate LFA-1–mediated adhesion, innate cells such as NK cells and neutrophils use LFA-1 directly to transmit activation signals.
Acknowledgments
We gratefully acknowledge Y. Bryceson, S. Rajagopalan, and C. Watzl for help in NK cell isolation, as well as M. Faure, C. Watzl, A. Cherukuri, and S. Pierce for helpful discussions.
Note added in proof: Consistent with the model proposed here for the regulation of NK cell cytotoxicity, phosphorylated Vav1 in NK cells is a specific substrate of tyrosine phosphatase SHP-1 during inhibition mediated by binding of inhibitory receptor to MHC class I on target cells (31).
References
- 1.Moretta, A., C. Bottino, M. Vitale, D. Pende, C. Cantoni, M.C. Mingari, R. Biassoni, and L. Moretta. 2001. Activating receptors and coreceptors involved in human natural killer cell-mediated cytolysis. Annu. Rev. Immunol. 19:197–223. [DOI] [PubMed] [Google Scholar]
- 2.Cerwenka, A., and L.L. Lanier. 2001. Natural killer cells, viruses, and cancer. Nat. Rev. Immunol. 1:41–49. [DOI] [PubMed] [Google Scholar]
- 3.Diefenbach, A., E. Tomasello, M. Lucas, A.M. Jamieson, J.K. Hsia, E. Vivier, and D.H. Raulet. 2002. Selective associations with signaling proteins determine stimulatory versus costimulatory activity of NKG2D. Nat. Immunol. 3:1142–1149. [DOI] [PubMed] [Google Scholar]
- 4.Gilfillan, S., E.L. Ho, M. Cella, W.M. Yokoyama, and M. Colonna. 2002. NKG2D recruits two distinct adapters to trigger NK cell activation and costimulation. Nat. Immunol. 3:1150–1155. [DOI] [PubMed] [Google Scholar]
- 5.Colucci, F., E. Schweighoffer, E. Tomasello, M. Turner, J.R. Ortaldo, E. Vivier, V.L. Tybulewicz, and J.P. Di Santo. 2002. Natural cytotoxicity uncoupled from the Syk and ZAP-70 intracellular kinases. Nat. Immunol. 3:288–294. [DOI] [PubMed] [Google Scholar]
- 6.Helander, T.S., O. Carpén, O. Turunen, P.E. Kovanen, A. Vaheri, and T. Timonen. 1996. ICAM-2 redistributed by ezrin as a target for killer cells. Nature. 382:265–268. [DOI] [PubMed] [Google Scholar]
- 7.Matsumoto, G., M.P. Nghiem, N. Nozaki, R. Schmits, and J.M. Penninger. 1998. Cooperation between CD44 and LFA-1/CD11a adhesion receptors in lymphokine-activated killer cell cytotoxicity. J. Immunol. 160:5781–5789. [PubMed] [Google Scholar]
- 8.Lanier, L.L. 2000. Turning on natural killer cells. J. Exp. Med. 191:1259–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bromley, S.K., W.R. Burack, K.G. Johnson, K. Somersalo, T.N. Sims, C. Sumen, M.M. Davis, A.S. Shaw, P.M. Allen, and M.L. Dustin. 2001. The immunological synapse. Annu. Rev. Immunol. 19:375–396. [DOI] [PubMed] [Google Scholar]
- 10.Penninger, J.M., and G.R. Crabtree. 1999. The actin cytoskeleton and lymphocyte activation. Cell. 96:9–12. [DOI] [PubMed] [Google Scholar]
- 11.Wülfing, C., A. Bauch, G.R. Crabtree, and M.M. Davis. 2000. The vav exchange factor is an essential regulator in actin-dependent receptor translocation to the lymphocyte-antigen-presenting cell interface. Proc. Natl. Acad. Sci. USA. 97:10150–10155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Villalba, M., K. Bi, F. Rodriguez, Y. Tanaka, S. Schoenberger, and A. Altman. 2001. Vav1/Rac-dependent actin cytoskeleton reorganization is required for lipid raft clustering in T cells. J. Cell Biol. 155:331–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bustelo, X.R. 2000. Regulatory and signaling properties of the Vav family. Mol. Cell. Biol. 20:1461–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Billadeau, D.D., K.M. Brumbaugh, C.J. Dick, R.A. Schoon, X.R. Bustelo, and P.J. Leibson. 1998. The Vav-Rac1 pathway in cytotoxic lymphocytes regulates the generation of cell-mediated killing. J. Exp. Med. 188:549–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Galandrini, R., G. Palmieri, M. Piccoli, L. Frati, and A. Santoni. 1999. Role for the Rac1 exchange factor Vav in the signaling pathways leading to NK cell cytotoxicity. J. Immunol. 162:3148–3152. [PubMed] [Google Scholar]
- 16.Jiang, K., B. Zhong, D.L. Gilvary, B.C. Corliss, E. Hong-Geller, S. Wei, and J.Y. Djeu. 2000. Pivotal role of phosphoinositide-3 kinase in regulation of cytotoxicity in natural killer cells. Nat. Immunol. 1:419–425. [DOI] [PubMed] [Google Scholar]
- 17.Colucci, F., E. Rosmaraki, S. Bregenholt, S.I. Samson, V. Di Bartolo, M. Turner, L. Vanes, V. Tybulewicz, and J.P. Di Santo. 2001. Functional dichotomy in natural killer cell signaling. Vav1-dependent and -independent mechanisms. J. Exp. Med. 193:1413–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Barber, D.F., and E.O. Long. 2003. Coexpression of CD58 or CD48 with intercellular adhesion molecule 1 on target cells enhances adhesion of resting NK cells. J. Immunol. 170:294–299. [DOI] [PubMed] [Google Scholar]
- 19.Cai, Z., A. Brunmark, M.R. Jackson, D. Loh, P.A. Peterson, and J. Sprent. 1996. Transfected Drosophila cells as a probe for defining the minimal requirements for stimulating unprimed CD8+ T cells. Proc. Natl. Acad. Sci. USA. 93:14736–14741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Watzl, C., and E.O. Long. 2003. Natural killer cell inhibitory receptors block actin cytoskeleton-dependent recruitment of 2B4 (CD244) to lipid rafts. J. Exp. Med. 197:77–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zheng, L., A. Sjolander, J. Eckerdal, and T. Andersson. 1996. Antibody-induced engagement of beta 2 integrins on adherent human neutrophils triggers activation of p21ras through tyrosine phosphorylation of the protooncogene product Vav. Proc. Natl. Acad. Sci. USA. 93:8431–8436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mainiero, F., A. Soriani, R. Strippoli, J. Jacobelli, A. Gismondi, M. Piccoli, L. Frati, and A. Santoni. 2000. RAC1/P38 MAPK signaling pathway controls β1 integrin-induced interleukin-8 production in human natural killer cells. Immunity. 12:7–16. [DOI] [PubMed] [Google Scholar]
- 23.Melero, I., M.A. Balboa, J.L. Alonso, E. Yague, J.P. Pivel, F. Sanchez-Madrid, and M. López-Botet. 1993. Signaling through the LFA-1 leucocyte integrin actively regulates intercellular adhesion and tumor necrosis factor-alpha production in natural killer cells. Eur. J. Immunol. 23:1859–1865. [DOI] [PubMed] [Google Scholar]
- 24.Huang, J., D. Tilly, A. Altman, K. Sugie, and H.M. Grey. 2000. T-cell receptor antagonists induce Vav phosphorylation by selective activation of Fyn kinase. Proc. Natl. Acad. Sci. USA. 97:10923–10929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mócsai, A., M.J. Zhou, F.Y. Meng, V.L. Tybulewicz, and C.A. Lowell. 2002. Syk is required for integrin signaling in neutrophils. Immunity. 16:547–558. [DOI] [PubMed] [Google Scholar]
- 26.Miranti, C.K., L. Leng, P. Maschberger, J.S. Brugge, and S.J. Shattil. 1998. Identification of a novel integrin signaling pathway involving the kinase Syk and the guanine nucleotide exchange factor Vav1. Curr. Biol. 8:1289–1299. [DOI] [PubMed] [Google Scholar]
- 27.Long, E.O., D.F. Barber, D.N. Burshtyn, M. Faure, M. Peterson, S. Rajagopalan, V. Renard, M. Sandusky, C.C. Stebbins, N. Wagtmann, et al. 2001. Inhibition of natural killer cell activation signals by killer cell immunoglobulin-like receptors (CD158). Immunol. Rev. 181:223–233. [DOI] [PubMed] [Google Scholar]
- 28.Lou, Z.K., D. Jevremovic, D.D. Billadeau, and P.J. Leibson. 2000. A balance between positive and negative signals in cytotoxic lymphocytes regulates the polarization of lipid rafts during the development of cell-mediated killing. J. Exp. Med. 191:347–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Valensin, S., S.R. Paccani, C. Ulivieri, D. Mercati, S. Pacini, L. Patrussi, T. Hirst, P. Lupetti, and C.T. Baldari. 2002. F-actin dynamics control segregation of the TCR signaling cascade to clustered lipid rafts. Eur. J. Immunol. 32:435–446. [DOI] [PubMed] [Google Scholar]
- 30.Porter, J.C., M. Bracke, A. Smith, D. Davies, and N. Hogg. 2002. Signaling through integrin LFA-1 leads to filamentous actin polymerization and remodeling, resulting in enhanced T cell adhesion. J. Immunol. 168:6330–6335. [DOI] [PubMed] [Google Scholar]
- 31.Stebbins, C.C., C. Watzl, D.D. Billadeau, P.J. Leibson, D.N. Burshtyn, and E.O. Long. 2003. Vav1 dephosphorylation by the phosphatase SHP-1 as a mechanism for inhibition of cellular cytotoxicity. Mol. Cell. Biol. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]