

Abstract
The production of interleukin (IL)-12 is critical for the development of interferon (IFN)-γ–dependent resistance to Toxoplasma gondii. Nevertheless, when this response is dysregulated, such as occurs in the absence of IL-10, the uncontrolled inflammation that results can have lethal consequences for the host. Recently, we demonstrated that lipoxin (LX)A4, an eicosanoid mediator that depends on 5-lipoxygenase (LO) for its biosynthesis, exerts a regulatory role on dendritic cell IL-12 production triggered artificially by a T. gondii extract. We now formally establish the physiological relevance of this pathway in the systemic control of IL-12 production induced by live T. gondii infection and demonstrate its function to be distinct from that of IL-10. Thus, T. gondii–exposed wild-type, but not 5-LO–deficient animals, produced high levels of serum LXA4 beginning at the onset of chronic infection. Moreover, 5-LO−/−, in contrast to wild-type mice, succumbed during the same period displaying a marked encephalitis. The increased mortality of the 5-LO−/− animals was also associated with significant elevations of IL-12 and IFN-γ and was completely prevented by the administration of a stable LXA4 analogue. Together, these findings demonstrate a new pathway involving the induction of host LXs for the in vivo regulation of proinflammatory responses during microbial infection.
Keywords: Toxoplasma gondii, interleukin-12, eicosanoid, lipoxygenase, lipoxin A4
Introduction
Cell-mediated immune responses to infectious agents must be carefully regulated to avoid damage to host tissues. Cytokines, such as IL-12, IFN-γ, and TNF, which are critical for host resistance to many intracellular pathogens, are also detrimental when expressed in an uncontrolled manner (1). Murine infection with the protozoan parasite Toxoplasma gondii provides an excellent example of this concept. IL-12–dependent production of IFN-γ is essential for the control of both acute and chronic T. gondii infection (2). At the same time, IL-10–deficient mice, which display normal resistance to infection, succumb to excessive tissue damage associated with the overproduction of the proinflammatory cytokines listed above (3, 4). Similar scenarios in which dysregulated pathogen-induced cytokine production results in inflammatory disease have been observed in a wide variety of parasitic (5) and bacterial (6, 7) infections.
In addition to down-regulatory cytokines, other endogenous mediators may play an important role in controlling excess production of host-protective proinflammatory cytokines. In the process of studying the response of murine dendritic cells (DCs)* to T. gondii, we recently uncovered a novel mechanism for the regulation of IL-12 production involving lipoxin (LX)A4, an eicosanoid product generated from arachidonic acid. Splenic DCs from mice injected with soluble tachyzoite antigen (STAg) of T. gondii produce high levels of IL-12 by a mechanism dependent on the cell-surface expression of the chemokine receptor CCR5 (8). However, this response is short-lived and cannot be restimulated with STAg for a period of ∼1 wk (9). We found that the observed loss of responsiveness was associated with reduced expression of CCR5 by DCs, a phenomenon dependent on the presence of an enzyme, 5-lypoxygenase (5-LO), critical in the generation of LXA4. Moreover, suppressed IL-12 production by DCs could be induced exogenously by the administration of a stable LXA4 mimetic (10). Other eicosanoids generated by the 5-LO pathway including leukotrienes (LTB4, LTC4, and LTD4) as well as 5-Oxo-ETE, 5-HETE, and a battery of related compounds were not able to mimic the activity of LXA4 when tested in the nanogram to microgram range. On the basis of the evidence above, we hypothesized that the induction of LXA4 by STAg is the mechanism responsible for the down-regulation of CCR5-dependent IL-12 production observed after the administration of this microbial stimulus.
Although LXA4 is able to potently suppress T. gondii–induced IL-12 production by DCs, our previous demonstration of this phenomenon involved a nonphysiological model system using STAg injection. A critical question remained as to whether the same mechanism functions in the regulation of IL-12 and IL-12–dependent IFN-γ synthesis during natural infection with T. gondii and whether it impacts on the outcome of disease. In this study, we have addressed this issue by investigating the induction of LXA4 after the infection of mice with an avirulent strain of the parasite and after the fate of such infections in 5-LO−/− animals incapable of generating the eicosanoid. Our findings reveal a critical role for T. gondii–induced LXA4, distinct from that of IL-10 in the regulation of IL-12–dependent IFN-γ production and tissue damage during infection with this protozoan. Thus, they demonstrate a previously unrecognized function for this lipid mediator in modulating the inflammatory response triggered in vivo by infectious agents.
Materials and Methods
Animals.
Wild-type controls (B6, 129S F2/J) and 5-LO–deficient (B6, 129S Alox-5) mice were obtained from The Jackson Laboratory and bred and maintained in an NIAID Association for the Assessment and Accreditation of Laboratory Animal Care–accredited animal facility. Previous studies with the experimental model used indicated no major differences in the susceptibility to T. gondii of B6 and B6 × 129 crosses over the first 3 mo of infection (11). Animals 5–7-wk-old of both sexes were used on a randomized basis in all experiments.
Infections and In Vivo Treatments.
T. gondii cysts (ME49 strain) were recovered from brain homogenates from chronically infected mice and suspended in PBS. Animals were experimentally infected by intraperitoneal injection with 20 cysts per mouse. In some experiments, animals also received five intraperitoneal injections in alternating days with vehicle (0.2 ml/animal), IL-10 (0.1 μg/animal), and 15-epi-16-phenoxy-parafluoro-LXA4-methyl ester (1 μg/animal/treatment, 0.025 mg/kg; Berlex Biosciences) from days 10–20 after infection. The LX analogue was prepared by total organic synthesis and isolated and characterized as previously described (12).
LXA4, Nitrite (NO2), and Cytokine Determinations.
Serum levels of LXA4 were measured according to the procedure of Levy et al. (13) using a commercial kit (Neogen). NO2 levels were evaluated in culture supernatants by means of the Griess reaction (3). IL-12p40, IFN-γ, and IL-10 serum levels were measured by ELISA as previously described (8, 14), whereas macrophage inflammatory protein (MIP)-1α, MIP-1β, and RANTES were quantitated using commercial ELISA kits (R&D Systems).
In Situ Staining and Flow Cytometry.
In situ immunostaining of CD4, CD8, CD11c, IL-12p40, and CCR5 was performed as previously described (8). In brief, acetone-fixed serial sections were incubated with biotin-conjugated antibodies against CD4, CD8, CD11c, or CCR5 (Becton Dickinson). After washing, sections were exposed to streptavidin-conjugated horseradish peroxidase (Jackson ImmunoResearch Laboratories) and the reaction was developed using a metal-conjugated horseradish peroxidase substrate (Pierce Chemical Co.). In some experiments, sections were simultaneously double stained with rat anti–IL-12p40 mAb and the reaction was developed with anti–rat IgG AlexaFluor 594 (Molecular Probes) followed by counterstaining with DAPI (Molecular Probes). After additional washing, the sections were examined microscopically and the images were recorded with a digital camera (×200). For flow cytometric analysis, brain mononuclear cell suspensions were prepared and stained with anti-CD11c, anti-CD11b, and anti-CCR5 (Becton Dickinson) as previously described (15).
Macrophage Cultures.
Thioglycollate-elicited macrophages were obtained from C57Bl/6 mice 5 d after injection. Cells were plated, incubated with different concentrations of LXA4, IL-10, or vehicle for 1 h, and infected with T. gondii tachyzoites (5:1 parasite/macrophage ratio). After 4 or 48 h the cultures were stopped, the cells were fixed, Wright-Giemsa stained, and the number of intracellular parasites were evaluated microscopically. In parallel cultures, 24-h supernatants were collected for later evaluation of NO2, cytokine, and chemokine levels.
Statistical Analysis.
The statistical significance of differences in mean values between experimental versus control or vehicle treated samples was evaluated by means of Student's t test. Differences were considered to be significant at P values of < 0.05.
Results
T. gondii Infection Induces LXA4 Production In Vivo.
To address whether the regulatory effects of LXA4 on IL-12 production play a role in the outcome of in vivo infection with T. gondii, we first determined whether the mediator is induced after natural exposure to the live parasite. As shown in Fig. 1 A, LXA4 was detected at substantial levels in sera of wild-type mice after intraperitoneal inoculation with T. gondii cysts beginning at 10 d and reaching a plateau 15 d after infection. Importantly, only baseline levels of the eicosanoid were detected in simultaneously infected 5-LO−/− mice, confirming the dependence of LXA4 production on 5-LO in vivo. In contrast, serum IL-10 levels peaked earlier (5–10 d after infection) and were only marginally affected by 5-LO deficiency (Fig. 1 B). These observations demonstrate that IL-10 and LXA4 are induced with different kinetics, suggesting that they may involve distinct pathways.
Figure 1.
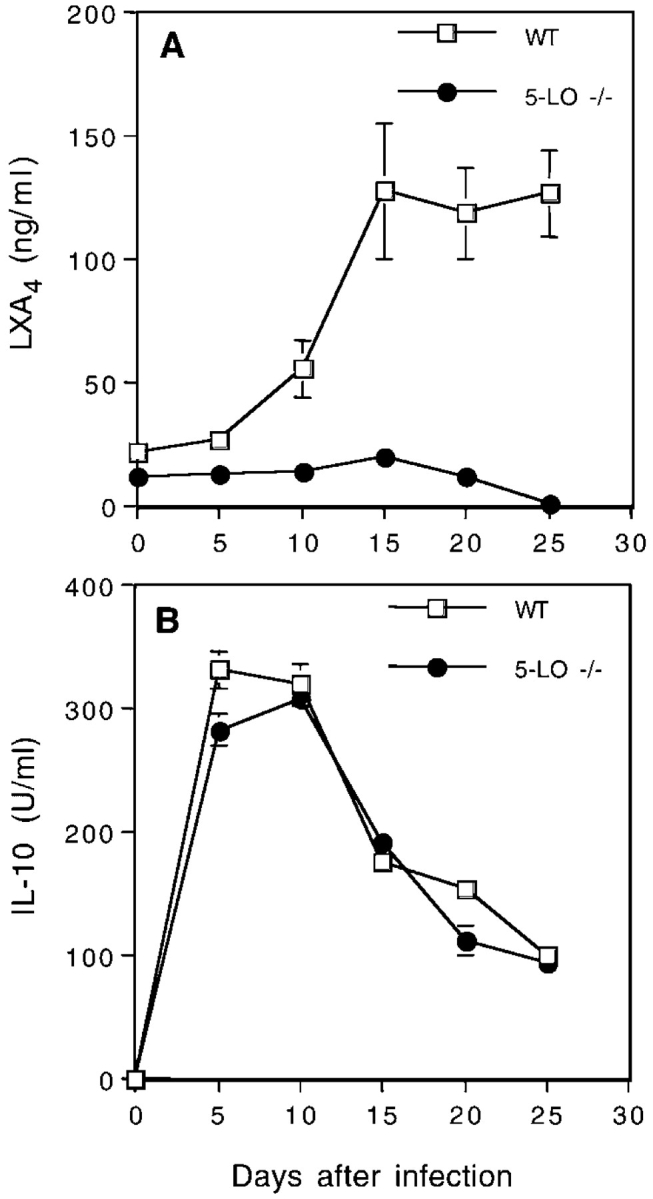
LXA4 and IL-10 are induced with different kinetics during T. gondii infection. Wild-type and 5-LO–deficient mice were infected intraperitoneally with 20 cysts of T. gondii (ME49 strain) and at the indicated times the animals were bled and LXA4 and IL-10 levels were assessed by ELISA in the serum. The values represent mean ± SD of triplicate samples of at least three animals per time point per group. The experiment shown is representative of two performed.
5-LO−/− Mice Display Increased Susceptibility to T. gondii Infection.
To assess whether the induction of LXA4 plays a role in host resistance, we compared the outcome of infection in wild-type and 5-LO−/− mice. As shown in Fig. 2, A and B , ME49-inoculated 5-LO–deficient mice exhibited an accelerated loss in body weight and abruptly succumbed between 27 and 31 d after infection in contrast to the long-term survival of wild-type animals. Importantly, this increased susceptibility to T. gondii was not the result of defective immune control of the parasite. Thus, infected 5-LO−/− mice consistently showed a lower rather than higher burden of T. gondii tissue cysts in the brain than the wild-type controls (Fig. 2 C). This finding suggested that parasite-induced immune responses are enhanced in the absence of LXA4 generation.
Figure 2.

5-LO−/− mice infected with T. gondii display profound weight loss and accelerated mortality despite the presence of lower parasite burdens and elevated proinflammatory cytokine production. Wild-type (B6, 129J F2) and 5-LO–deficient (B6, 129J Alox-5) animals (males and females) were infected as described in Fig. 1 and (A) body weight and (B) survival rates were monitored during the times indicated (n = 15 animals per group, per experiment). (C) To evaluate parasite load, the animals were killed and cysts were counted in brain homogenates. For systemic cytokine production, mice were bled at the indicated time points and the levels of (D) IL-12p40, (E) IFN-γ, and (F) TNF were measured in serum by ELISA. The values presented are the mean ± SD of at least three animals per time point per group. The experiments shown are representative of at least two performed that gave similar results. *, statistically significant (P < 0.05) differences between the means of the values obtained with 5-LO–deficient versus wild-type control mice.
T. gondii–infected Mice Show Augmented Proinflammatory Cytokine Production and Tissue Pathology in the Absence of 5-LO.
To determine whether the absence of 5-LO affects parasite-induced proinflammatory cytokine responses, we studied the kinetics of serum IL-12, IFN-γ, and TNF production in T. gondii–infected animals. Consistent with the observation of lower parasite burdens, serum levels of IL-12 and IFN-γ were found to be significantly elevated in infected 5-LO−/− mice compared with their wild-type counterparts (Fig. 2, D and E). Particularly striking was the uncontrolled increase in IL-12 production observed with time in the knockout animals. Although serum TNF levels also appeared to be elevated in the 5-LO−/− mice, the observed increases were not statistically significant (Fig. 2 F).
The appearance of elevated proinflammatory cytokine production in the presence of decreased parasite numbers suggested that the increased mortality of 5-LO−/− mice results from cytokine-induced tissue damage. In support of this hypothesis, brain sections from the knockout animals 25 d after infection showed markedly enhanced meningitis when compared with equivalent sections from infected wild-type control mice (unpublished data). In addition, the 5-LO–deficient brain tissue displayed increased encephalitis with CD4+ and CD8+ cell infiltration (Fig. 3 A, d and e, and B). As expected from our recent findings demonstrating a suppressive influence of 5-LO on CCR5 expression (10), staining for this chemokine receptor was also enhanced in the same sections from infected knockout mice (Fig. 3 A, f, and C and D). In contrast to the brain, no evidence of enhanced tissue damage was observed in the liver and spleen of the same infected 5-LO−/− animals (unpublished data).
Figure 3.
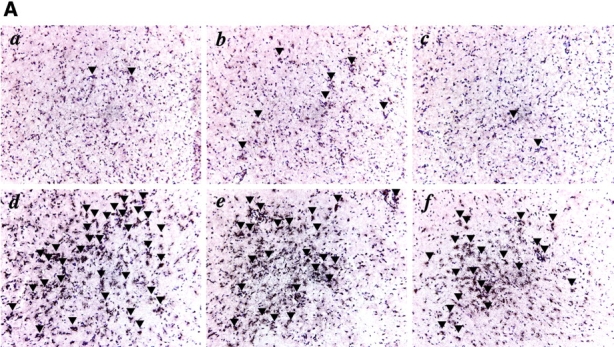

Uncontrolled encephalitis and leukocyte infiltration in the brains of 5-LO−/− mice chronically infected with T. gondii. (A) Frozen sections of brains of wild-type controls (a, b, and c) and 5-LO−/− (d, e, and f) mice infected for 25 d with T. gondii stained with anti-CD4 (a and d), anti-CD8 (b and e), or anti-CCR5 (c and f). Representative micrographs (×200) from at least three animals per group are presented. Arrows indicate reactive cells in the section. The mean ± SD of the total counts of (B) CD3+ CD4+, CD3+ CD8+ cells or of the (C) CD11c+ CD11blo, CD11c+ CD11bhi cells and the intensity of the (D) latter cells obtained from brain homogenates (n = 3) of the same experimental groups is also shown. *, statistically significant (P < 0.05) differences between group means.
When brain sections from the same infected wild-type (Fig. 4 a) and 5-LO−/− (Fig. 4 b) mice were stained for IL-12p40 (red), a massive increase in the intensity of expression and frequency of positive cells was observed in the latter samples. Some, but not all of this staining was associated with CD11c+ cells (green), suggesting that in this model 5-LO regulates IL-12 expression in both DCs as well as other cells in the tissue infiltrates.
Figure 4.

Enhanced IL-12p40 production by CD11c+ and CD11c− cells in the brains of 5-LO−/− mice chronically infected with T. gondii. (a) The same frozen sections of brain from wild-type and (b) 5-LO−/− mice analyzed in Fig. 3 were double stained with anti-CD11c (green) or anti–IL-12p40 (red) and counterstained with DAPI (blue). Representative micrographs (×200) from at least three animals per group are presented.
In Vivo Administration of a Stable LXA4 Analogue Rescues 5-LO−/− Mice from T. gondii–induced Mortality.
Although 5-LO−/− mice are clearly unable to produce LXA4 in response to T. gondii infection, it was still possible that the absence of a different 5-LO–dependent metabolic product accounts for the increased IL-12 production and mortality seen in the knockout animals. To attempt to confirm the role of LXA4 in the 5-LO–dependent suppression observed, we reconstituted T. gondii–infected 5-LO–deficient animals by injecting them every other day with a stable analogue of LXA4 (15-epi-16-phenoxy-parafluor-LXA4-methyl ester). To additionally examine possible redundancies in the activities of LXA4 and IL-10, a second group of infected 5-LO−/− mice was injected with IL-10 using the same regimen. As expected, nonreconstituted 5-LO−/− animals succumbed ∼25–28 d after infection (Fig. 5 A). However, treatment with 0.25 mg/kg LX analogue rescued these animals from death increasing their survival time to >80 d (Fig. 5 A and unpublished data). In contrast, IL-10–treated 5-LO−/− mice succumbed much earlier, between 43 and 50 d after infection. Interestingly, when brain cysts were enumerated on day 25, an increased frequency (P < 0.05) was observed in the IL-10–treated wild-type and 5-LO−/− animal groups when compared with those assayed in untreated control groups (Fig. 5 B). In contrast, LX analogue treatment caused a smaller but significant elevation in the cyst counts in the brains of 5-LO−/− mice but did not affect cyst numbers in wild-type animals. Nevertheless, LX treatment restored parasite burdens in the 5-LO−/− tissues to the levels observed in the 5-LO–sufficient controls (Fig. 5 B).
Figure 5.

In vivo administration of a stable analogue of LXA4 rescues 5-LO−/− animals from mortality induced by T. gondii infection. Wild-type controls or 5-LO−/− mice were infected with T. gondii as described above and treated every other day from days 10 to 20 with vehicle (0.2 ml/animal/time), LXA4 analogue (1 μg/animal/time), or IL-10 (0.1 μg/animal/time). (A) Survival rates, (B) cysts counts, (C) serum IL-12, and (D) serum IFN-γ levels were assessed as previously described. Values presented are the mean ± SD of triplicate samples of five animals per group. *, statistically significant (P < 0.05) differences between group means.
To additionally analyze the effects of LX and IL-10 reconstitution, we evaluated serum levels of IL-12, IL-10, and IFN-γ 20 d after infection, a time point at which IL-12 production in wild-type animals has stabilized but remains elevated with respect to uninfected mice. As previously described (Fig. 2), nonreconstituted infected 5-LO−/− mice displayed significantly higher levels of IL-12 and IFN-γ than these infected control animals. Importantly, LX treatment significantly lowered the concentrations of both cytokines and in the case of IFN-γ, restored its concentration to the level observed in wild-type controls (Fig. 5, C and D). In contrast, LX analogue administration had no detectable effect on IL-10 levels (Fig. 5 E), arguing that the activity of the compound is not due to the stimulation of IL-10 production. On the other hand, IL-10 treatment of both wild-type and knockout animals caused decreases in both IL-12 and IFN-γ as well as an increase in endogenous IL-10 (Fig. 5, C–E). Neither IL-10 or LX analogue treatment resulted in significant changes in LXA4 levels in sera of either wild-type or 5-LO–deficient animals (unpublished data). The above experiments argue that the increased susceptibility of 5-LO−/− mice to T. gondii is the result of the loss of LX production and that LX and IL-10 regulate cytokine production and host mortality by different mechanisms.
Distinct Roles of IL-10 and LXA4 in the Regulation of Inflammation and Macrophage Effector Function.
To additionally investigate the immunosuppressive functions of LXA4 with respect to those of IL-10, we compared the efficacy of the two mediators in a variety of assays of microbicidal and proinflammatory response. To measure their effects on lymphokine-mediated intracellular control of parasite growth, we preincubated elicited peritoneal macrophages with LXA4 or IL-10 and then infected the cells with tachyzoites of T. gondii in the presence of IFN-γ. After 48 h, the macrophages were Giemsa stained and the number of intracellular parasites was evaluated by microscopy. As shown in Fig. 6 A, IL-10, but not LXA4 suppressed the microbiostatic effects of IFN-γ on intracellular T. gondii growth. Consistent with this observation, IL-10, but not LXA4 inhibited nitric oxide production by the same cells (Fig. 6 B). In contrast, both IL-10 and LXA4 pretreatment suppressed TNF (Fig. 6 C), MIP-1β, and RANTES (unpublished data) production whereas IL-10 slightly enhanced and LXA4 markedly inhibited MIP-1α secretion (Fig. 6 D) in these cultures. Thus, although LXA4 lacks the suppressive effects on macrophage effector function displayed by IL-10, it shares with IL-10 the ability to inhibit the proinflammatory cytokine and chemokine responses generated by these cells upon lymphokine activation.
Figure 6.

Redundant and nonredundant roles of IL-10 and LXA4 in the regulation of proinflammatory function. Thioglycollate-elicited macrophages were incubated with medium alone, 100 ng/ml IL-10, or 100 ng/ml LXA4. After 1 h, cells were infected with tachyzoites of the RH strain of T. gondii (5:1 parasite/macrophage ratio) with or without the addition of 100 ng/ml IFN-γ. (A) After an additional 48-h incubation, cells were fixed, Giemsa stained, and the number of intracellular parasites was determined by microscopic examination. In a parallel set of macro-phage cultures, supernatants were harvested at 24 h and (B) NO2, (C) TNF, and (D) MIP-1α production was determined as described in Materials and Methods. Bars represent mean ± SD of triplicate measurements performed on cells pooled from five animals. The results shown are representative of two independent experiments performed. *, statistically significant (P < 0.05) differences between group means.
Discussion
IL-12 plays a fundamental role in the induction of both innate and adaptive resistance to intracellular pathogens and as a product of APC is a primary mediator of immune function. Because of its potential toxicity when produced in excess, IL-12 production is known to be carefully regulated by a remarkably large number of different mechanisms (16). The major mediators previously implicated in the control of pathogen-induced IL-12 synthesis are the down-regulatory cytokines IL-10 and TGF-β. The results presented here reveal a new in vivo pathway for the regulation of IL-12–dependent antimicrobial immune responses involving the eicosanoid LXA4.
LXA4 was originally described as a selective regulator of leukotriene function (17) biosynthesized by leukocytes, endothelial cells, and platelets by means of transcellular pathway(s) (18). In later studies, LXA4 was shown to have potent inhibitory effects on several inflammatory mechanisms, including NK cell cytotoxicity (19), leukocyte responses to cytokines (e.g., TNF; reference 20), or to microbial stimulation (21), neutrophil and eosinophil migration (22, 23), as well as cell surface expression of P selectin and LFA-1 (24, 25).
The possibility that LXA4 centrally regulates immune responses was suggested by our recent findings indicating that the eicosanoid suppresses STAg-induced production of IL-12 by DCs both in vitro and in vivo (10). Although the molecular mechanism for this inhibition was not elucidated we proposed that it involves the down-regulation of CCR5 expression as a consequence of interaction of LXA4 with the formyl-peptide receptor-like receptor (FPRL) 1, a phenomenon previously described when FPRL-1 on human monocytes interacts with a peptide from HIV Gp120 (26). Although it is not yet clear whether FPRL-1 is the receptor responsible for these biological effects of LXA4, a recent study in which the transfection of HeLa cells with FPRL-1 led to LXA4 responsiveness as assessed by the inhibition of nuclear factor κB activation (27) supports the existence of this pathway. Regardless of the precise mechanism involved, it is clear from both the latter study and our own previous observations that exogenous LXA4 can have profound global effects on the immune response to microbial stimulation inhibiting DC migration and function as well as the induction of proinflammatory gene expression (10, 27).
Although our previous experiments also suggested that STAg itself induces LXA4-mediated inhibition of DC function in vivo, they did not address the physiological relevance of this response during natural infection. This study establishes that LXA4 is induced by T. gondii in vivo and this response plays a major host protective role in preventing parasite-induced inflammation and mortality. Upon infection with the protozoan, 5-LO−/− mice succumbed during the early chronic phase but displayed a significantly reduced parasite load. Instead, mortality appeared to be associated with profound weight loss, elevated proinflammatory cytokine production, and enhanced inflammation as reflected in an increased but diffuse infiltration of CD4+, CD8+, and CCR5+ cells in brain tissue. The latter observation may reflect a loss of regulation of chemokine and chemokine receptor expression as a consequence of impaired LX production. Interestingly, in direct contrast to our previously published findings on the effects of IL-10 deficiency in T. gondii infection (3, 9), the infected 5-LO−/− mice showed no obvious increase in hepatic inflammation/necrosis when compared with infected control animals.
In situ staining of brain sections from infected 5-LO−/− animals revealed a striking increase in IL-12 expression relative to sections from wild-type mice, suggesting that the enhanced production of the cytokine might be a major factor in the increased pathology and mortality observed. Although this observation supports the results of our previous study, indicating increased production of IL-12 in the spleens of STAg-injected 5-LO−/− mice (10), the findings differ in that both CD11c+ and CD11c− cells contribute to the response seen in infected brain in the experiments reported here. The latter discrepancy may simply reflect the dominance of DCs in the innate response to STAg in the spleen versus the more complex population of IL-12–producing cells generated during chronic infection.
Because 5-LO controls the production of a number of different eicosanoids (e.g., LTB4), it was necessary to confirm that the effects of 5-LO deficiency were the direct result of the failure to produce LXA4. Importantly, the reconstitution of 5-LO−/− mice with LX analogue early in infection resulted in complete protection against parasite-induced mortality and restored both IL-12 and IFN-γ to levels comparable to those in infected wild-type animals. Although this observation does not rule out the possible participation of other 5-LO dependent eicosanoids, it argues strongly that LXA4 is sufficient to mediate the protection against cytokine-associated mortality revealed by the behavior of T. gondii–exposed 5-LO–deficient animals.
A major question raised by our findings concerns the relative roles of IL-10 and LXA4 in regulating IL-12–dependent IFN-γ production during T. gondii infection. The observations that IL-10–deficient mice succumb 10–14 d after infection whereas 5-LO−/− mice survive for an additional 10–15 d suggests that these two down-regulatory mediators may have distinct functions but work in synergy to protect the host against excess proinflammatory cytokine production. Consistent with this hypothesis is our observation that the reconstitution of 5-LO−/− mice with IL-10 at a time when endogenous IL-10 levels have waned protects the animals for a short period of only 15–20 d, in contrast to the long-lasting protection induced by LXA4 reconstitution. These findings suggest that although IL-10 is sufficient to down-regulate IL-12 and other proinflammatory cytokines during initial acute infection, LXA4 is necessary to sustain long-term stabilization of these responses during chronic infection. Because it is now clear that the continued IL-12 production is required for the maintenance of host resistance to the parasite (14), it is possible that LXA4 may provide a mechanism for fine-tuning this response not afforded by IL-10 alone. In this regard, our observation that LXA4 lacks the suppressive activities on macrophage effector functions displayed by IL-10 (Fig. 5) supports the concept that its key role in T. gondii infection may be to regulate IL-12–dependent IFN-γ responses.
Although the results of this study point to a major role for endogenous LXA4 in the regulation of IL-12 responsiveness to T. gondii infection, it is important to note that the IL-12 response to this parasite is unusual in its dependence on CCR5 signaling. Thus, it is presently unclear whether this LX may exert the same regulatory effects on proinflammatory cytokine production induced by other pathogens. Nevertheless, the observation that exogenous LXA4 dramatically inhibits proinflammatory cytokine gene expression in murine epithelial cells exposed to Salmonella typhimurium (27) as well as our own observation that LXA4-treated mice show highly defective DC migration in response to LPS as well as STAg (10) argues that if induced by microbial stimulation, LXA4 can have broad effects on the host response to infection. Therefore, a key question is whether other pathogens, in addition to T. gondii, trigger endogenous LXA4 production. The identification of the specific microbial products that stimulate LXA4 biosynthesis and the signaling pathway(s) involved should yield information important for our understanding of the immunoregulation of the response to pathogens as well as the development of new strategies for manipulating the induction of this potent antiinflammatory mediator.
Acknowledgments
We thank Dr. Allen Cheever for histopathologic analysis and Dr. David Sacks and James Johndrow for helpful discussions.
This work was supported in part by NIH grants GM 38765 and PO1-DE 13499 to C. Serhan.
Footnotes
Abbreviations used in this paper: 5-LO, 5-lypoxygenase; DC, dendritic cell; FPRL, formyl-peptide receptor-like receptor; LX, lipoxin; MIP, macrophage inflammatory protein; NO2, nitrite; STAg, soluble tachyzoite antigen.
References
- 1.Shevach, E.M., J.T. Chang, and B.M. Segal. 1999. The critical role of IL-12 and the IL-12R beta 2 subunit in the generation of pathogenic autoreactive Th1 cells. Springer Semin. Immunopathol. 21:249–262. [DOI] [PubMed] [Google Scholar]
- 2.Yap, G.S., and A. Sher. 1999. Cell-mediated immunity to Toxoplasma gondii: initiation, regulation and effector function. Immunobiology. 201:240–247. [DOI] [PubMed] [Google Scholar]
- 3.Gazzinelli, R.T., M. Wysocka, S. Hieny, T. Scharton-Kersten, A. Cheever, R. Kuhn, W. Muller, G. Trinchieri, and A. Sher. 1996. In the absence of endogenous IL-10, mice acutely infected with Toxoplasma gondii succumb to a lethal immune response dependent on CD4+ T cells and accompanied by overproduction of IL-12, IFN-gamma and TNF-alpha. J. Immunol. 157:798–805. [PubMed] [Google Scholar]
- 4.Suzuki, Y., A. Sher, G. Yap, D. Park, L.E. Neyer, O. Liesenfeld, M. Fort, H. Kang, and E. Gufwoli. 2000. IL-10 is required for prevention of necrosis in the small intestine and mortality in both genetically resistant BALB/c and susceptible C57BL/6 mice following peroral infection with Toxoplasma gondii. J. Immunol. 164:5375–5382. [DOI] [PubMed] [Google Scholar]
- 5.Hoffmann, K.F., A.W. Cheever, and T.A. Wynn. 2000. IL-10 and the dangers of immune polarization: excessive type 1 and type 2 cytokine responses induce distinct forms of lethal immunopathology in murine schistosomiasis. J. Immunol. 164:6406–6416. [DOI] [PubMed] [Google Scholar]
- 6.Deckert, M., S. Soltek, G. Geginat, S. Lutjen, M. Montesinos-Rongen, H. Hof, and D. Schluter. 2001. Endogenous interleukin-10 is required for prevention of a hyperinflammatory intracerebral immune response in Listeria monocytogenes meningoencephalitis. Infect. Immun. 69:4561–4571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Silva, R.A., and R. Appelberg. 2001. Blocking the receptor for interleukin 10 protects mice from lethal listeriosis. Antimicrob. Agents Chemother. 45:1312–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aliberti, J., C. Reis e Sousa, M. Schito, S. Hieny, T. Wells, G.B. Huffnagle, and A. Sher. 2000. CCR5 provides a signal for microbial induced production of IL-12 by CD8 alpha+ dendritic cells. Nat. Immunol. 1:83–87. [DOI] [PubMed] [Google Scholar]
- 9.Reis e Sousa, C., G. Yap, O. Schulz, N. Rogers, M. Schito, J. Aliberti, S. Hieny, and A. Sher. 1999. Paralysis of dendritic cell IL-12 production by microbial products prevents infection-induced immunopathology. Immunity. 11:637–647. [DOI] [PubMed] [Google Scholar]
- 10.Aliberti, J., S. Hieny, C. Reis e Sousa, C.N. Serhan, and A. Sher. 2002. Lipoxin-mediated inhibition of IL-12 production by DCs: a mechanism for regulation of microbial immunity. Nat. Immunol. 3:76–82. [DOI] [PubMed] [Google Scholar]
- 11.Scharton-Kersten, T.M., G. Yap, J. Magram, and A. Sher. 1997. Inducible nitric oxide is essential for host control of persistent but not acute infection with the intracellular pathogen Toxoplasma gondii. J. Exp. Med. 185:1261–1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Serhan, C.N., J.F. Maddox, N.A. Petasis, I. Akritopoulou-Zanze, A. Papayianni, H.R. Brady, S.P. Colgan, and J.L. Madara. 1995. Design of lipoxin A4 stable analogs that block transmigration and adhesion of human neutrophils. Biochemistry. 34:14609–14615. [DOI] [PubMed] [Google Scholar]
- 13.Levy, B.D., S. Bertram, H.H. Tai, E. Israel, A. Fischer, J.M. Drazen, and C.N. Serhan. 1993. Agonist-induced lipoxin A4 generation: detection by a novel lipoxin A4-ELISA. Lipids. 28:1047–1053. [DOI] [PubMed] [Google Scholar]
- 14.Yap, G., M. Pesin, and A. Sher. 2000. Cutting edge: IL-12 is required for the maintenance of IFN-gamma production in T cells mediating chronic resistance to the intracellular pathogen, Toxoplasma gondii. J. Immunol. 165:628–631. [DOI] [PubMed] [Google Scholar]
- 15.Reichmann, G., E.N. Villegas, L. Craig, R. Peach, and C.A. Hunter. 1999. The CD28/B7 interaction is not required for resistance to Toxoplasma gondii in the brain but contributes to the development of immunopathology. J. Immunol. 163:3354–3362. [PubMed] [Google Scholar]
- 16.Braun, M.C., and B.L. Kelsall. 2001. Regulation of interleukin-12 production by G-protein-coupled receptors. Microbes Infect. 3:99–107. [DOI] [PubMed] [Google Scholar]
- 17.Serhan, C.N., M. Hamberg, and B. Samuelsson. 1984. Lipoxins: novel series of biologically active compounds formed from arachidonic acid in human leukocytes. Proc. Natl. Acad. Sci. USA. 81:5335–5339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Serhan, C.N. 1994. Lipoxin biosynthesis and its impact in inflammatory and vascular events. Biochim. Biophys. Acta. 1212:1–25. [DOI] [PubMed] [Google Scholar]
- 19.Ramstedt, U., C.N. Serhan, K.C. Nicolaou, S.E. Webber, H. Wigzell, and B. Samuelsson. 1987. Lipoxin A-induced inhibition of human natural killer cell cytotoxicity: studies on stereospecificity of inhibition and mode of action. J. Immunol. 138:266–270. [PubMed] [Google Scholar]
- 20.Pouliot, M., and C.N. Serhan. 1999. Lipoxin A4 and aspirin-triggered 15-epi-LXA4 inhibit tumor necrosis factor-alpha-initiated neutrophil responses and trafficking: novel regulators of a cytokine-chemokine axis relevant to periodontal diseases. J. Periodontal Res. 34:370–373. [DOI] [PubMed] [Google Scholar]
- 21.Gewirtz, A.T., B. McCormick, A.S. Neish, N.A. Petasis, K. Gronert, C.N. Serhan, and J.L. Madara. 1998. Pathogen-induced chemokine secretion from model intestinal epithelium is inhibited by lipoxin A4 analogs. J. Clin. Invest. 101:1860–1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bandeira-Melo, C., P.T. Bozza, B.L. Diaz, R.S. Cordeiro, P.J. Jose, M.A. Martins, and C.N. Serhan. 2000. Cutting edge: lipoxin (LX) A4 and aspirin-triggered 15-epi-LXA4 block allergen-induced eosinophil trafficking. J. Immunol. 164:2267–2271. [DOI] [PubMed] [Google Scholar]
- 23.Clish, C.B., J.A. O'Brien, K. Gronert, G.L. Stahl, N.A. Petasis, and C.N. Serhan. 1999. Local and systemic delivery of a stable aspirin-triggered lipoxin prevents neutrophil recruitment in vivo. Proc. Natl. Acad. Sci. USA. 96:8247–8252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fiore, S., and C.N. Serhan. 1995. Lipoxin A4 receptor activation is distinct from that of the formyl peptide receptor in myeloid cells: inhibition of CD11/18 expression by lipoxin A4-lipoxin A4 receptor interaction. Biochemistry. 34:16678–16686. [DOI] [PubMed] [Google Scholar]
- 25.Scalia, R., J. Gefen, N.A. Petasis, C.N. Serhan, and A.M. Lefer. 1997. Lipoxin A4 stable analogs inhibit leukocyte rolling and adherence in the rat mesenteric microvasculature: role of P-selectin. Proc. Natl. Acad. Sci. USA. 94:9967–9972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Deng, X., H. Ueda, S.B. Su, W. Gong, N.M. Dunlop, J.L. Gao, P.M. Murphy, and J.M. Wang. 1999. A synthetic peptide derived from human immunodeficiency virus type 1 gp120 downregulates the expression and function of chemokine receptors CCR5 and CXCR4 in monocytes by activating the 7-transmembrane G-protein-coupled receptor FPRL1/LXA4R. Blood. 94:1165–1173. [PubMed] [Google Scholar]
- 27.Gewirtz, A.T., L.S. Collier-Hyams, A.N. Young, T. Kucharzik, W.J. Guilford, J.F. Parkinson, I.R. Williams, A.S. Neish, and J.L. Madara. 2002. Lipoxin a4 analogs attenuate induction of intestinal epithelial proinflammatory gene expression and reduce the severity of dextran sodium sulfate-induced colitis. J. Immunol. 168:5260–5267. [DOI] [PubMed] [Google Scholar]