

Abstract
Type 1 diabetes and other organ-specific autoimmune diseases often cluster together in human families and in congenic strains of NOD (nonobese diabetic) mice, but the inherited immunoregulatory defects responsible for these diseases are unknown. Here we track the fate of high avidity CD4 T cells recognizing a self-antigen expressed in pancreatic islet β cells using a transgenic mouse model. T cells of identical specificity, recognizing a dominant peptide from the same islet antigen and major histocompatibility complex (MHC)-presenting molecule, were followed on autoimmune susceptible and resistant genetic backgrounds. We show that non-MHC genes from the NOD strain cause a failure to delete these high avidity autoreactive T cells during their development in the thymus, with subsequent spontaneous breakdown of CD4 cell tolerance to the islet antigen, formation of intra-islet germinal centers, and high titre immunoglobulin G1 autoantibody production. In mixed bone marrow chimeric animals, defective thymic deletion was intrinsic to T cells carrying diabetes susceptibility genes. These results demonstrate a primary failure to censor forbidden clones of self-reactive T cells in inherited susceptibility to organ-specific autoimmune disease, and highlight the importance of thymic mechanisms of tolerance in organ-specific tolerance.
Keywords: autoimmune disease, diabetes mellitus type I, clonal deletion, T lymphocytes, genetic predisposition to disease
Introduction
Autoimmune diseases that target specific organs such as the pancreatic islets, thyroid, adrenal glands, stomach, or nervous system, often cluster together in either different family relatives or occur simultaneously in one individual. High concordance among identical twins indicates a large inherited component in susceptibility to these diseases, and genetic studies reveal a complex pattern of interactions between MHC and non-MHC chromosomal regions (1). The nature of the inherited immunological defect(s) that leads to these diseases nevertheless remains obscure. On the one hand, a variety of lines of evidence suggest that these diseases may reflect excessive stimulation of autoreactive T cells by viruses, costimulatory molecules, or antigen-presenting cells that promote Th1 T cells (2–4). Alternatively these diseases may be related to observed heritable decreases in Th2 cells (5), regulatory T cells (3, 6), NKT cells (7, 8), CD4+CD25+ regulatory T cells (9), or diminished thymocyte signaling (10) or thymocyte apoptotic responses measured in vitro (11). Defects in antigen presentation and peripheral censoring of autoreactive T cells by processes such as clonal anergy or activation-induced death have also been proposed (12–16). One of the principal barriers to identifying the primary inherited defects, however, has been the inability to track the fate of organ-reactive T cells directly in vivo and identify primary inherited aberrations in this fate in autoimmune susceptible individuals.
Inherited susceptibility to autoimmune disease in the NOD (nonobese diabetic)* mouse strain represents a key model to begin identifying the primary immunological lesions underlying the clustering of different organ-specific autoimmune diseases (17). NOD mice spontaneously develop type 1 diabetes due to autoreactive T cell destruction of pancreatic islet β cells. A particular allelic product of the NOD strain MHC, I-Ag7, is a key diabetes susceptibility gene and shares unique sequence and structural motifs with diabetes-susceptible MHC gene products in humans (18, 19). Like humans, diabetes in NOD mice nevertheless requires many other susceptibility genes other than the MHC, and elegant mapping and congenic breeding strategies have delineated the complex nature of inherited susceptibility (1, 20, 21).
C57BL/6 or C57BL/10 strain mice, which are not especially prone to organ-specific autoimmunity, do not develop insulitis or diabetes when the NOD MHC haplotype is introduced by congenic breeding, underscoring the importance of other non-MHC genes. In fact, substituting a number of different non-MHC chromosomal regions from the C57BL/10 strain into the NOD strain by congenic breeding, either individually or in combination, is sufficient to partially or completely prevent development of insulitis or diabetes despite the continued presence of the NOD MHC alleles (1, 21, 22). Conversely, when a different MHC type is introduced into the NOD strain, as in the NOD.H-2k congenic derivative, insulitis and diabetes are prevented but autoimmune thyroiditis is exaggerated (21, 23, 24). Introducing a defective B7.2 (CD86) costimulatory gene into NOD suppresses insulitis and diabetes but leads instead to spontaneous autoimmune damage of the peripheral nerves (25). These results point to a general susceptibility to organ-specific autoimmunity in NOD caused by a complex set of non-MHC genes, with alleles of MHC or costimulatory molecules influencing the specific organ targets of this general immunological defect (1, 21).
In addition to the complicated inheritance of autoimmune susceptibility, the complexity and heterogeneity of the normal T cell repertoire also represents a barrier to visualizing the primary immunological defects underlying autoimmune susceptibility. Self-reactive T cells can be readily demonstrated in the circulation and peripheral tissues of animals and individuals with no apparent predisposition to autoimmune disease, but the heterogeneity in TCR specificity and affinity and the need to use functional response assays to measure these cells makes it difficult to interpret the presence of these cells. Censoring of self-reactive T cells through the induction of clonal deletion or clonal anergy in the thymus and in peripheral lymphoid tissues requires a critical threshold of antigen/MHC and TCR affinity to be triggered. For organ-specific antigens that are present in only trace amounts in the thymus or circulation, many T cells fall below this threshold and escape thymic deletion (26–32) because either the T cells have lower affinity or TCR density, the peptide/MHC combinations they recognize are less efficiently presented, or they recognize unique epitopes that are only generated in the peripheral organ.
Three different TCR transgenic models that employ TCRs from islet reactive clones that could be grown in vitro from autoimmune mice represent examples of clones that fall below this thymic selection threshold, although the islet antigens recognized by these TCRs are not yet known (33, 34). Peripheral deletion or other regulatory mechanisms may normally prevent T cells of this type from becoming activated (15, 35). Nevertheless, many organ-specific antigens do reach the circulation and thymus in low amounts (eg. insulin, thyroglobulin), and a great number appear to be made there by rare thymic medullary epithelial cells (36, 37). Efficiently presented dominant peptides from these self-antigens can be shown to censor reactive T cells by thymic deletion and by causing selective export of anergic CD4+CD25+ cells that may be responsible for regulating the clones that fall below these thymic selection screens (32, 38–46). The relative roles of thymic and peripheral mechanisms for achieving organ-specific tolerance and the pathogenesis of autoimmune disease thus remains a key unresolved issue.
To study how the (non-MHC) general autoimmune susceptibility genes in NOD mice affect the acquisition of tolerance by organ-specific CD4 T cells, we sought to compare the fate of islet-reactive CD4 T cells bearing a well-defined high avidity TCR in a TCR transgenic mouse model, when bred to H-2 matched strains with an underlying autoimmune susceptible or resistant background. By holding the TCR, the islet antigen and the H-2–presenting molecules constant, and varying the non-MHC background genes, we show here that non-MHC genes from the NOD strain act within high avidity autoreactive CD4 T cells to cause them to escape clonal deletion in the thymus and reach the peripheral lymphoid organs and pancreatic islets in greatly elevated numbers.
Materials and Methods
Mice, Antibodies, Histology.
3A9 TCR transgenic (47) and ILK-3 Ins-HEL transgenic mice (32) produced in C57BL/6J were backcrossed to B10.Br/SgSnJ (JAX) or NOD.H2k (48) (gift from L. Wicker, Cambridge University, Cambridge, UK). Data presented are from N5-N10 mice. TCR, transgenic mice expressing HEL under the rat insulin promoter (insHEL), and H-2 genotype of each mouse was tested twice by PCR (32). Urine glucose was tested using Testape at biweekly intervals or when cage was wet. Mice with two successive positive Testape were called diabetic, euthanized, and diabetes confirmed by blood glucose measurement. Nondiabetic mice were culled at 6 mo. From each mouse, one-half pancreas was fixed in 10% formalin, paraffin embedded, and stained with H&E, while the other half was snapped frozen in OTC (Tissue Tek) in liquid nitrogen. Frozen sections were stained with peanut agglutinin biotin/streptavidin-alkaline phosphatase/Fast Blue (all from Vector Labs) and with sheep anti–mouse IgD/anti–sheep HRP (both from The Binding Site)/DAB. HEL-binding IgG was measured in serum by ELISA on Nunc maxisorp plates coated with 100 μg/ml HEL, developed with sheep anti–mouse IgG-alkaline phosphatase (Southern Biotech Associates, Inc.) and Sigma Substrate 104. A reference pool of immune sera from HEL primed and boosted B6 mice was set to contain 100 arbitrary units. Radiation chimeras were constructed as described previously (32), using a total of 2 × 106 bone marrow cells to reconstitute recipients irradiated with 5.5Gy twice separated by 3 h. Recipients were analyzed 6–10 wk after reconstitution, therefore diabetes was not monitored in these mice.
Flow Cytometry.
6–12-wk old nondiabetic mice (Testape negative) were analyzed. Thymus and spleen were passed through a sterile cell strainer. Lymphocytes from pancreatic lymph nodes were obtained by pressing between two glass slides. Liberase R1 (Boehringer) was injected into the common bile duct of double transgenic mice to inflate the pancreas. The pancreas was then incubated 20 min at 37°C, and washed with a solution containing FCS. Islets were handpicked and dissociated using a protein gel-loading pipette tip. The following antibody stains were used: mouse IgG1 anti-clonotypic 1G12 antibody (49) (gift of E. Unanue and D. Peterson, Washington University, St. Louis, MO) as culture supernatant followed by rat monoclonal anti–mouse IgG1 conjugated to FITC or allo-phycocyanin (BD PharMingen); anti–CD8-PE (Caltag); anti-CD4 allo-phycocyanin, anti-CD3, anti-CD69, anti-Va2, anti-CD45.1-biotin, and anti-CD45.2-biotin (all from BD PharMingen), anti-CD24 biotin (Caltag), anti-Vb8 biotin (clone F23.1), and streptavidin-PerCP (BD). Data was collected on a Becton Dickinson LSR, and analyzed with WinList™, CELLQuest™ PRO, and FlowJo™.
Results
To track how the acquisition of CD4 T cell tolerance to organ-specific self-antigens is altered by the general autoimmune susceptibility genes from NOD, we used an experimental model involving a cross between two transgenic mouse strains. In the 3A9 TCR transgenic strain (47) many CD4 T cells carry the same TCR recognizing a normally foreign antigen, hen egg lysozyme (HEL) peptide 46–61 bound to I-Ak (50). HEL 46–61 is the dominant peptide epitope presented by I-Ak, and its high avidity binding to I-Ak and recognition by the 3A9 TCR (Vb8.2/Va3) are very well characterized (50). T cells bearing the 3A9 TCR can be readily detected in vivo with a clonotypic anti-TCR mAb developed by Peterson and Unanue (reference 49, Fig. 2). The insHEL transgenic strain synthesizes high concentrations of membrane-bound HEL as a self-antigen in pancreatic islet β cells under control of the rat insulin promoter (32). Trace quantities of cleaved HEL are present in the circulation, mirroring the distribution of insulin itself (32). When the two transgenic strains are intercrossed to make TCR/insHEL double-transgenic progeny, the large population of HEL-reactive T cells now reacts with the islet-specific self-antigen, and the fate of these T cells can be readily studied.
Figure 2.
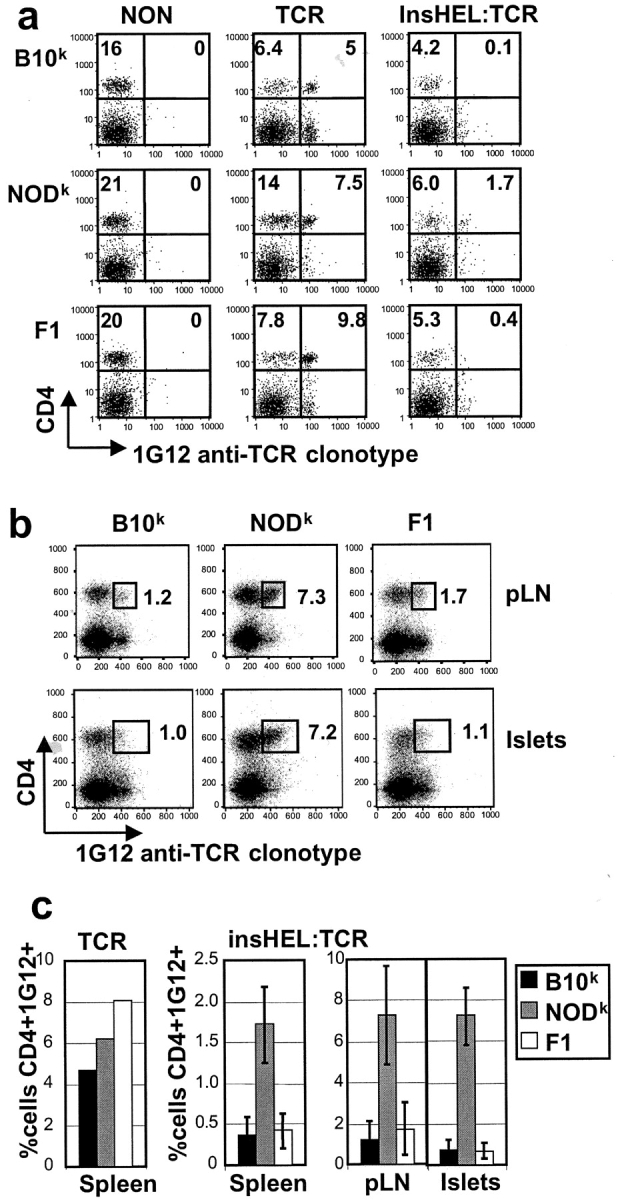
Failure to eliminate islet-reactive CD4 T cells in NODk mice. (a) Spleen cells from 6–12-wk old nondiabetic female mice of the indicated genotypes were stained with 1G12 anti-TCR clonotype and antibody to CD4. The percentage of lymphocytes falling within the quadrants is shown. (b) The same analysis performed on cells from pancreatic lymph nodes (pLN) and pancreatic islets of 6–12-wk old nondiabetic TCR/insHEL mice. (c) Data collected as above compiled from multiple animals of the indicated genotypes.
Previously we showed that TCR/insHEL double transgenic mice on the (B6×B10.BR)F1 background remain free of diabetes, but develop early and extensive insulitis that remains subclinical or “benign” (32). The insulitis was explained by thymic export of large numbers of CD4 T cells that had reduced but not lost their responsiveness to HEL so that high concentrations of HEL such as exist in the islet could elicit a weak response by most of the cells (32). The weakened responsiveness of the T cells reflected two changes: (i) selective downregulation of the transgene-encode TCR Va3/Vb8.2 pairs, while still retaining a sufficient density of TCRs to allow survival by coexpressing a second TCR-α chain; and (ii) an independent diminished responsiveness of the T cells to TCR stimulation revealed by stimulating all the surface TCRs with anti-Vb8.2 antibody (32).
Spontaneous Progression of Insulitis to Form Germinal Centers and Autoantibody.
We backcrossed the TCR and insHEL transgenic strains to two H-2k matched congenic partner strains: NOD.H-2k (reference 48; referred to below as NODk) which carries all the autoimmune susceptibility alleles of NOD except the H-2, and hence normally lacks insulitis or diabetes but is susceptible to autoimmune thyroiditis and sialitis; and B10.BR (referred to below as B10k), which carries diabetes resistance alleles at most non-MHC diabetes loci (51). Then, we intercrossed mice carrying TCR and insHEL transgenes to produce TCR/insHEL double transgenic mice of three genetic makeups: NODk, B10k, and (NODk × B10k)F1.
The effects of the different strain backgrounds were first determined by studying large cohorts of female TCR/insHEL animals and littermate controls until 24 wk old. No autoimmune diabetes or insulitis developed in NODk, B10k, or F1 hybrid progeny carrying either the TCR or insHEL transgenes alone, or neither transgene. The lack of insulitis or diabetes in the NODk animals reflects the inhibitory effects of this H-2 haplotype on islet-specific autoimmunity in NOD mice (but not on thyroid or salivary autoimmunity; references 21 and 51). By contrast all TCR/insHEL double transgenic mice had extensive lymphocytic infiltrates in pancreatic islets by 6 wk old regardless of the strain background, and this inflammation persisted to the 24-wk time point (Fig. 1 A). As discussed above, the spontaneous insulitis in double-transgenic mice on the autoimmune resistant strain backgrounds (B10k and F1) can be explained by the presence of HEL-reactive TCRs on most of the peripheral CD4s, albeit expressed at reduced cell surface density and weak responsiveness to antigen. It is important to note, however, that the populations of T cells and IgD+ resting B lymphocytes in these chronically inflamed islets reflects the lymphocyte populations in the circulation (Fig. 1, and data not shown), and the majority probably accumulate in these sites secondary to chronic inflammation and independently from activation or any specificity for the inciting HEL antigen. The presence of infiltrating cells therefore give little information about the frequency or responsiveness of the minority of HEL-specific Th cells among them.
Figure 1.

Islet-reactive T cells cause germinal center formation, autoantibodies, and diabetes in NODk mice. (a) Pancreatic islets stained with H&E from TCR/InsHEL mice of the indicated genotypes. Right panel is a frozen section stained with peanut agglutinin (blue) and anti-IgD (brown). GC, germinal center. (b) Onset of diabetes in TCR/insHEL female mice of the indicated genotypes. Data represent >20 mice per group. (c) HEL-binding IgG titre in serum from diabetic double transgenic mice, collected within 2 wk of diabetes onset. Titres are expressed in arbitrary units relative to a reference immune sera from HEL-immunized nontransgenic mice set at 100 units.
In contrast to the absence of autoimmune diabetes in insHEL or TCR animals on the NODk background, 90% of TCR/insHEL double transgenic mice developed diabetes on this background. Diabetes onset occurred over a broad age range from 8–24 wk old (Fig. 1 B) and was accompanied by formation of large germinal centers in the pancreatic islets (Fig. 1 A) and spontaneous secretion of high titre anti-HEL IgG1 autoantibodies in the serum (Fig. 1 C). Only 20% of B10k TCR/insHEL animals developed diabetes, and this appeared to reflect a distinct disorder because it developed only during a narrow age window in mice <12 wk old, and none of these animals produced measurable anti-HEL IgG autoantibody in their serum (Fig. 1 C) or had discernible germinal centers in the islets. Consistent with a different genetic basis, the B10k form of low-incidence/antibody-free diabetes was completely suppressed by one full set of non-MHC genes from NOD in F1-hybrids, and likewise the NODk form of high incidence/high autoantibody disease was fully suppressed by non-MHC B10 genes (Fig. 1 B).
Defective Censoring of Islet Reactive CD4 Cells in NODk.
The progression to diabetes, formation of germinal centers, and secretion of anti-HEL IgG in NODk double transgenic mice indicated that the 3A9 HEL-reactive CD4 T cells either failed to be tolerized or broke out of tolerance as a result of non-MHC NOD genes. To explore this issue, CD4 T cells bearing the 3A9 anti-HEL TCR were measured in different tissues of young mice before the onset of diabetes by flow cytometric staining with the 1G12 anti-clonotypic antibody (Fig. 2). In TCR-transgenic mice lacking insHEL antigen, a high frequency of CD4+ TCR clonotype+ cells was present in the spleen and lymph nodes regardless of the strain background. By contrast, while the frequency of peripheral CD4+ TCR clonotype+ cells was reduced in all TCR/insHEL double-transgenic mice, much higher numbers of islet reactive T cells were present in the spleen, pancreatic lymph nodes, and pancreatic islets in the NODk animals. The failure to eliminate islet reactive T cells in NOD appears to be a recessive defect as it was fully corrected in (NODk × B10k)F1 hybrid animals.
Receptor downregulation is an important means of diminishing antigen reactivity, and almost all CD4+ T cells in the periphery of TCR/insHEL B10k mice display half the surface density of CD3 and Vβ8 that are found on TCR transgenic mice without the negative-selecting antigen (Fig. 3 A and Fig. 4). Most of these T cells carry the transgenic Vα3 chain on their surface, but at 10-fold reduced levels compared with TCR cells (Fig. 3 B) so that they are difficult to detect by staining with the clonotypic 1G12 antibody (Fig. 3 A). Since transgenic Vβ8 chains on these cells are only reduced by 50%, the remaining receptors must contain a second TCR-α chain. This conclusion is supported by the fact that Vα2 chains are expressed by a fraction of these cells, and by the finding that the level of Vα2 displayed on their surface is about half that found on nontransgenic mice (Fig. 3 C). By contrast, very little decrease in CD3 or Vβ8 occurred on peripheral CD4 T cells from NODk TCR/insHEL mice, and a large fraction still express high surface densities of clonotypic receptor 1G12 (Fig. 3 A). Thus, insHEL caused a dramatic reduction in the avidity of peripheral T cells reacting with HEL on the B10k background, but induced little change compared with TCR-transgenic animals without HEL on the NODk background.
Figure 3.

Failure to downregulate TCR expression in splenocytes of NODk animals. (a) Spleen cells from nondiabetic female mice of various genotypes were stained with antibodies to CD4, CD3, Vβ8, and 1G12 clonotype. Histograms show profiles of CD4-positive cells from mice of the indicated genotypes. (b) Vα3 expression on CD4+Vβ8+ spleen cells from B10k mice of the indicated genotypes. (c) Staining for an endogenous TCR-α chain, Vα2, on CD4+ spleen cells. The percentage of CD4+ cells that are Vα2+ is indicated.
Figure 4.

High TCR levels on lymphocytes in the islets of NODk mice. Islets from nondiabetic female mice were purified and analyzed as in Fig. 3 a.
A similar difference in HEL-reactive TCR expression was exhibited by the lymphocytes that infiltrated pancreatic islets of NODk and B10k mice. Like the circulating T cells, infiltrating CD4+ T cells show less reduction in CD3 or Vb3 in NODk TCR/insHEL animals compared with the B10k counterparts (Fig. 4). Consistent with a difference in responsiveness to HEL made in the pancreatic islets, clonotype-positive CD4 cells specifically increased expression of CD69 in the islets relative to their levels in lymph nodes or spleen (Fig. 5). Clonotype-negative cells in the same islet infiltrates showed no induction of CD69 (Fig. 5). Likewise, no specific increase in CD69 occurred on clonotype-positive CD4 cells in the islets of B10k double transgenic mice (Fig. 5). While these data indicate a difference in the number of high avidity autoreactive T cells homing to the islet, the majority of islet infiltrating cells are nevertheless likely to have been attracted independently of their antigen specificity, since the infiltrate is broadly composed of B cells, clonotype-positive and -negative CD4 and CD8 cells (Fig. 1, and data not shown).
Figure 5.
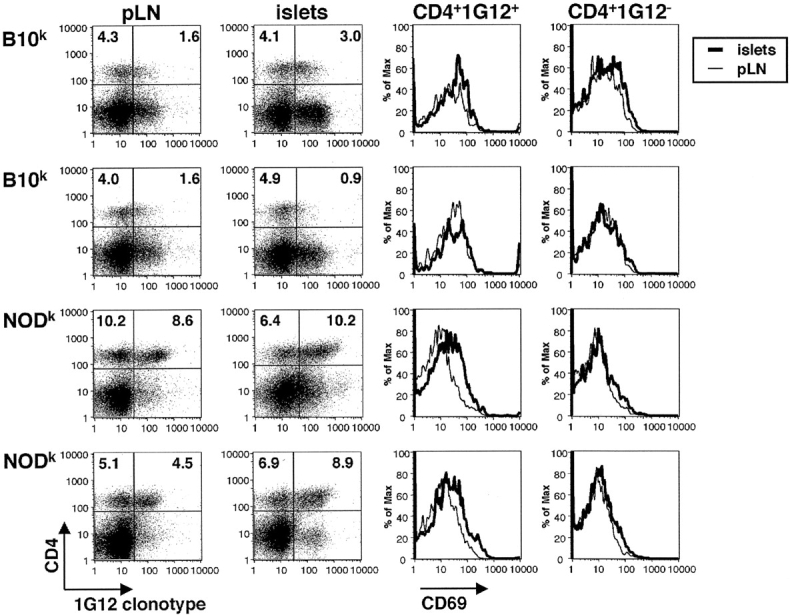
T cell activation in the islets is specific to CD4+clonotype+ NODk T cells. Lymphocytes from pancreatic lymph nodes and islets of two nondiabetic TCR/insHEL female mice of B10k or NODk background were stained with CD4, 1G12 clonotype, and the activation marker CD69. The percentage of lymphocytes falling within the quadrants is indicated. Histograms show profiles of CD69 expression on clonotype-positive or clonotype-negative CD4 cells, overlaying cells from the islet and pancreatic lymph node of the same animal.
Defective Thymic Deletion of Islet Autoreactive T Cells.
The presence of large numbers of high avidity T cells in NODk insHEL/TCR animals could reflect expansion of these cells as a result of dysregulated peripheral tolerance mechanisms, or failure to delete these cells in the thymus. Analysis of thymocytes from the double-transgenic mice showed a failure to eliminate high avidity islet reactive T cells during their development in the thymus (Fig. 6).
Figure 6.

Failure to delete islet-reactive T cells in the thymus of NODk animals. (a) Thymocytes from 6–12-wk old nondiabetic female mice of the indicated genotypes were stained with 1G12 anti-TCR clonotype, and antibodies to CD4 and CD8. The percentage of CD4+CD8− and CD4+CD8+ thymocytes are shown. (b) The 1G12 profiles are shown for CD4+CD8− mature thymocytes. 1G12 staining cannot be detected on CD4+CD8+ thymocytes. (c) Absolute number of CD4+CD8+ thymocytes was calculated for each group. From left to right, there were exactly 4, 6, 16, 3, 5, 13, and 10 mice, respectively. (d) Compilation of the number of CD4+1G12+ thymocytes in each group.
In TCR-transgenic mice lacking insHEL, strong positive selection is evidenced by the skewing to form a large population of CD4+8− single-positive thymocytes (Fig. 6 A). In both NODk or B10k backgrounds most of these single-positive thymocytes bear high surface densities of the 3A9 TCR based on 1G12 staining (Fig. 6 B). In double-transgenic B10k mice expressing insHEL, high avidity HEL-reactive T cells are negatively selected during the CD4+8+ double-positive and early single-positive stages. Thus, the total number of double-positive cells is reduced ∼50% and the number of CD4 single-positive cells is dramatically decreased (Fig. 6, A and C). The CD4 single positives that mature in these thymi carry greatly reduced clonotypic TCR (Fig. 6 B and Fig. 7) and a smaller reduction in surface CD3 and Vβ8 levels (Fig. 7), comparable to the CD4 cells found in the periphery of these mice (Fig. 3).
Figure 7.

Failure to downregulate TCR expression in CD4+ thymocytes of NODk animals. (a) Thymocytes from nondiabetic female mice of various genotypes were stained with antibodies to CD4, CD8, and 1G12 clonotype, together with either CD3 or Vβ8. All histograms show profiles of CD4+CD8− cells, except for the graphs on the right which are gated on CD4+CD8+ (DP) thymocytes.
In double transgenic mice on the NODk background, however, there is no reduction in the numbers of double-positive cells and CD4 single-positive cells continue to be formed in approximately the same frequency as in TCR littermates (Fig. 6 A). These autoreactive single positive thymocytes show no evidence of selection for low avidity cells, as they continue to express high densities of clonotypic TCR (Fig. 6 B) and high surface levels of CD3 and Vβ8 (Fig. 7). As a result, whereas almost no high avidity CD4 T cells recognizing islet HEL are formed in the thymus of B10k double transgenic mice, these cells continue to be formed in NODk double transgenic thymi in numbers that are only slightly reduced compared with animals lacking the autoantigen (Fig. 6 D).
In theory, the presence of CD4+1G12+ cells in the thymus of TCR/insHEL NODk mice could have resulted from a peripheral tolerance defect, allowing recirculation of mature CD4+ cells back to the thymus after activation and expansion in the periphery. Three lines of evidence exclude this possibility. First, negative selection is evident within the double-positive population in B10k but not in NODk thymi. Second, high avidity autoreactive cells are present in much higher numbers in NODk thymi of mice of various ages, and they do not accumulate with age as would be expected for peripheral expansion (Fig. 8 A). Third, all of the clonotype-high single positive thymocytes are CD24 (heat stable antigen)hi, indicating that they are immature recently formed T cells that have failed to be deleted, and not mature cells that have homed back (Fig. 8 B).
Figure 8.

Increased CD4+1G12+ thymocytes in NODk insHEL/TCR mice is not due to mature T cell recirculation to the thymus. (a) Absolute number of CD4+1G12+ mature thymocytes in insHEL/TCR double transgenic on B10k, NODk, and F1 background plotted versus age. (b) Thymocytes from insHEL/TCR double transgenic mice were stained with 1G12, CD4, CD8, and CD24. CD24 profiles are shown for 1G12-positive or -negative CD4+CD8− mature thymocytes.
T Cell Intrinsic Action of NOD Genes on Thymic Deletion to Islet Antigen.
The failure to delete islet-HEL specific T cells in the NODk double-transgenic mice could reflect differences in the expression or presentation of insHEL to T cells in the thymus, or differences in the way the immature T cells respond to self-antigen. To distinguish between these possibilities, we constructed radiation chimeras using mixtures of bone marrow from TCR-transgenic mice of B10k and NODk backgrounds to reconstitute lethally irradiated insHEL or nontransgenic recipients (Fig. 9 A). Leukocytes from the two donor strains could be distinguished by expression of different allelic forms of the Ly5 (CD45) surface protein. F1 hybrid recipients were used in most experiments, thus allowing any radioresistant host T cells to be distinguished as Ly5ab double-positive, compared with the donor-derived T cell populations which carry only one or other marker. In preliminary studies using nontransgenic donor bone marrow, we found that NODk-derived T cells preferentially expand within the CD4+8+ double-positive population of mixed chimeras (Fig. 9 B). The preferential expansion of NODk-derived cells could be offset by reconstituting with bone marrow mixtures containing a 4:1 ratio of B10k to NODk marrow (Fig. 9 C). Using 4:1 mixtures of TCR transgenic marrow, chimeras were obtained that formed comparably sized populations of single-positive T cells and peripheral CD4+ 1G12+ T cells in the absence of insHEL (InsHEL−; Fig. 9, D and E).
Figure 9.

Failure to delete islet-reactive cells in NODk is T cell intrinsic. (a) Design of bone marrow chimera experiment. (b) Nontransgenic Ly5ab recipients were reconstituted with a 1:1 mixture of bone marrow from B10k and NODk nontransgenic mice. The thymocytes were stained with CD4, CD8, Ly5a and Ly5b. Percentage of each thymocyte subsets is shown for two mice. (c) Percentage is as in b, except that the ratio of B10k/NODk bone marrow cells is 4:1. (d) InsHEL-positive or -negative recipients were reconstituted with a 4:1 ratio of B10k/NODk marrow. Percentage of thymocyte subsets is shown from analysis of four nontransgenic and eight insHEL-transgenic recipients. (e) Percentage of peripheral lymphocytes bearing CD4 and the HEL- reactive TCR from each donor genotype from the mice in days.
When the same mixtures reconstituted negatively selecting thymuses of insHEL-transgenic recipients, by contrast, B10k-derived T cells were selectively reduced in frequency in the double-positive compartment and very few B10k single-positive T cells or CD4+clonotype+ T cells remained. NODk-derived DP and single-positive T cells in the same thymuses were not markedly deleted despite bearing the same TCR and recognizing the same antigen/MHC combination (Fig. 9 D). As a result, despite the chimeric mice being reconstituted with a fourfold excess of B10k bone marrow, islet-reactive CD4 T cells in spleen and pancreatic lymph node of the chimeric mice were almost entirely of NODk origin (Fig. 9 E). Thus, non-MHC genes from NOD act cell autonomously within high avidity autoreactive T cells to make them insensitive to thymic negative selection.
Discussion
This study shows that self-reactive CD4 T cells bearing a high affinity TCR for a dominant epitope of HEL fail to be effectively censored during their development in the thymus of NODk mice that are making HEL in pancreatic islet β cells. The primary effect of the non-MHC genes from NOD is within the high affinity autoreactive T cells, causing them to be insensitive to clonal deletion stimuli in the thymus at the double-positive and CD4+CD24high single-positive stages. In conjunction with this NOD defect in thymic deletion, HEL-specific T cells specifically respond to antigen in the pancreatic islets where T cell help becomes available, after a variable time-lag of weeks or months, as evidenced by spontaneous development of germinal centers and large amounts of HEL-binding IgG1 autoantibody, and gradual progression to diabetes. These results define an important primary defect in the thymic selection and acquisition of T cell self-tolerance that is likely to play an important part in the general autoimmune susceptibility of NOD mice. The findings focus attention on thymic selection mechanisms for organ specific tolerance, and raise subsequent questions about how this defect might arise and how it is connected to other genetic and immunological defects described in mice and humans with organ-specific autoimmune disease.
Negative selection of self-reactive thymocytes is strikingly altered by non-MHC NOD genes acting within the T cells. In the presence of insHEL self-antigen, HEL-specific T cells bearing B10 genes showed a marked deletion in their numbers at the double-positive stage and relatively few reached the single-positive stage (Figs. 6 and 7). By contrast, T cells bearing the NOD genes showed little reduction in double-positive thymocytes and only a modest decrease in single-positive thymocytes bearing the HEL-specific TCR. Three lines of evidence establish that thymic negative selection is the primary defect, and exclude the possibility that the elevated numbers of CD4 single-positive T cells in the NODk thymus represent mature T cells that have expanded in the periphery and then homed back to the thymus. Foremost, the CD4+1G12+ cells of NODk InsHEL/TCR thymus are entirely immature CD24hi cells, indicating they are recently differentiated rather than mature cells that have returned (Fig. 8). Second, deletion is evident within the less mature CD4+8+ cells in the B10k animals but not in NODk (Fig. 6). Third, the numbers of escaped CD4+1G12+ thymocytes is highest in very young mice an diminishes with age (Fig. 8), whereas the opposite might be expected if the cells must first be expanded in the periphery. The mixed bone marrow chimera experiments exclude the possibility that differences in the types of antigen-presenting cells or efficiency or types of antigens presented to the T cells account for the failure to censor the thymic and peripheral autoreactive T cell populations, and demonstrate that NODk genes act within individual immature T cells to dramatically change the way they respond to TCR engagement by negative selecting autoantigen.
The thymic selection defects defined here are paralleled by results from Kishimoto and Sprent (11) who have shown that thymocytes from NOD mice are selectively resistant to in vitro or in vivo negative selection induced by anti-TCR and anti-CD28 antibodies or by bacterial su-perantigens, when compared with B6-H-2nod congenic controls. Since it is unclear how well anti-TCR or superantigen responses mirror the acquisition of tolerance to self-antigens, our findings clearly establish that NOD genes dramatically interfere with tolerance by negative selection in vivo, as well as showing that these inherited defects act cell autonomously within individual autoreactive CD4 cells.
The chief difference between the two analyses is the finding by Kishimoto and Sprent that NOD thymocyte apoptosis defects are only detectable in the DP-SP transitional cell population in vitro, with normal apoptosis of double-positive thymocytes. By contrast, NOD genes prevented islet antigen induced deletion in DP and SP populations in vivo. This may reflect differences in the strength and quality of signals provided by plate-bound antibodies compared with antigen-presenting cells bearing trace amounts of self-antigen. Of potentially greater significance, however, is the finding here that peripheral autoreactive T cells with the NOD genes fail to exhibit the modulation of surface TCR density that is maintained by B10 T cells (Fig. 3). This result is at odds with the conclusion that NOD genes selectively altered in vitro responses of thymocytes but not of mature peripheral T cells (11) and indicates that the inherited NOD T cell defect in sensitivity to autoantigen may carry through to alter peripheral T cell regulation as well. In this regard, non-MHC NOD genes have been shown to render thymocytes, peripheral T cells and B cells resistant to dexamethasone or cyclophosphamide-induced apoptosis (12, 52, 53).
Biochemical differences in TCR signaling in thymocytes from NOD mice have been previously shown by Delovitch and colleagues (10). In particular they showed that, compared with BALB/c thymocytes, the TCR on NOD thymocytes is less efficient at activating the ZAP70/ras/ERK pathway, which is known to be important for inducing T cell-positive and -negative selection. It will be important in future work to investigate whether inefficient activation of this pathway explains the selection defects identified here, and whether these signaling differences reflect inherited differences in components of the TCR pathway itself or in modulating factors such as costimulatory or inhibitory receptors. The latter are interesting candidates since the genes for CD28 and CTLA-4 lie in chromosomal regions of inherited diabetes susceptibility in NOD (12, 54).
CD28 signaling is a potent enhancer of negative selection in double-positive thymocytes, and complete deficiency of CD28 or its B7.1 ligand greatly accelerates the onset of diabetes in NOD mice while deficiency of the B7.2 ligand triggers autoimmune neuritis (9, 25). The CTLA-4 receptor for B7 ligands also modulates TCR signaling apparently acting at the level of TCR phosphorylation upstream of ZAP70 (55). In future experiments it will be important to map the inheritance of the thymic deletion defect by intercrosses and use of congenic strains carrying individual resistance loci.
It is interesting to consider that the intrinsic differences in NOD thymocyte response to TCR signals may also account for reduced activity of regulatory cells (3, 6), NKT cells (7, 8), and CD4+CD25+ regulatory T cells (9) in this strain. NKT cells are selected from double-positive thymocytes to carry a particular TCR in response to an unknown self-antigen presented by CD1 (56). Similarly, anergic CD4+CD25+ regulatory T cells are selected by self-antigen during the double-positive to single-positive transition (43), at the same stage that negative selection occurs. The intrinsic defects in thymocyte response to self-antigen observed here might therefore also diminish the selection of NKT and CD4+CD25+ cells. Thus, a single primary defect in thymocyte selection could conceivably create two separate secondary abnormalities contributing to autoimmune susceptibility: increased numbers of high avidity T cells recognizing organ-specific self-antigens and decreased numbers of NKT cells or anergic CD4+25+ cells capable of regulating the autoreactive population.
The thymic selection defects described here could account for the observed increase in T cells capable of proliferating to self antigens in NOD mice (13, 14) by allowing more autoreactive T cells to leave the thymus. Because the actual frequency of high avidity autoreactive T cells is not measured by proliferation assays, it is nevertheless also possible that enhanced proliferation in these assays reflects the deficiencies of NKT cells or CD4+25+ regulatory cells discussed above. In the Ridway and Kanagawa studies, inefficient negative selection of thymic or peripheral T cells was proposed but via a different mechanism, namely reduced presentation of self-antigens by the NOD I-Ag7 molecule. The defect in negative selection defined here is clearly established to reside in the responding T cells and not in the antigen-presenting cells, but it is interesting to consider that the two heritable deficits may be additive or multiplicative: an MHC-linked deficit in presentation of islet autoantigens by I-Ag7 may compound the non-MHC linked deficit in TCR-induced deletion in response to those antigens.
The insensitivity of NOD T cells to autoantigen may apply both during thymic development and after emigration to the periphery (see above). If this can be confirmed, the intrinsic T cell insensitivity caused by non-MHC NOD genes may also diminish peripheral deletion of CD8 T cells that recognize organ specific autoantigens (16, 57). Interestingly, Verdegauer found that diabetes resistance genes from NOR caused fewer CD8 T cells with an islet-reactive TCR to accumulate in the periphery (15). It is unclear if the two results are related, however, since Verdaguer et al. found no evidence for thymic deletion in the resistant background, homozygosity for resistance genes was required, and the reduced number of islet reactive cells may have been secondary to absence of helper cells promoting expansion or to diminished presentation of the islet autoantigen.
Is the failure to delete HEL-reactive T cells in the thymus the sole cause of progression to diabetes in the NODk double-transgenic mice? It is a straightforward extrapolation to connect the increased number of high avidity HEL-reactive T cells in the peripheral lymphoid tissues and pancreas to the formation of high titred IgG1 against HEL and pancreatic germinal centers in these mice, since both of these processes depend on cognate T cell help and no autoantibody is made in insHEL animals without the HEL-specific TCR transgene. It is also a reasonable extrapolation to assume that β cell destruction also results from the increased numbers of HEL-reactive CD4 cells, since diabetes in the NODk female mice requires both the TCR and insHEL genes. Nontolerant T cells from 3A9 TCR transgenic are capable of rapid β cell destruction, because they induce diabetes within 14 d after adoptive transfer into unirradiated B10k insHEL recipients provided the recipients are immunized with HEL in adjuvant (unpublished data). Nevertheless, it is striking that diabetes and anti-HEL IgG antibody production in NODk double transgenic mice occurs stochastically between 3 and 6 mo of age, despite all of the mice having increased numbers of HEL-reactive T cells and extensive insulitis at 6–8 wk old. Breakage of tolerance to HEL may take a variable period of time simply because the high avidity HEL-reactive T cells must first organize the insulitis into supportive structures like germinal centers, or it may require a separate deficit such as the regulatory abnormalities discussed above or an unknown exogenous insult.
It is interesting that progression to diabetes occurs by an apparently distinct genetic and cellular mechanism in a subset of B10k double transgenic mice (Fig. 1). In these animals, diabetes develops only at an 8–12 wk age window and is not accompanied by any IgG1 secretion to insHEL, and this form of diabetes is fully suppressed by a single set of NODk chromosomes in F1 hybrids. The early onset disease linked to B10 genes has parallels to the early onset diabetes that occurs in BDC2.5 TCR transgenic mice on the C57BL/6.H-2NOD strain background (35). A region on chromosomes 7 from the related B6 or B10 strains has been shown to contain a diabetes susceptibility genes, but it is not yet known what gene or defect may be involved (35, 58, 59).
Despite the conceptually attractive idea that peripherally induced mechanisms are critical for acquiring self-tolerance to organ-specific antigens, the primary defect in thymic negative selection found here indicates that thymic selection may play a more critical role than has been thought. Indeed a growing body of evidence implicates thymic selection as a critical step for organ-specific tolerance. In quail/chicken chimeras, tolerance to a transplanted quail wing bud is only acquired if some of the T cells develop in a quail thymus (60, 61). Neonatal thymectomy in rodents has long been known to lead to organ-specific autoimmune diseases, notably autoimmune gastritis and oophoritis. Thymic-derived T cells, notably a CD4+25+ subset, have been shown to be capable of suppressing autoimmune and immunopathologic reactions in thymectomized or T cell lymphopenic recipients (43–46). Deficiency of CD4+25+ cells and autoimmune disease in mice without IL-2 or IL-2R may reflect a primarily thymic function of IL-2 in tolerance, because the IL-2Rb deficiency disorder is corrected when expression of this receptor is limited to developing thymocytes (62). A growing list of organ-specific genes has been shown to be expressed in the thymus, especially in thymic medullary epithelial cells (36, 37), and inheritance of an insulin gene allele that causes lower thymic expression is associated with diabetes susceptibility in humans (63, 64). Finally, multiple organ-specific autoimmune diseases develop in people with the inherited Mendelian disorder, Autoimmune Polyendocrinopathy, resulting from loss of a putative transcription factor primarily expressed in subsets of thymic epithelial or dendritic cells (65–67).
Our results have implications for diabetes prevention and therapy efforts. If similar defects in thymocyte or peripheral T cell responses to self-antigens contribute to human type 1 diabetes, then agents that further lessen TCR signaling such as cyclosporin or CD28/B7 blockade may be contraindicated. These maneuvers may exaggerate the failure to censor self-reactive T cells and lead to worsening of disease or development of other autoimmune diseases, as occurs when NOD mice are made deficient for B7 or CD28 molecules (9, 25). Agents that nonspecifically augment costimulation, stimulate negative selection selectively, or that stimulate formation of regulatory T cells may be better at correcting the primary defect in individuals with organ-specific autoimmunity.
Acknowledgments
We thank Drs. L. Wicker, E. Unanue, and D. Peterson for their generous gifts of mice and antibody, Adrienne McKenzie, Katherine Sullivan, and the staff of Medical Genome Centre for curating the mouse colony, Aisling Murtagh and Suzanne Ewing for genotyping, and Stephen Martin for IV injections.
This work was supported by a grant from the NIH and the Juvenile Diabetes Research Foundation. S. Lesage is a recipient of a CIHR scholarship.
Footnotes
Abbreviations used in this paper: HEL, hen egg lysozyme; insHEL, transgenic mice expressing HEL under the rat insulin promoter; NOD, nonobese diabetic.
References
- 1.Todd, J.A., and L.S. Wicker. 2001. Genetic protection from the inflammatory disease type 1 diabetes in humans and animal models. Immunity. 15:387–395. [DOI] [PubMed] [Google Scholar]
- 2.Sinha, A.A., M.T. Lopez, and H.O. McDevitt. 1990. Autoimmune diseases: the failure of self tolerance. Science. 248:1380–1388. [DOI] [PubMed] [Google Scholar]
- 3.Bach, J.F., and L. Chatenoud. 2001. Tolerance to islet autoantigens in Type 1 diabetes. Annu. Rev. Immunol. 19:131–161. [DOI] [PubMed] [Google Scholar]
- 4.Adorini, L., S. Gregori, and L.C. Harrison. 2002. Understanding autoimmune diabetes: insights from mouse models. Trends Mol. Med. 8:31–38. [DOI] [PubMed] [Google Scholar]
- 5.Delovitch, T.L., and B. Singh. 1997. The nonobese diabetic mouse as a model of autoimmune diabetes: immune dysregulation gets the NOD. Immunity. 7:727–738. [DOI] [PubMed] [Google Scholar]
- 6.Serreze, D.V., and E.H. Leiter. 1988. Defective activation of T suppressor cell function in nonobese diabetic mice. Potential relation to cytokine deficiencies. J. Exp. Med. 140:3801–3807. [PubMed] [Google Scholar]
- 7.Gombert, J.M., A. Herbelin, E. Tancrede-Bohin, M. Dy, C. Carnaud, and J.F. Bach. 1996. Early quantitative and functional deficiency of NK1+-like thymocytes in the NOD mouse. Eur. J. Immunol. 26:2989–2998. [DOI] [PubMed] [Google Scholar]
- 8.Baxter, A.G., S.J. Kinder, K.J.L. Hammond, R. Scollay, and D.I. Godfrey. 1997. Association between αβTCR+CD4− CD8− T cell deficiency and IDDM in NOD/Lt mice. Diabetes. 46:572–582. [DOI] [PubMed] [Google Scholar]
- 9.Salomon, B., D.J. Lenschow, L. Rhee, N. Ashourian, B. Singh, A. Sharpe, and J.A. Bluestone. 2000. B7/CD28 costimulation is essential for the homeostasis of the CD4+ CD25+ immunoregulatory T cells that control autoimmune diabetes. Immunity. 12:431–440. [DOI] [PubMed] [Google Scholar]
- 10.Salojin, K., J. Zhang, M. Cameron, B. Gill, G. Arreaza, A. Ochi, and T.L. Delovitch. 1997. Impaired plasma membrane targeting of Grb2-murine son of sevenless (mSOS) complex and differential activation of the Fyn-T cell receptor (TCR)-ζ-Cbl pathway mediate T cell hyporesponsiveness in autoimmune nonobese diabetic mice. J. Exp. Med. 186:887–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kishimoto, H., and J. Sprent. 2001. A defect in central tolerance in NOD mice. Nat. Immunol. 2:1025–1031. [DOI] [PubMed] [Google Scholar]
- 12.Colucci, F., M.L. Bergman, C. Penha-Goncalves, C.M. Cilio, and D. Holmberg. 1997. Apoptosis resistance of nonobese diabetic peripheral lymphocytes linked to the Idd5 diabetes susceptibility region. Proc. Natl. Acad. Sci. USA. 94:8670–8674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ridgway, W.M., H. Ito, M. Fasso, C. Yu, and C.G. Fathman. 1998. Analysis of the role of variation of major histocompatibility complex class II expression on nonobese diabetic (NOD) peripheral T cell response. J. Exp. Med. 188:2267–2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kanagawa, O., S.M. Martin, B.A. Vaupel, E. Carrasco-Marin, and E.R. Unanue. 1998. Autoreactivity of T cells from nonobese diabetic mice: an I-Ag7-dependent reaction. Proc. Natl. Acad. Sci. USA. 95:1721–1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Verdaguer, J., A. Amrani, B. Anderson, D. Schmidt, and P. Santamaria. 1999. Two mechanisms for the non-MHC-linked resistance to spontaneous autoimmunity. J. Immunol. 162:4614–4626. [PubMed] [Google Scholar]
- 16.Kreuwel, H.T., J.A. Biggs, I.M. Pilip, E.G. Pamer, D. Lo, and L.A. Sherman. 2001. Defective CD8+ T cell peripheral tolerance in nonobese diabetic mice. J. Immunol. 167:1112–1117. [DOI] [PubMed] [Google Scholar]
- 17.Kikutani, H., and S. Makino. 1992. The murine autoimmune diabetes model: NOD and related strains. Adv. Immunol. 51:285–322. [DOI] [PubMed] [Google Scholar]
- 18.Hattori, M., J.B. Buse, R.A. Jackson, L. Glimcher, M.E. Dorf, M. Minami, S. Makino, K. Moriwaki, H. Kuzuya, H. Imura, et al. 1986. The NOD mouse: recessive diabetogenic gene in the major histocompatibility complex. Science. 231:733–735. [DOI] [PubMed] [Google Scholar]
- 19.Chao, C.C., H.K. Sytwu, E.L. Chen, J. Toma, and H.O. McDevitt. 1999. The role of MHC class II molecules in susceptibility to type I diabetes: identification of peptide epitopes and characterization of the T cell repertoire. Proc. Natl. Acad. Sci. USA. 96:9299–9304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Serreze, D.V., and E.H. Leiter. 1994. Genetic and pathogenic basis of autoimmune diabetes in NOD mice. Curr. Opin. Immunol. 6:900–906. [DOI] [PubMed] [Google Scholar]
- 21.Wicker, L.S., J.A. Todd, and L.B. Peterson. 1995. Genetic control of autoimmune diabetes in the NOD mouse. Annu. Rev. Immunol. 13:179–200. [DOI] [PubMed] [Google Scholar]
- 22.Serreze, D.V., M. Bridgett, H.D. Chapman, E. Chen, S.D. Richard, and E.H. Leiter. 1998. Subcongenic analysis of the Idd13 locus in NOD/Lt mice: evidence for several susceptibility genes including a possible diabetogenic role for β2-microglobulin. J. Immunol. 160:1472–1478. [PubMed] [Google Scholar]
- 23.Rasooly, L., C.L. Burek, and N.R. Rose. 1996. Iodine-induced autoimmune thyroiditis in NOD-H-2h4 mice. Clin. Immunol. Immunopathol. 81:287–292. [DOI] [PubMed] [Google Scholar]
- 24.Damotte, D., E. Colomb, C. Cailleau, N. Brousse, J. Charreire, and C. Carnaud. 1997. Analysis of susceptibility of NOD mice to spontaneous and experimentally induced thyroiditis. Eur. J. Immunol. 27:2854–2862. [DOI] [PubMed] [Google Scholar]
- 25.Salomon, B., L. Rhee, H. Bour-Jordan, H. Hsin, A. Montag, B. Soliven, J. Arcella, A.M. Girvin, J. Padilla, S.D. Miller, and J.A. Bluestone. 2001. Development of spontaneous autoimmune peripheral polyneuropathy in B7-2-deficient NOD mice. J. Exp. Med. 194:677–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ohashi, P.S., S. Oehen, K. Buerki, H. Pircher, C.T. Ohashi, B. Odermatt, B. Malissen, R.M. Zinkernagel, and H. Hengartner. 1991. Ablation of “tolerance” and induction of diabetes by virus infection in viral antigen transgenic mice. Cell. 65:305–317. [DOI] [PubMed] [Google Scholar]
- 27.Heath, W.R., J. Allison, M.W. Hoffmann, G. Schonrich, G. Hammerling, B. Arnold, and J.F. Miller. 1992. Autoimmune diabetes as a consequence of locally produced interleukin-2. Nature. 359:547–549. [DOI] [PubMed] [Google Scholar]
- 28.Goverman, J., A. Woods, L. Larson, L.P. Weiner, L. Hood, and D.M. Zaller. 1993. Transgenic mice that express a myelin basic protein-specific T cell receptor develop spontaneous autoimmunity. Cell. 72:551–560. [DOI] [PubMed] [Google Scholar]
- 29.Lafaille, J.J., K. Nagashima, M. Katsuki, and S. Tonegawa. 1994. High incidence of spontaneous autoimmune encephalomyelitis in immunodeficient anti-myelin basic protein T cell receptor transgenic mice. Cell. 78:399–408. [DOI] [PubMed] [Google Scholar]
- 30.Scott, B., R. Liblau, S. Degermann, L.A. Marconi, L. Ogata, A.J. Caton, H.O. McDevitt, and D. Lo. 1994. A role for non-MHC genetic polymorphism in susceptibility to spontaneous autoimmunity. Immunity. 1:73–83. [DOI] [PubMed] [Google Scholar]
- 31.Degermann, S., C. Reilly, B. Scott, L. Ogata, H. von Boehmer, and D. Lo. 1994. On the various manifestations of spontaneous autoimmune diabetes in rodent models. Eur. J. Immunol. 24:3155–3160. [DOI] [PubMed] [Google Scholar]
- 32.Akkaraju, S., W.Y. Ho, D. Leong, K. Canaan, M.M. Davis, and C.C. Goodnow. 1997. A range of CD4 T cell tolerance: partial inactivation to organ-specific antigen allows nondestructive thyroiditis or insulitis. Immunity. 7:255–271. [DOI] [PubMed] [Google Scholar]
- 33.Katz, J.D., B. Wang, K. Haskins, C. Benoist, and D. Mathis. 1993. Following a diabetogenic T cell from genesis through pathogenesis. Cell. 74:1089–1100. [DOI] [PubMed] [Google Scholar]
- 34.Verdaguer, J., D. Schmidt, A. Amrani, B. Anderson, N. Averill, and P. Santamaria. 1997. Spontaneous autoimmune diabetes in monoclonal T cell nonobese diabetic mice. J. Exp. Med. 186:1663–1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gonzalez, A., J.D. Katz, M.G. Mattei, H. Kikutani, C. Benoist, and D. Mathis. 1997. Genetic control of diabetes progression. Immunity. 7:873–883. [DOI] [PubMed] [Google Scholar]
- 36.Jolicoeur, C., D. Hanahan, and K.M. Smith. 1994. T-cell tolerance toward a transgenic β-cell antigen and transcription of endogenous pancreatic genes in thymus. Proc. Natl. Acad. Sci. USA. 91:6707–6711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Derbinski, J., A. Schulte, B. Kyewski, and L. Klein. 2001. Promiscuous gene expression in medullary thymic epithelial cells mirrors the peripheral self. Nat. Immunol. 2:1032–1039. [DOI] [PubMed] [Google Scholar]
- 38.Smith, K.M., D.C. Olson, R. Hirose, and D. Hanahan. 1997. Pancreatic gene expression in rare cells of thymic medulla: evidence for functional contribution to T cell tolerance. Int. Immunol. 9:1355–1365. [DOI] [PubMed] [Google Scholar]
- 39.Harrington, C.J., A. Paez, T. Hunkapiller, V. Mannikko, T. Brabb, M. Ahearn, C. Beeson, and J. Goverman. 1998. Differential tolerance is induced in T cells recognizing distinct epitopes of myelin basic protein. Immunity. 8:571–580. [DOI] [PubMed] [Google Scholar]
- 40.Targoni, O.S., and P.V. Lehmann. 1998. Endogenous myelin basic protein inactivates the high avidity T cell repertoire. J. Exp. Med. 187:2055–2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Klein, L., T. Klein, U. Ruther, and B. Kyewski. 1998. CD4 T cell tolerance to human C-reactive protein, an inducible serum protein, is mediated by medullary thymic epithelium. J. Exp. Med. 188:5–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Klein, L., M. Klugmann, K.A. Nave, V.K. Tuohy, and B. Kyewski. 2000. Shaping of the autoreactive T-cell repertoire by a splice variant of self protein expressed in thymic epithelial cells. Nat. Med. 6:56–61. [DOI] [PubMed] [Google Scholar]
- 43.Jordan, M.S., A. Boesteanu, A.J. Reed, A.L. Petrone, A.E. Holenbeck, M.A. Lerman, A. Naji, and A.J. Caton. 2001. Thymic selection of CD4+CD25+ regulatory T cells induced by an agonist self-peptide. Nat. Immunol. 2:301–306. [DOI] [PubMed] [Google Scholar]
- 44.Fowell, D., and D. Mason. 1993. Evidence that the T cell repertoire of normal rats contains cells with the potential to cause diabetes. Characterization of the CD4+ T cell subset that inhibits this autoimmune potential. J. Exp. Med. 177:627–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Itoh, M., T. Takahashi, N. Sakaguchi, Y. Kuniyasu, J. Shimizu, F. Otsuka, and S. Sakaguchi. 1999. Thymus and autoimmunity: production of CD25+CD4+ naturally anergic and suppressive T cells as a key function of the thymus in maintaining immunologic self-tolerance. J. Immunol. 162:5317–5326. [PubMed] [Google Scholar]
- 46.Suri-Payer, E., A.Z. Amar, A.M. Thornton, and E.M. Shevach. 1998. CD4+CD25+ T cells inhibit both the induction and effector function of autoreactive T cells and represent a unique lineage of immunoregulatory cells. J. Immunol. 160:1212–1218. [PubMed] [Google Scholar]
- 47.Ho, W.Y., M.P. Cooke, C.C. Goodnow, and M.M. Davis. 1994. Resting and anergic B cells are defective in CD28-dependent costimulation of naive CD4+ T cells. J. Exp. Med. 179:1539–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Podolin, P.L., A. Pressey, N.H. DeLarato, P.A. Fischer, L.B. Peterson, and L.S. Wicker. 1993. I-E+ nonobese diabetic mice develop insulitis and diabetes. J. Exp. Med. 178:793–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Van Parijs, L., D.A. Peterson, and A.K. Abbas. 1998. The Fas/Fas ligand pathway and Bcl-2 regulate T cell responses to model self and foreign antigens. Immunity. 8:265–274. [DOI] [PubMed] [Google Scholar]
- 50.Allen, P.M., G.R. Matsueda, R.J. Evans, J.B. Dunbar, Jr., G.R. Marshall, and E.R. Unanue. 1987. Identification of the T-cell and Ia contact residues of a T-cell antigenic epitope. Nature. 327:713–715. [DOI] [PubMed] [Google Scholar]
- 51.Todd, J.A., T.J. Aitman, R.J. Cornall, S. Ghosh, Jr., S. Hall, C.M. Hearne, A.M. Knight, J.M. Love, M.A. McAleer, J.-B. Prins, N. Rodrigues, M. Lathrop, A. Pressey, N.H. DeLarato, L.B. Peterson, and L.S. Wicker. 1991. Genetic analysis of autoimmune type 1 diabetes mellitus in mice. Nature. 351:542–547. [DOI] [PubMed] [Google Scholar]
- 52.Penha-Goncalves, C., K. Leijon, L. Persson, and D. Holmberg. 1995. Type 1 diabetes and the control of dexamethazone-induced apoptosis in mice maps to the same region on chromosome 6. Genomics. 28:398–404. [DOI] [PubMed] [Google Scholar]
- 53.Colucci, F., C.M. Cilio, K. Lejon, C.P. Goncalves, M.L. Bergman, and D. Holmberg. 1996. Programmed cell death in the pathogenesis of murine IDDM: resistance to apoptosis induced in lymphocytes by cyclophosphamide. J. Autoimmun. 9:271–276. [DOI] [PubMed] [Google Scholar]
- 54.Hill, N.J., P.A. Lyons, N. Armitage, J.A. Todd, L.S. Wicker, and L.B. Peterson. 2000. NOD Idd5 locus controls insulitis and diabetes and overlaps the orthologous CTLA4/IDDM12 and NRAMP1 loci in humans. Diabetes. 49:1744–1747. [DOI] [PubMed] [Google Scholar]
- 55.Chambers, C.A., and J.P. Allison. 1999. Costimulatory regulation of T cell function. Curr. Opin. Cell Biol. 11:203–210. [DOI] [PubMed] [Google Scholar]
- 56.Gapin, L., J.L. Matsuda, C.D. Surh, and M. Kronenberg. 2001. NKT cells derive from double-positive thymocytes that are positively selected by CD1d. Nat. Immunol. 2:971–978. [DOI] [PubMed] [Google Scholar]
- 57.Morgan, D.J., H.T. Kreuwel, and L.A. Sherman. 1999. Antigen concentration and precursor frequency determine the rate of CD8+ T cell tolerance to peripherally expressed antigens. J. Immunol. 163:723–727. [PubMed] [Google Scholar]
- 58.Ghosh, S., S.M. Palmer, N.R. Rodrigues, H.J. Cordell, C.M. Hearne, R.J. Cornall, J.B. Prins, P. McShane, G.M. Lathrop, L.B. Peterson, et al. 1993. Polygenic control of autoimmune diabetes in nonobese diabetic mice. Nat. Genet. 4:404–409. [DOI] [PubMed] [Google Scholar]
- 59.McAleer, M.A., P. Reifsnyder, S.M. Palmer, M. Prochazka, J.M. Love, J.B. Copeman, E.E. Powell, N.R. Rodrigues, J.B. Prins, D.V. Serreze, et al. 1995. Crosses of NOD mice with the related NON strain. A polygenic model for IDDM. Diabetes. 44:1186–1195. [DOI] [PubMed] [Google Scholar]
- 60.Ohki, H., C. Martin, C. Corbel, M. Coltey, and N.M. Le Douarin. 1987. Tolerance induced by thymic epithelial grafts in birds. Science. 237:1032–1035. [DOI] [PubMed] [Google Scholar]
- 61.Le Douarin, N., C. Corbel, A. Bandeira, V. Thomas-Vaslin, Y. Modigliani, A. Coutinho, and J. Salaun. 1996. Evidence for a thymus-dependent form of tolerance that is not based on elimination or anergy of reactive T cells. Immunol. Rev. 149:35–53. [DOI] [PubMed] [Google Scholar]
- 62.Malek, T.R., B.O. Porter, E.K. Codias, P. Scibelli, and A. Yu. 2000. Normal lymphoid homeostasis and lack of lethal autoimmunity in mice containing mature T cells with severely impaired IL-2 receptors. J. Immunol. 164:2905–2914. [DOI] [PubMed] [Google Scholar]
- 63.Pugliese, A., M. Zeller, A. Fernandez, Jr., L.J. Zalcberg, R.J. Bartlett, C. Ricordi, M. Pietropaolo, G.S. Eisenbarth, S.T. Bennett, and D.D. Patel. 1997. The insulin gene is transcribed in the human thymus and transcription levels correlated with allelic variation at the INS VNTR-IDDM2 susceptibility locus for type 1 diabetes. Nat. Genet. 15:293–297. [DOI] [PubMed] [Google Scholar]
- 64.Vafiadis, P., S.T. Bennett, J.A. Todd, J. Nadeau, R. Grabs, C.G. Goodyer, S. Wickramasinghe, E. Colle, and C. Polychronakos. 1997. Insulin expression in human thymus is modulated by INS VNTR alleles at the IDDM2 locus. Nat. Genet. 15:289–292. [DOI] [PubMed] [Google Scholar]
- 65.Finnish-German Consortium. 1997. An autoimmune disease, APECED, caused by mutations in a novel gene featuring two PHD-type zinc-finger domains. Nat. Genet. 17:399–403. [DOI] [PubMed] [Google Scholar]
- 66.Nagamine, K., P. Peterson, H.S. Scott, J. Kudoh, S. Minoshima, M. Heino, K.J. Krohn, M.D. Lalioti, P.E. Mullis, S.E. Antonarakis, et al. 1997. Positional cloning of the APECED gene. Nat. Genet. 17:393–398. [DOI] [PubMed] [Google Scholar]
- 67.Heino, M., P. Peterson, N. Sillanpaa, S. Guerin, L. Wu, G. Anderson, H.S. Scott, S.E. Antonarakis, J. Kudoh, N. Shimizu, et al. 2000. RNA and protein expression of the murine autoimmune regulator gene (Aire) in normal, RelB-deficient and in NOD mouse. Eur. J. Immunol. 30:1884–1893. [DOI] [PubMed] [Google Scholar]