

Abstract
Human immunodeficiency virus type 1 (HIV-1) fuses with cells after sequential interactions between its envelope glycoproteins, CD4 and a coreceptor, usually CC chemokine receptor 5 (CCR5) or CXC receptor 4 (CXCR4). CMPD 167 is a CCR5-specific small molecule with potent antiviral activity in vitro. We show that CMPD 167 caused a rapid and substantial (4–200-fold) decrease in plasma viremia in six rhesus macaques chronically infected with simian immunodeficiency virus (SIV) strains SIVmac251 or SIVB670, but not in an animal infected with the X4 simian–human immunodeficiency virus (SHIV), SHIV-89.6P. In three of the SIV-infected animals, viremia reduction was sustained. In one, there was a rapid, but partial, rebound and in another, there was a rapid and complete rebound. There was a substantial delay (>21 d) between the end of therapy and the onset of full viremia rebound in two animals. We also evaluated whether vaginal administration of gel-formulated CMPD 167 could prevent vaginal transmission of the R5 virus, SHIV-162P4. Complete protection occurred in only 2 of 11 animals, but early viral replication was significantly less in the 11 CMPD 167-recipients than in 9 controls receiving carrier gel. These findings support the development of small molecule CCR5 inhibitors as antiviral therapies, and possibly as components of a topical microbicide to prevent HIV-1 sexual transmission.
Keywords: HIV, AIDS, antiretroviral therapy, microbicide, chemokine receptor
Introduction
The primate immunodeficiency viruses HIV-1 and simian immunodeficiency virus (SIV), and the chimeric, engineered viruses known as simian–human immunodeficiency viruses (SHIVs), all fuse with their target cells in a process triggered by the sequential interaction of the viral envelope glycoprotein complex with the CD4 receptor and a coreceptor (1, 2). The most important HIV-1 coreceptors are CCR5 and CXCR4, with CCR5 being most commonly used by viruses that dominate the early stages of infection (3–6). Indeed, the absence of CCR5 (homozygosity for the defective CCR5-Δ32 allele) is strongly protective against acquisition of HIV-1 infection, sexually or otherwise (7–10). Several small molecules that bind to CCR5, including SCH-C, SCH-D, and UK-427,857 are now in clinical development to treat HIV-1 infection (11–17) (C. Hitchcock, personal communication). The licensing of enfuvirtide (T-20) strongly supports the overall concept of using entry inhibitors for this purpose (18, 19).
Entry inhibitors may also be useful as topically applied, vaginal or rectal microbicides, or even for systemically applied prophylaxis, to prevent HIV-1 infection. There is an urgent need to develop chemical methods of prevention, particularly for covert use by women (20–22). The rationale for including a CCR5-specific inhibitor in a microbicide formulation is theoretically strong, because of the importance of CCR5 for establishment of HIV-1 infection (3–10). Therefore, we have studied the effect of a small molecule CCR5 inhibitor on both established viral infection in rhesus macaques and on the transmission of a new infection.
CMPD 167 is a CCR5-specific receptor antagonist that is no longer being developed as an antiviral drug for humans. However, it is a potent inhibitor of HIV-1 infection in vitro and its toxicology and pharmacology properties are appropriate for monkey analyses (23, 24). We used SIVmac251 and SIVB670 as the test viruses to study the effect of CMPD 167 on established viral infection. None of the available CCR5-using SHIVs consistently generates an adequately sustained level of plasma viremia during chronic infection (25–27). SIV strains use CCR5 as their paramount coreceptor in primary T cells in vitro (28, 29), but their coreceptor usage profile is broader on coreceptor-transfected cell lines compared with HIV-1 (3–6). For vaginal challenge studies, we used SHIV-162P4, which exclusively uses CCR5 for entry in vitro (30, 31). We have reported that a vaginally applied anti-gp120 mAb, b12, can protect macaques against vaginal transmission of SHIV-162P4 (25).
We show that CMPD 167 has antiviral activity against SIVmac251 and SIVB670 replication in vivo, by causing marked, and sometimes sustained, reductions in plasma viremia. However, even concentrations of CMPD 167 in the millimolar range were generally unable to prevent vaginal transmission of SHIV-162P4, although they did reduce the extent of viremia after infection. These results support the development of CCR5 inhibitors as antiviral drugs. They may also be useful as vaginal microbicides if they can be successfully combined with other entry inhibitors.
Materials and Methods
Inhibition of SIVmac251, SIVB670, and SHIV-162P4 Replication In Vitro.
Uncloned isolates of SIVmac251 (32), SIVB670 (33), and SHIV-89.6P (34) were provided by P.A. Marx, L. Martin (both affiliated with Tulane University, New Orleans, LA), and N. Letvin (Harvard University, Boston, MA), respectively. SHIV-162P4 was a gift from J. Harouse and C. Cheng-Mayer (both affiliated with Rockefeller University, New York, NY) (30, 31). Replication of test viruses in human and macaque PBMCs in the presence and absence of CMPD 167 was determined as described previously (29, 35).
Infusion of CMPD 167 into SIV-infected Macaques.
Six adult rhesus macaques (Macacca mulatta) of Indian origin ranging from 6 to 10 yr of age were infected with SIV for 116–293 d before CMPD 167 therapy. Two macaques (AE55 and V744) had been inoculated intravenously with SIVmac251 and four (T246, T682, N217, and K299) had been inoculated intravaginally with SIVB670. A seventh rhesus macaque of Chinese origin was inoculated intravenously with SHIV-89.6P (a CXCR4-using virus).
CMPD 167 (10 mg/kg in PBS) was administered intravenously by bolus injection twice daily for 14 d and once daily for an additional 14 d. Plasma viral loads were monitored 1–2 times daily during treatment, and at least weekly for 6 wk after therapy ceased. Plasma viral load was determined by quantifying SIV gag RNA using a real-time RT-PCR assay (36). As used in the present analysis, the assay has a sensitivity threshold of 60 RNA copies/ml for all of the viruses tested, with an interassay coefficient variation of <25%. CD4+ T cell counts were measured using a whole-blood staining method and allophycocyanin-conjugated anti-CD4 mAb RPA-T4 (Becton Dickinson). The percentage of CD4+ T cells was determined by flow cytometry using a FACSCalibur™ flow cytometer and CELLQuest™ software, as described previously (36). Absolute numbers of lymphocytes were determined using a hematology analyzer system (Advia 120; Bayer Diagnostics, Inc). Absolute CD4+ T cell counts were calculated by multiplying the percentage of lymphocytes that were CD4+ by the absolute number of lymphocytes per microliter of blood. All studies adhered to the Guide for the Care and Use of Laboratory Animals, prepared by the National Research Council, National Institutes of Health, and with the Guidelines of the Tulane National Primate Research Center Institutional Animal Care and Use Committee.
Vaginal Administration of CMPD 167 and Vaginal Challenge with SHIV-162P4.
Normal, cycling rhesus macaques 5–19 yr of age were used. To thin the vaginal epithelium, macaques were treated with a single 30-mg intramuscular injection of depo-medroxyprogesterone acetate (Depo-Provera®; Pfizer) for 30–33 d, as described previously (25). The macaques were sedated with ketamine, placed in ventral recumbency, and 4–5 ml of either 2.5% hydroxymethyl cellulose (HMC) gel or CMPD 167 (0.6 mg/ml and 1 mM) in HMC was atraumatically introduced into the vaginal vault using a pliable French catheter, followed 15 min later by 300 TCID50 of SHIV-162P4 in 1 ml of RPMI 1640 medium. Blood was collected weekly for at least 50 d. Plasma viremia was quantified using either the bDNA assay (Bayer Diagnostics Inc.) which has a sensitivity limit of 500 RNA copies/ml plasma (see Fig. 4, top, challenge stock A), or by RT-PCR (sensitivity limit, 60 RNA copies/ml) (see Fig. 4, bottom, challenge stock B) as described in the preceding paragraph (37).
Figure 4.

After the infection, viral loads in macaques were challenged vaginally with SHIV-162P4 after vaginal administration of HMC gel or CMPD 167 formulated in HMC at ∼1 mM. Two separate, but similar, experiments are shown, each performed using a different SHIV-162P4 challenge stock and a different method of quantifying viral RNA. Hence, the results are plotted separately.
Data Analysis.
In the treatment experiments, the viremic t 1/2 was calculated by fitting an exponential function to the data derived either from the period from the viremia peak during acute infection, or from the period between treatment initiation and the first local minimum after the rapid decline. From the equation
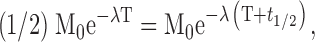 |
where M0 is the maximum viremia and T = 0, we derive t 1/2 = (ln2)/λ. In the microbicide studies, the effect of vaginal CMPD 167 administration on the extent of initial SHIV-162P4 replication was determined by comparing the maximum viral loads recorded (log RNA copies/ml) in the control and CMPD 167 recipient groups. For the two uninfected CMPD 167 recipients (see Fig. 4, challenge stock A), the assay detection limit was considered to be the maximum value. We also compared the area under the curve (AUC) of the log viral loads as a function of time for the two groups. The AUC values were derived by approximating the curves to histograms describing the log RNA copies/ml for the time intervals between days 0 and 35. For the two uninfected animals, viral load was considered to be equal to the detection limit (500 RNA copies/ml), rather than zero; this procedure errs on the side of underestimating statistical differences between the CMPD 167 and control groups. The Student's t test (one tail, unequal variance) included in Excel (Microsoft) was used to assess the statistical significance of observed differences in means between groups. Standard deviations given are for n − 1 degrees of freedom.
Results
CMPD 167 Is a Small Molecule CCR5 Inhibitor of SIV Replication In Vitro
A program at Merck Research Labs to identify new ways to treat HIV-1 infection led to the development of the CCR5 inhibitor CMPD 167 (Fig. 1) , designated previously MRK-1 (23, 24). CMPD 167 is a potent receptor antagonist for both human and macaque CCR5; it inhibits chemokine ligand binding to macaque CCR5 with Ki values of 1 and 5 nM in buffer and whole blood, respectively. Although it is not being developed for human use due to a marginal therapeutic index (the highest concentration with no adverse effect is ∼10–30 mg/kg), the pharmacokinetic profile of CMPD 167 in macaques renders it useful for evaluating the antiviral potential of CCR5 ligands in vivo (23).
Figure 1.

Structure of the CCR5 inhibitor, CMPD 167.
First, we confirmed that CMPD 167 inhibits the replication of SIVmac251, SIVB670, and SHIV-162P4 in PBMCs from different, uninfected macaques (Table I). Preinfection PBMC samples from the SIV-infected test animals were not available for this purpose because the animals were acquired for this work only after they had already been infected with SIV for other reasons. We have shown previously that the potency of a different small molecule CCR5 inhibitor, TAK-779, against SIVmac251 and SIVmac239 in vitro varies considerably in a donor-dependent manner (23). The same finding was made with CMPD 167, its 50% inhibitory concentration (IC50) values against SIVmac251 and SIVB670 varying throughout a >250-fold range in PBMCs from different macaques (Table I). Whatever the IC50 value, high concentrations of CMPD 167 (up to 10 μM) caused complete or almost complete (i.e., >90%) inhibition of the replication of the test viruses in cells from most, but not all, of the donor macaques (Table I), similar to what was observed using TAK-779 (23).
Table I.
Inhibition of SIV, SHIV and HIV-1 Replication by CMPD 167 In Vitro
| Virus | PBMC type |
Donor | IC50 | Max Inhibition |
|---|---|---|---|---|
| nM | % | |||
| SIVmac251 | macaque | A | 130 | 91 |
| B | 85 | 77 | ||
| C | 8.0 | 100 | ||
| D | 36 | 96 | ||
| E | 0.47 | 100 | ||
| SIVB670 | macaque | B | 9.3 | 55 |
| C | 5.3 | 98 | ||
| SHIV-162P4 | macaque | A | 61 | 86 |
| B | 3.4 | 100 | ||
| C | 5.2 | 100 | ||
| F | 30 | 100 | ||
| SHIV-89.6P | macaque | G | >10,000 | – |
| H | >10,000 | – | ||
| SHIV-162P4 | human | I | 0.030 | 100 |
| J | 0.24 | 98 | ||
| HIV-1 JR-FL | human | I | 0.27 | 100 |
| J | 0.20 | 97 | ||
| HIV-1 CC1/85 | human | I | 0.31 | 100 |
| J | 0.15 | 100 |
IC50 values derived from individual experiments using PBMC from different uninfected macaques (A–H ) or humans (I and J) are presented, along with the maximum percentage inhibition achieved with higher concentrations of CMPD 167 (up to 10 μM). Macques A–H do not correspond to animals described in Figs. 2–4.
CMPD 167 also inhibited HIV-1 replication in human PBMCs, with IC50 values usually in the low nanomolar range (Table I). The range of IC50 values for this and other CCR5 inhibitors was much less in human than in macaque PBMCs (Table I and not depicted). We have observed this with small molecule CCR5 inhibitors in general. SHIV-162P4 replication was also inhibited by CMPD 167 in both human and macaque PBMCs, again to a variable extent (Table I). However, the extent of the variation in SHIV-162P4 sensitivity in macaque PBMCs was less than observed with SIVmac251 and SIVB670. CMPD 167 did not inhibit SHIV-89.6P replication in macaque PBMCs (Table I), although this virus was sensitive to the CXCR4-specific inhibitor AMD3100, with IC50 values <1 nM (23) (not depicted). Hence, as concluded previously from studies using TAK-779, SHIV-89.6P behaves as an X4 virus in macaque PBMCs, with no detectable sensitivity to CCR5-specific inhibitors (30).
We again interpret these observations to mean that CCR5 is the paramount coreceptor used by SIVmac251 and SIVB670 to enter macaque PBMCs in vitro (30). However, unknown factors significantly influence the efficiency of action of CCR5-targeted inhibitors in these cells, more than in human PBMCs. A more extensive analysis is now in progress to understand this variation.
Treatment of SIV-infected Rhesus Macaques with CMPD 167
Six rhesus macaques of Indian origin infected with uncloned SIV isolates were monitored for 116–240 d until a stable viral set point was achieved. Some of the test animals were obtained only very shortly before therapy was commenced, limiting the amount of pretherapy viral load data that could be obtained.
To ensure consistency in dosing and to minimize animal-to-animal variability in pharmacokinetic profiles, we elected to deliver CMPD 167 by intravenous infusion. Except for animal K299, the monkeys received 10 mg/kg of CMPD 167 twice daily for 14 d followed by a switch to once-daily dosing for a further 14 d, to reduce the cumulative stress on the animals caused by drug infusion under anesthesia. This regimen provided the maximum possible receptor coverage while staying acceptably below the toxic level of CMPD 167 for macaques. The peak concentration of CMPD 167 in macaque plasma during twice-daily dosing is ∼15 μM. Because the plasma half life of CMPD 167 is ∼2 h, this dosing regimen maintained a trough concentration of ∼70 nM (at 12 h), which is >90% inhibitory concentration value for inhibiting the binding of chemokines to human and macaque CCR5 (reference 30; unpublished data). The trough level is also above the IC50 value for inhibition of SIV replication in PBMCs from some of the uninfected donor macaques (Table I). However, this concentration may not be sufficient to completely block SIV replication in all the infected animals if the variation in their sensitivity to CMPD 167 is similar to that observed in vitro using PBMCs from different, uninfected macaques (Table I). After the change to once-daily dosing, the trough concentration of CMPD 167 in plasma (5–10 nM at 24 h) approximates the IC50 value for inhibition of chemokine binding in vitro (24). This concentration is unlikely to be high enough to completely block macaque CCR5, particularly in any animals whose cells are at the less sensitive end of the range observed in vitro (Table I). Thus, some SIV entry via CCR5 is to be expected over part or all of the dosing cycle in some of the test animals.
The administration of CMPD 167 caused rapid, exponential reductions in plasma viremia in all six SIVmac251- or SIVB670-infected animals, as discussed further in the next section (Fig. 2) . Significant declines in viral load were detectable within 24 h, with an initial nadir being reached after 3–6 d. At the nadir, the viral load reduction from the pretherapy value varied from 4- to 200-fold in different animals; the mean was 55-fold (Fig. 2 and Table II). The mean t 1/2 for the viral load decline was 0.94 ± 0.18 d, significantly shorter than the corresponding value for the decline during primary infection in the same animals (6.4 ± 2.7 d) (Table II, P = 0.0020, Student's t test). Hence, the drug-induced decline in viral load was rapid with little variation between animals.
Figure 2.

Effect of CMPD 167 infusion into macaques infected with SIVmac251, SIVB670, or SHIV-89.6P. Twice-daily CMPD 167 therapy was commenced on day 0 (indicated by vertical line). After 14 d, therapy was administered once daily (arrowhead, 1X) for an additional 14 d, until day 28, when treatment was stopped (arrowhead, END). The dotted line indicates the viral load “baseline” at the time therapy was initiated. Each panel represents the study of an individual macaque.
Table II.
Summary of the Outcome of CMPD 167 Infusion into Different Macaques Infected with SIVmac251, SIVB670, or SHIV89.6P
| Animal | V744 | AE55 | T246 | T682 | N217 | K299 | M244 |
|---|---|---|---|---|---|---|---|
| Virus | SIVmac251 | SIVmac251 | SIVB670 | SIVB670 | SIVB670 | SIVB670 | SHIV89.6P |
| Start of therapy (days after challenge) | 116 | 116 | 240 | 240 | 240 | 132 | 55 |
|
t
1/2 (days) for VL decline in primary infection, [R2] |
10 [0.90] | 5.3 [0.84] | 2.9 [0.82] | 5.2 [0.78] | 8.8 [0.78] | 5.9 [0.59] | 3.7 [0.69] |
| VL at start of therapy (RNA copies/ml) | 1.3 × 106 | 2.9 × 105 | 1.1 × 106 | 4.4 × 104 | 2.3 × 106 | 1.4 × 106 | 6.2 × 103 |
| VL at initial treatment-induced nadir (RNA copies/ml) |
a5.6 × 104 | a7.0 × 104 | 1.1 × 105 | 5.5 × 103 | 2.1 × 105 | 7.1 × 103 | NAe |
| Fold reduction in VL at initial nadir | 23 | 4 | 10 | 80 | 11 | 200 | 7 |
| Time to reach nadir (days) | 3.5 | 4 | 3 | 7 | 4 | 6 | NAe |
|
t
1/2 (days) for VL decline to initial treatment-induced nadir, [R2] |
0.70 [0.93] | 1.1 [0.82] | 0.89 [0.98] | 1.1 [0.95] | 1.1 [0.96] | 0.77 [0.96] | 7.4 [0.27]f |
| VL reboundb (days from end of treatment) | 43 | 2 | 36 | 29 | −14c | NAd | NA |
R2 describes the fit of the exponential function to the data. A value >0.25 indicates a good correlation with the logarithmically transformed linear function. NA, Not applicable.
Lower values were recorded subsequently; the lowest value for AE55 was 1.1 × 104 RNA copies/ml on day 21; for V744, 1.9 × 104 on day 17.
Rebound indicates the number of days from the end of CMPD 167 treatment until viral load met or exceeded the pretreatment, baseline value.
Rebound occurred 14 d before the end of therapy, not after it.
This animal was removed from the study after 10 d of therapy.
A full description is provided on the next page.
Although no treatment effect was detectable, t1/2 for the spontaneous VL decline was measured over the relevant period.
We also studied the effect of CMPD 167 on viral load induced by SHIV-89.6P, to see whether the lack of sensitivity of this virus to this inhibitor in vitro (Table I) was also true in vivo. Because SHIV-89.6P is highly aggressive and usually causes very rapid disease progression in macaques of Indian origin (38, 39), we used a macaque from China. Viral loads are generally lower and the disease course slower in these animals (39). We also elected to begin therapy of the SHIV-89.6P–infected animal, M244, on an earlier day after infection (day 55) than for the SIV-infected animals, to minimize the chances of it progressing to disease before the end of our work. The pretherapy viral load in M244 had not fully stabilized by day 55, as shown by the markedly downward slope of the viremia plot in the period immediately before CMPD 167 was infused. In contrast, the viral loads in the SIV-infected animals T246 and T682 were slowly rising in the last 40 d before therapy, and in V744 and AE55, they were slowly falling (minimal pretherapy data were available from animals N217 and K299).
CMPD 167 had a negligible effect on plasma viral load in M244 (Fig. 2). Indeed, the viral load marginally increased in the first 5 d of therapy, leading to a twofold reduction in the slope of the viral load plot (Fig. 2 and Table II). In contrast, the viral load plots in the SIV-infected animals T246, T682, V744, and AE55 all showed a steep, downward slope in the first 5 d after CMPD 167 infusion that was >10-fold steeper than the corresponding pretherapy slope. The much poorer fit of an exponential function to the viral load after treatment in M244 than for the SIV-infected animals also suggests that M244 alone did not undergo a drug-induced reduction in viremia (Table II). Viral loads fluctuated only moderately in M244 during therapy, with no clear downward trend over and above the natural decline that was in progress before therapy (Fig. 2). Furthermore, when therapy was stopped, there was no viral load rebound. Hence we conclude that a CCR5 inhibitor has a negligible effect on SHIV-89.6P replication in macaque cells in vivo, just as it does in vitro (Table I). SHIV-89.6P is functionally an X4 virus (30). Moreover, the lack of effect of CMPD 167 on SHIV-89.6P replication suggests that the viral load declines in the SIVmac251- and SIVB670-infected animals were not attributable merely to the infusion of the inhibitor under anesthesia.
The Viral Load Responses to CMPD 167 Infusion in Individual Macaques
All six SIV-infected animals responded differently to CMPD 167 infusion (Fig. 2).
Macaque V744 (SIVmac251 Infected).
A significant drop in viremia from the baseline value of 1.3 × 106 RNA copies/ml was detectable within 36 h, and a nadir of 56,000 copies/ml, a 23-fold decline, was reached after 3.5 d. Viral load rebounded slightly, fluctuating in the range 19,000–390,000 copies/ml for the remainder of the treatment period, even after the switch to once-daily dosing. Indeed, the lowest plasma viremia level (19,000 copies/ml) occurred 2 d after the dosing reduction. Viremia was 160,000 copies/ml when therapy ended. It remained suppressed at close to that level for a further 21 d, before steadily rebounding to reach values from 106 to 2 × 106 copies/ml by 5–8 wk after the end of therapy.
Macaque AE55 (SIVmac251 Infected).
Baseline viral load was 290,000 RNA copies/ml. A decline was detected after 2 d, and a nadir of 70,000 copies/ml was reached after 4 d, a 4.1-fold decline. Viremia hovered from 110,000 to 190,000 copies/ml for the next 10 d, before declining again (coincidentally or otherwise) to fluctuate from 11,000 to 370,000 copies/ml, during the period of halved CMPD 167 dosage. The nadir during that period, 11,000 copies/ml, was 26-fold below baseline, but viral load gradually rebounded. The viral load after therapy was from 150,000 to 430,000 copies/ml for 8 wk, similar to the pretherapy value.
Macaque T246 (SIVB670 Infected).
The baseline viral load of 1.1 × 106 RNA copies/ml dropped by 10-fold to 110,000 copies/ml within 3 d. Viremia remained fairly steady in the range 130,000–350,000 copies/ml until therapy was discontinued after 27 d, when it was 200,000 copies/ml. There was no evidence of any viral load rebound during therapy, but a transient, partial increase to 750,000 copies/ml took place 5–11 d after the end of therapy. By day 22 after therapy, viral load was 450,000 copies/ml, approximately twice the level on the day of discontinuation. Viremia rebounded fully over the next 2 wk, eventually reaching 1.5 × 106 copies/ml 36 d after the end of therapy, similar to the pretherapy level.
Macaque T682 (SIVB670 Infected).
A rapid, 80-fold decline in plasma viremia from a baseline level of 440,000 RNA copies/ml to a nadir of 5,500 copies/ml occurred during the first 7 d. A slow but steady rebound to 62,000 copies/ml took place over the next 8 d, after which, the viral load gradually, but slightly, declined again. Viral load was 32,000 copies/ml at the end of therapy, 14-fold below baseline. A gradual rebound was detectable within 3–5 d, and viremia steadily increased thereafter, reaching 750,000 copies/ml by 36 d after the end of therapy, 1.7-fold higher than baseline.
Macaque N217 (SIVB670 Infected).
The response of this animal to CMPD 167 was unusual. Within 4 d, viremia had dropped by 11-fold from the baseline value of 2.3 × 106 to a nadir of 210,000 copies/ml. A modest, short-lived, transient blip occurred over the next 5 d, but viremia was still depressed (280,000 copies/ml) on day 9 of therapy. However, a strong rebound was seen during the next 10 d, viremia rising as high as 1.1 × 107 copies/ml by day 21, fivefold above the pretherapy value. A further decline took place over the remaining 4 d, so that the level of viremia at the end of therapy (2.6 × 106 copies/ml) was almost the same as the pretherapy value (2.3 × 106 copies/ml). Viral load did not increase after therapy was discontinued; indeed, 36 d later, plasma viremia (940,000 copies/ml) was 2.8-fold lower than on the day therapy was stopped.
Macaque K299 (SIVB670 Infected).
This animal was included in the work despite its availability on short notice. A limited amount of information could be obtained as K299 became ill and died after 10 d of therapy from “fatal fasting syndrome,” which is a common disease of obese macaques (40). The death was unrelated to CMPD 167 infusion. The viral load at the onset of therapy (1.4 × 106 RNA copies/ml) declined very rapidly after CMPD 167 was infused, halving within 24 h, and reached a nadir of 7,100 copies/ml (a 200-fold decline) within 6 d. A partial rebound to 90,000 copies/ml had occurred by the time the animal died after a further 4 d.
CD4 T Cell Responses to CMPD 167
CD4+ T cell counts were measured in three of the test animals (N217, T246, and T682) before and during the course of CMPD 167 therapy (Fig. 3) . Because we were examining viral loads twice daily in the initial studies, insufficient blood volumes were available for CD4 counts to be measured for the other three animals. There was a marked, two- to threefold increase in the CD4+ T cell count in T246 and T682 within 7 d of therapy that coincided with the initial decrease in viral load, followed by a partial decline (Figs. 2 and 3). In N217, the initial increase in the CD4+ count occurred more slowly, peaking after 21 d at approximately twice the pretherapy level (Fig. 3). In all three macaques, the CD4+ T cell count was greater at the end of CMPD 167 therapy than at the beginning, and it was still elevated 35 d later (Fig. 3).
Figure 3.

Absolute CD4+ T cell counts in the blood of three SIV-infected macaques before, during, and after treatment with CMPD 167. The pretreatment values shown were determined 164–184 d before therapy (84–104 d after infection).
Evaluating CMPD 167 for Its Ability to Prevent the Vaginal Transmission of an R5 SHIV
We also studied whether CMPD 167 could be used as a topical microbicide to prevent the vaginal transmission of SHIV-162P4 to uninfected macaques (20, 21). SHIV-162P4 rarely causes sustained plasma viremia; the usual pattern is a transient, high level of replication followed by a decline to minimal viremia levels by 5–8 wk (25). Hence, we are using this virus only to determine whether a candidate microbicide can have a short-term protective effect against virus transmission and/or its early replication.
In preliminary experiments, saline solutions of CMPD 167 in the micromolar or low millimolar range failed to protect any macaques against SHIV-162P4 (unpublished data). Therefore, we formulated CMPD 167 in 2.5% HMC gel at 0.6 mg/ml (∼1 mM) and applied 5 ml to the vagina of each test macaque, followed by vaginal SHIV-162P4 challenge 15 min later. HMC gel itself had no protective effect compared with saline (unpublished data). Moreover, we confirmed that in vitro formulation in HMC gel did not interfere with the antiviral activity of CMPD 167 in either human or macaque PBMCs (unpublished data).
In two experiments involving a total of 20 macaques and two different SHIV-162P4 challenge stocks, we saw limited evidence of full protection by CMPD 167 when the data were pooled and analyzed. Thus, all 9 animals that received HMC gel alone became infected, as did 9 of 11 animals given CMPD 167 in HMC (Fig. 4) . The difference in the infection rate between the two groups was not significant. However, plasma viremia after infection was significantly less, on average, in the CMPD 167 group than in the control group. The mean peak log viral load values were lower (4.4 ± 1.1) for the CMPD 167 recipients than in the HMC controls (6.1 ± 0.93) (P = 0.0011). An approximation of cumulative plasma viremia was also estimated by calculating the mean AUC (±SD) for log RNA copies per ml as a function of time over the first 35 d after challenge. The AUC also was significantly lower for the 11 CMPD 167 recipients (120 ± 22) than for the 9 control animals (150 ± 25) (P = 0.0080).
Discussion
We have studied how a small molecule, a CCR5-specific inhibitor, affects SIV replication in vivo. Although CMPD 167 is not now being developed as an antiviral drug, similar CCR5 inhibitors are (41). The initial rate of viral load decline in response to CMPD 167 (t 1/2, 0.94 ± 0.18 d) was as fast as rates observed during protease inhibitor monotherapy in humans (42, 43), and comparable to that induced by the combination of Tenofovir and β-2′,3′-dideoxy-3′-thia-5-fluorocytidine in one of two SIV-infected pig-tailed macaques (44). Direct comparisons involving different test viruses and species are difficult, but CMPD 167 clearly has significant antiviral activity in vivo.
There was considerable variation in the responses of individual animals to CMPD 167. The mean viremia reduction at the initial nadir was 55-fold but ranged from 4.1-fold (AE55) to 200-fold (K299). Moreover, the level of residual viral replication in each test animal was both substantial and variable, with nadir values from 5,500 to 200,000 RNA copies/ml (Table II). Neither the rate nor the extent of the initial decline was correlated with the viral load at the onset of therapy (4.4 × 104 to 2.3 × 106 RNA copies/ml), rather higher than usually found in HIV-1–infected humans (39, 42, 43). One plausible explanation for animal-to-animal variation is that it is related to factors that influence the antiviral potency of CMPD 167 and another CCR5 inhibitor, TAK-779, in vitro (Table I; reference 29). Clearly, if the IC50 for inhibition of SIV replication in vivo spanned the 275-fold range (0.47–130 nM) observed in PBMCs from different, uninfected macaques in vitro, a considerable variation in viral load reduction would be expected. With predicted trough levels of 70 nM during the twice-daily dosing phase, lower during once-daily dosing, plasma drug levels may simply not have been high enough to substantially inhibit SIV replication in some of the animals. Unfortunately, because the macaques were only acquired after they had been infected with SIV, we could not study the inherent sensitivity of their PBMCs to CMPD 167 before commencing therapy. Future studies will be designed accordingly.
The molecular mechanisms underlying animal-to-animal variation in response to CMPD 167 are unclear. Differences in the levels of CCR5 expression on the target cells of different animals, or in the extent of CC-chemokine secretion, could affect the potency of any CCR5 inhibitor (41, 45–52). The variable responses could also reflect the use by SIV of alternative coreceptors (such as BOB, CXCR6, etc.), which would be insensitive to CMPD 167. The expression levels of these potential coreceptors on various cell types is host dependent (53).
Factors applying only under in vivo conditions could also influence how different macaques responded to CMPD 167. Animal-to-animal differences in pharmacokinetics could be relevant, although we deliberately used intravenous dosing to minimize such variation. Drug levels may be lower in the lymphoid compartments, where SIV replicates, than in the plasma. Due to the toxicity profile of CMPD 167, we were not able to perform dose-escalation studies. Finally, although all the macaques were infected with SIVmac251 or the related SIVB670 virus, host immune responses are likely to create viral diversity during the period between infection and the onset of therapy; therefore, partially resistant viruses could be present in some animals. Additional studies, some now commenced, will be required to resolve these questions.
Many of these factors might also apply to understanding variation in the antiviral effects of CCR5 inhibitors in humans (13, 14). Host-dependent variable responses have been observed in early human trials of SCH-C; viral load declines after 10-d dosing varied from 0 to >1.5 log in different individuals all infected with SCH-C–sensitive R5 HIV-1 isolates (13, 14). Extrapolations from SIV-infected macaques to HIV-1–infected humans are problematic, but the use of alternative coreceptors by HIV-1 is much less common than by SIV, at least in vitro (28, 29, 53–56). Hence, alternative coreceptor usage may not contribute to the variable responses to CCR5 inhibitors in humans, and it is only one possible explanation of what we observed in the macaques.
A notable observation was that animals V744 and T246 had no detectable plasma viremia rebound throughout the 28 d of CMPD 167 therapy despite high residual viral loads of 105–106 RNA copies/ml. Hence, SIV might not be able to generate escape mutants easily in response to a CCR5 inhibitor, at least in some infected animals. In vitro, the development of HIV-1 escape mutants in response to different small molecule CCR5 inhibitors is a slow and complex process requiring the accumulation of multiple sequence changes in gp120, without coreceptor switching to CXCR4 (reference 35; unpublished data). Switching to CXCR4 use as an escape pathway is probably less likely with SIV than HIV-1. However, plasma viremia did rebound rapidly during therapy of animal N217, rapidly but only partially in T682, and slowly in AE55 (Fig. 2). Indeed, in N217, there was a net increase in viral load during the third week of therapy, and the increase in CD4+ T cell count in N217 was delayed compared with animals T246 and T682 (Fig. 4). We are now investigating whether a CMPD 167 escape mutant developed in N217 within the first 5–10 d of therapy that was not just resistant to the inhibitor but that was also able to briefly replicate in a new population of target cells, perhaps because of a change in coreceptor usage. In principle, this could have adverse pathogenic consequences. However, the upward spike in virus replication was temporary; viral load in N217 had dropped back to baseline by the time therapy was discontinued and continued to decline modestly, so whatever occurred was a transient event.
The viremia rebound in animal T682 during therapy was only partial, but it was notable that when therapy was stopped, viral load returned to baseline rather more rapidly than it did in V744 and T246. Partially resistant mutant viruses may have developed during therapy in T682, and also in AE55. Genotypic and phenotypic studies of the viral strains present in the test animals before, during, and after CMPD 167 therapy are now underway.
A particularly unexpected finding was that, in V744, no rebound in viremia occurred for 3 wk after CMPD 167 therapy ceased; only later did viremia rise gradually to the baseline level. Furthermore, in T246, full rebound to the pretherapy level only took place 22–36 d after the end of therapy. When conventional highly active antiretroviral therapy is discontinued in humans, viral load usually rebounds within ∼7–14 d, even from levels <500 RNA copies/ml (57, 58). In macaques V744 and T246, plasma viremia at the end of CMPD 167 therapy was much higher (up to 2 × 106 and 1.5 × 106 RNA copies/ml, respectively), yet the rebound was still markedly delayed. Therefore, a CCR5 inhibitor can have a sustained effect on viral replication after therapy is stopped, at least in some test animals. Delays of a few days before viremia rebounded after the completion of a 10-d dosing protocol have also been observed in some HIV-1–infected patients during early human trials of SCH-C (13). Hence, whatever happened in animal V744 might not be unique to SIV infection of a monkey. We are now investigating whether CMPD 167 and other CCR5 inhibitors can have a sustained effect on the expression and recycling of functional CCR5 receptors in macaque and human PBMCs.
In summary, CMPD 167 monotherapy caused a rapid decrease in viremia in all 6 SIV-infected animals, and an increase in CD4+ T cell count in all three animals in which this was measured. The reduced viral load could be maintained throughout the 28 d of therapy (and sometimes well beyond) in some monkeys, even in the face of high viral replication and despite reducing CMPD 167 input to what may have been suboptimal levels. Together with early evidence of other CCR5 inhibitors being effective in HIV-1–infected humans (13) (C. Hitchcock, personal communication), these findings should encourage the practical development of CCR5 blockers as antiviral drugs. These compounds should be combined with other entry inhibitors or more conventional antiviral drugs, based on in vitro studies (11–15, 17, 59, 60).
We also evaluated whether CMPD 167 could prevent vaginal transmission of the R5 virus SHIV-162P4. The use of a SHIV rather than a SIV permitted comparison of CMPD 167 with other compounds specific for the HIV-1 envelope glycoproteins, such as the anti-gp120 mAb b12, which can protect macaques against vaginal transmission of SHIV-162P4 (25). We found that CMPD 167, delivered vaginally at 1 mM in HMC gel, was not consistently protective. Hence, a CCR5-specific compound that inhibits SHIV-162P4 replication in the nanomolar range in vitro was ineffective against the same challenge virus in vivo, even when used at a million-fold higher concentration. mAb b12 could only protect macaques against SHIV-162P4 when applied vaginally at 1 mg/ml, a concentration 100- to 1,000-fold more than neutralized the same virus in vitro (25). Clearly, the quantitative aspects of achieving protection using a CCR5-specific inhibitor are even more daunting. A higher concentration of CMPD 167, or a different formulation of it, might be more effective, but 1 mM was the highest concentration we could achieve at neutral pH.
It is possible that a topically applied small molecule CCR5 inhibitor cannot be fully effective against vaginal (or rectal) transmission, because it may not penetrate to the sites where CCR5-mediated fusion occurs during the early stages of infection. These events may happen at or close to the vaginal epithelium where a CCR5 inhibitor could intervene (61–65). However, an alternative or additional route of transmission involves transport of virus to lymph nodes within dendritic cells, where it only fuses with CD4+ CCR5+ T cells upon arrival (63, 64, 66, 67). If so, a topically applied, receptor-blocking compound has little chance to intervene successfully when used alone. We did observe partial protection in that viral load after infection was reduced in the CMPD 167 recipients. The simplest explanation is that CMPD 167 successfully prevented the transmission of some of the virus particles that normally establish infection, thereby diminishing SHIV-162P4 replication during primary infection. However, other routes of transmission remained unimpeded.
The goal of a microbicide must be to fully prevent the establishment of infection; a modest reduction in viral load after infection would not be of significant benefit. In that sense, the results obtained with CMPD 167 are not encouraging. However, the SHIV-162P4 inoculum used to challenge the progesterone-treated macaques is ∼108 RNA copies/ml, sufficient to cause infection of almost 100% of the test animals (25). Therefore, this model is a stringent test of a vaginal microbicide. The infectious HIV-1 dose for women is not known, so it is hard to predict how CMPD 167 would fare in naturally exposed women, given the many complex assumptions that must be made. However, it is reasonable to assume that the lower the inoculum to be countered, the easier the task for a receptor-blocker (25, 41). Combinations of entry inhibitors that include a CCR5-specific small molecule should, therefore, be further evaluated in macaques (25, 59). CCR5 inhibitors that act differently from CMPD 167 (i.e., a chemokine derivative that caused sustained CCR5 down-regulation) should also be pursued (52, 68).
Acknowledgments
We thank J. LeBlanc, M. Dodd, W. Cyprian, K. Callahan, M. Barrilleaux, A. Gallego, and A. Hoffman for technical assistance.
This work was supported by Public Health Service grants (AI52048, AI41420, AI49080, HD36310, and RR00164) and in part by federal funds from the National Cancer Institute, National Institutes of Health under contract no. NO1-CO-124000. M.S. Springer, J.A. DeMartino, S.J. Siciliano, and K. Lyons are employees of Merck Research Labs. The other authors declare no financial interest.
Abbreviations used in this paper: AUC, area under the curve; HMC, hydroxymethyl cellulose; IC50, 50% inhibitory concentration; SHIV, simian–human immunodeficiency virus; SIV, simian immunodeficiency virus.
References
- 1.Wyatt, R., and J. Sodroski. 1998. The HIV-1 envelope glycoproteins: fusogens, antigens, and immunogens. Science. 280:1884–1888. [DOI] [PubMed] [Google Scholar]
- 2.Doms, R.W., and J.P. Moore. 2000. HIV-1 membrane fusion: targets of opportunity. J. Cell Biol. 151:F9–F14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Berger, E.A., P.M. Murphy, and J.M. Farber. 1999. Chemokine receptors as HIV-1 coreceptors: roles in viral entry, tropism, and disease. Annu. Rev. Immunol. 17:657–700. [DOI] [PubMed] [Google Scholar]
- 4.Doms, R.W. 2001. Chemokine receptors and HIV entry. AIDS. 15:S34–S35. [DOI] [PubMed] [Google Scholar]
- 5.Dragic, T. 2001. An overview of the determinants of CCR5 and CXCR4 co-receptor function. J. Gen. Virol. 82:1807–1814. [DOI] [PubMed] [Google Scholar]
- 6.Clapham, P.R., and A. McKnight. 2002. Cell surface receptors, virus entry and tropism of primate lentiviruses. J. Gen. Virol. 83:1809–1829. [DOI] [PubMed] [Google Scholar]
- 7.Liu, R., W.A. Paxton, S. Choe, D. Ceredini, S.R. Martin, R. Horuk, M.E. MacDonald, H. Stuhlmann, R.A. Koup, and N.R. Landau. 1996. Homozygous defect in HIV-1 coreceptor accounts for resistance of some multiply-exposed individuals to HIV-1 infection. Cell. 86:367–377. [DOI] [PubMed] [Google Scholar]
- 8.Samson, M., F. Libert, B.J. Doranz, J. Rucker, C. Liesnard, C.M. Farber, S. Saragosti, C. Lapoumeroulie, J. Cognaux, C. Forceille, et al. 1996. Resistance to HIV-1 infection in caucasian individuals bearing mutant alleles of the CCR-5 chemokine receptor gene. Nature. 382:722–725. [DOI] [PubMed] [Google Scholar]
- 9.Dean, M., M. Carrington, C. Winkler, G.A. Huttley, M.W. Smith, R. Allikmets, J.J. Goedert, S.P. Buchbinder, E. Vittinghoff, E. Gomperts, et al. 1996. Genetic restriction of HIV-1 infection and progression to AIDS by a deletion allele of the CKR5 structural gene. Hemophilia Growth and Development Study, Multicenter AIDS Cohort Study, Multicenter Hemophilia Cohort Study, San Francisco City Cohort, ALIVE Study. Science. 273:1856–1862. (published erratum appears in Science. 1996. 274:1069) [DOI] [PubMed]
- 10.O'Brien, S.J., and J.P. Moore. 2000. The effect of genetic variation in chemokines and their receptors on HIV transmission and progression to AIDS. Immunol. Rev. 177:99–111. [DOI] [PubMed] [Google Scholar]
- 11.Moore, J.P., and M. Stevenson. 2000. New targets for inhibitors of HIV-1 replication. Nat. Rev. Mol. Cell Biol. 1:40–49. [DOI] [PubMed] [Google Scholar]
- 12.Richman, D.D. 2001. HIV chemotherapy. Nature. 410:995–1001. [DOI] [PubMed] [Google Scholar]
- 13.Baroudy, B. 2002. AIDS-14th International Conference (Part I), New Drugs, New Targets Symposium. 24.
- 14.Reynes, J., R. Rouzier, T. Kanouni, V. Baillat, B. Baroudy, A. Keung, C. Hogan, M. Markowitz, and M. Laughlin. 2002. SCH C: Safety and antiviral effects of a CCR5 receptor antagonist in HIV-1 infected subjects. 9th Conference on Retroviruses and Opportunistic Infections. 53.
- 15.Schwarz, M.K., and T.N. Wells. 2002. New therapeutics that modulate chemokine networks. Nat. Rev. Drug Disc. 1:347–358. [DOI] [PubMed] [Google Scholar]
- 16.Kazmierski, W., N. Bifulco, H. Yang, L. Boone, F. DeAnda, C. Watson, and T. Kenakin. 2003. Recent progress in discovery of small-molecule CCR5 chemokine receptor ligands as HIV-1 inhibitors. Bioorg. Med. Chem. 11:2663–2676. [DOI] [PubMed] [Google Scholar]
- 17.Kilby, J.M., and J.J. Eron. 2003. Novel therapies based on mechanisms of HIV-1 cell entry. N. Engl. J. Med. 348:2228–2238. [DOI] [PubMed] [Google Scholar]
- 18.Lalezari, J.P., K. Henry, M. O'Hearn, J.S. Montaner, P.J. Piliero, B. Trottier, S. Walmsley, C. Cohen, D.R. Kuritzkes, J.J. Eron, Jr., et al. 2003. Enfuvirtide, an HIV-1 fusion inhibitor, for drug-resistant HIV infection in North and South America. N. Engl. J. Med. 348:2175–2185. [DOI] [PubMed] [Google Scholar]
- 19.Lazzarin, A., B. Clotet, D. Cooper, J. Reynes, K. Arasteh, M. Nelson, C. Katlama, H.J. Stellbrink, J.F. Delfraissy, J. Lange, et al. 2003. Efficacy of enfuvirtide in patients infected with drug-resistant HIV-1 in Europe and Australia. N. Engl. J. Med. 348:2186–2195. [DOI] [PubMed] [Google Scholar]
- 20.Turpin, J.A. 2002. Considerations and development of topical microbicides to inhibit the sexual transmission of HIV. Expert Opin. Investig. Drugs. 11:1077–1097. [DOI] [PubMed] [Google Scholar]
- 21.Stone, A. 2002. Microbicides: a new approach to preventing HIV and other sexually transmitted infections. Nat. Rev. Drug Discov. 1:977–985. [DOI] [PubMed] [Google Scholar]
- 22.Shattock, R.A., and J.P. Moore. 2003. Inhibiting HIV-1 sexual transmission. Nat. Rev. Microbiol. 1:25–34. [DOI] [PubMed] [Google Scholar]
- 23.Finke, P.E., B. Oates, L.C. Meurer, J.L. Loebach, K.A. Parker, J.J. Hale, C.L. Lynch, R.J. Budhu, A.L. Gentry, C.G. Caldwell, et al. 2002. The discovery of potent human CCR5 antagonists based on a cyclopentane scaffold. Inflammation Research Association 11th National Convention Proceedings. A56:S130.
- 24.Kumar, S., G.Y. Kwei, G.K. Pook, S.A. Iliff, Y. Wang, Q. Chen, R.B. Franklin, V. Didolkar, R.W. Wang, M. Yamazaki, et al. 2003. Pharmacokinetics and interactions of a novel antagonist of chemokine receptor 5 (CCR5) with Ritonavir in rats and monkeys. J. Pharm. Exp. Therapeutics. 304:1161–1171. [DOI] [PubMed] [Google Scholar]
- 25.Veazey, R.S., R.J. Shattock, M. Pope, J.C. Kirijan, J. Jones, Q. Hu, T. Ketas, P.A. Marx, P.J. Klaase, D.R. Burton, and J.P. Moore. 2003. Prevention of virus transmission to macaque monkeys by a vaginally applied monoclonal antibody to HIV gp120. Nat. Med. 9:343–346. [DOI] [PubMed] [Google Scholar]
- 26.Pal, R., B. Taylor, S. Foulke, R. Woodward, M. Merges, R. Praschunus, A. Gibson, and M. Reitz. 2003. Characterization of a simian human immunodeficiency virus encoding the envelope gene from the CCR5-tropic HIV-1 Ba-L. J. Acquir. Immune Defic. Syndr. 33:300–307. [DOI] [PubMed] [Google Scholar]
- 27.Enose, Y., A. Miyake, E. Ido, and M. Hayami. 2003. Infection of a chimeric simian and human immunodeficiency virus with CCR5-specific HIV-1 envelope to rhesus macaques. J. Vet. Med. Sci. 65:283–286. [DOI] [PubMed] [Google Scholar]
- 28.Edinger, A.L., T.L. Hoffman, M. Sharron, B. Lee, B. O'Dowd, and R.W. Doms. 1998. Use of GPR1, GPR15, and STRL33 as coreceptors by diverse human immunodeficiency virus type 1 and simian immunodeficiency virus envelope proteins. Virol. 249:367–378. [DOI] [PubMed] [Google Scholar]
- 29.Zhang, Y., B. Lou, R.B. Lal, A. Gettie, P.A. Marx, and J.P. Moore. 2000. Use of inhibitors to evaluate coreceptor usage by simian and simian/human immunodeficiency viruses and human immunodeficiency virus type 2 in primary cells. J. Virol. 74:6893–6910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Harouse, J.M., A. Gettie, R.C. Tan, J. Blanchard, and C. Cheng-Mayer. 1999. Distinct pathogenic sequela in rhesus macaques infected with CCR5 or CXCR4 utilizing SHIVs. Science. 284:816–819. [DOI] [PubMed] [Google Scholar]
- 31.Harouse, J.M., A. Gettie, T. Eshetu, R.C. Tan, R. Bohm, J. Blanchard, G. Baskin, and C. Cheng-Mayer. 2001. Mucosal transmission and induction of simian AIDS by CCR5-specific simian/human immunodeficiency virus SHIV(SF162P3). J. Virol. 75:1990–1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marx, P.A., A.I. Spira, A. Gettie, P.J. Dailey, R.S. Veazey, A.A. Lackner, C.J. Mahoney, C.J. Miller, L.E. Claypool, D.D. Ho, and N.J. Alexander. 1996. Progesterone implants enhance SIV vaginal transmission and early virus load. Nat. Med. 2:1084–1089. [DOI] [PubMed] [Google Scholar]
- 33.Trichel, A.M., E.D. Roberts, L.A. Wilson, L.N. Martin, R.M. Ruprecht, and M. Murphey-Corb. 1997. SIV/DeltaB670 transmission across oral, colonic, and vaginal mucosae in the macaque. J. Med. Primatol. 26:3–10. [DOI] [PubMed] [Google Scholar]
- 34.Reimann, K.A., J.T. Li, R. Veazey, M. Halloran, I.W. Park, G.B. Karlsson, J. Sodroski, and N.L. Letvin. 1996. A chimeric simian/human immunodeficiency virus expressing a primary patient human immunodeficiency virus type 1 isolate env causes an AIDS-like disease after in vivo passage in rhesus monkeys. J. Virol. 70:6922–6928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Trkola, A., S.E. Kuhmann, J.M. Strizki, E. Maxwell, T. Ketas, T. Morgan, P. Pugach, S. Xu, L. Wojcik, J. Tagat, et al. 2002. HIV-1 escape from a small molecule, CCR5-specific entry inhibitor does not involve CXCR4 use. Proc. Natl. Acad. Sci. USA. 99:395–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Veazey, R.S., B. Ling, I. Pandrea, H. McClure, A.A. Lackner, and P.A. Marx. 2003. Decreased CCR5 expression on CD4+ T cells of SIV-infected sooty mangabeys. AIDS Res. Hum. Retroviruses. 19:227–233. [DOI] [PubMed] [Google Scholar]
- 37.Lifson, J.D., J.L. Rossio, M. Piatak, Jr., T. Parks, L. Li, R. Kiser, V. Coalter, B. Fisher, B.M. Flynn, S. Czajak, et al. 2001. Role of CD8(+) lymphocytes in control of simian immunodeficiency virus infection and resistance to rechallenge after transient early antiretroviral treatment. J. Virol. 75:10187–10199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Feinberg, M.B., J.M. McCune, F. Miedema, J.P. Moore, and H. Schuitemaker. 2002. HIV tropism and CD4+ T-cell depletion. Nat. Med. 8:537. [DOI] [PubMed] [Google Scholar]
- 39.Ling, B., R.S. Veazey, A. Luckay, C. Penedo, K. Xu, J.D. Lifson, and P.A. Marx. 2002. SIV(mac) pathogenesis in rhesus macaques of Chinese and Indian origin compared with primary HIV infections in humans. AIDS. 16:1489–1496. [DOI] [PubMed] [Google Scholar]
- 40.Bronson, R.T., M. O'Connell, N. Klepper-Kilgore, L.V. Chalifoux, and P. Sehgal. 1982. Fatal fasting syndrome of obese macaques. Lab. Anim. Sci. 32:187–192. [PubMed] [Google Scholar]
- 41.Moore, J.P., and R.W. Doms. 2003. The entry of entry inhibitors; a fusion of science and medicine. Proc. Natl. Acad. Sci. USA. 100:10598–10602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bonhoeffer, S., R.M. May, G.M. Shaw, and M.A. Nowak. 1997. Virus dynamics and drug therapy. Proc. Natl. Acad. Sci. USA. 94:6971–6976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Perelson, A.S. 2002. Modelling viral and immune system dynamics. Nat. Rev. Immunol. 2:28–36. [DOI] [PubMed] [Google Scholar]
- 44.Shen, A., M.C. Zink, J.L. Mankowski, K. Chadwick, J.B. Margolick, L.M. Carruth, M. Li, J.E. Clements, and R.F. Siliciano. 2003. Resting CD4+ T lymphocytes but not thymocytes provide a latent viral reservoir in a simian immunodeficiency virus-Macaca nemestrina model of human immunodeficiency virus type 1-infected patients on highly active antiretroviral therapy. J. Virol. 77:4938–4949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Paxton, W.A., S.R. Martin, D. Tse, T.R. O'Brien, J. Skurnick, N.L. VanDevanter, N. Padian, J.F. Braun, D.P. Kotler, S.M. Wolinsky, and R.A. Koup. 1996. Relative resistance to HIV-1 infection of CD4 lymphocytes from persons who remain uninfected despite multiple high-risk sexual exposure. Nat. Med. 2:412–417. [DOI] [PubMed] [Google Scholar]
- 46.Paxton, W.A., A.U. Neumann, S. Kang, L. Deutch, R.C. Brown, R.A. Koup, and S.M. Wolinsky. 2001. RANTES production from CD4+ lymphocytes correlates with host genotype and rates of human immunodeficiency virus type 1 disease progression. J. Infect. Dis. 183:1678–1681. [DOI] [PubMed] [Google Scholar]
- 47.Reeves, J.D., S.A. Gallo, N. Ahmad, J.L. Miamidian, P.E. Harvey, M. Sharron, S. Pohlmann, J.N. Sfakianos, C.A. Derdeyn, R. Blumenthal, et al. 2002. Sensitivity of HIV-1 to entry inhibitors correlates with envelope/coreceptor affinity, receptor density, and fusion kinetics. Proc. Natl. Acad. Sci. USA. 99:16249–16254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Moore, J.P. 1997. Coreceptors: implications for HIV pathogenesis and therapy. Science. 276:51–52. [DOI] [PubMed] [Google Scholar]
- 49.Wu, L., W.A. Paxton, N. Kassam, N. Ruffing, J.B. Rottman, N. Sullivan, H. Choe, J. Sodroski, W. Newman, R.A. Koup, and C.R. Mackay. 1997. CCR5 levels and expression pattern correlate with infectability by macrophage-tropic HIV-1, in vitro. J. Exp. Med. 185:1681–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cocchi, F., A.L. DeVico, R. Yarchoan, R. Redfield, F. Cleghorn, W.A. Blattner, A. Garzino-Demo, S. Colombini-Hatch, D. Margolis, and R.C. Gallo. 2000. Higher macrophage inflammatory protein (MIP)-1alpha and MIP-1beta levels from CD8+ T cells are associated with asymptomatic HIV-1 infection. Proc. Natl. Acad. Sci. USA. 97:13812–13817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lee, B., M. Sharron, L.J. Montaner, D. Weissman, and R.W. Doms. 1999. Quantification of CD4, CCR5, and CXCR4 levels on lymphocyte subsets, dendritic cells, and differentially conditioned monocyte-derived macrophages. Proc. Natl. Acad. Sci. USA. 96:5215–5220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sabbe, R., G.R. Picchio, C. Pastore, O. Chaloin, O. Hartley, R. Offord, and D.E. Mosier. 2001. Donor- and ligand-dependent differences in C-C chemokine receptor 5 reexpression. J. Virol. 75:661–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sharron, M., S. Pohlmann, K. Price, E. Lolis, M. Tsang, F. Kirchhoff, R.W. Doms, and B. Lee. 2000. Expression and coreceptor activity of STRL33/Bonzo on primary peripheral blood lymphocytes. Blood. 96:41–49. [PubMed] [Google Scholar]
- 54.Zhang, Y.J., T. Dragic, Y. Cao, L. Kostrikis, D.S. Kwon, D.R. Littman, V.N. KewalRamani, and J.P. Moore. 1998. Use of coreceptors other than CCR5 by non-syncytium-inducing adult and pediatric isolates of human immunodeficiency virus type 1 is rare in vitro. J. Virol. 72:9337–9344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang, Y.J., and J.P. Moore. 1999. Will multiple coreceptors need to be targeted by inhibitors of human immunodeficiency virus type 1 entry? J. Virol. 73:3443–3448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pohlmann, S., B. Lee, S. Meister, M. Krumbiegel, G. Leslie, R.W. Doms, and F. Kirchhoff. 2000. Simian immunodeficiency virus utilizes human and sooty mangabey but not rhesus macaque STRL33 for efficient entry. J. Virol. 74:5075–5082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Davey, R.T., Jr., N. Bhat, C. Yoder, T.W. Chun, J.A. Metcalf, R. Dewar, V. Natarajan, R.A. Lempicki, J.W. Adelsberger, K.D. Miller, et al. 1999. HIV-1 and T cell dynamics after interruption of highly active antiretroviral therapy (HAART) in patients with a history of sustained viral suppression. Proc. Natl. Acad. Sci. USA. 96:15109–15114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chun, T.W., R.T. Davey, Jr., M. Ostrowski, J. Shawn Justement, D. Engel, J.I. Mullins, and A.S. Fauci. 2000. Relationship between pre-existing viral reservoirs and the re-emergence of plasma viremia after discontinuation of highly active anti-retroviral therapy. Nat. Med. 6:757–761. [DOI] [PubMed] [Google Scholar]
- 59.Tremblay, C.L., F. Giguel, C. Kollmann, Y. Guan, T.C. Chou, B.M. Baroudy, and M.S. Hirsch. 2002. Anti-human immunodeficiency virus interactions of SCH-C (SCH 351125), a CCR5 antagonist, with other antiretroviral agents in vitro. Antimicrob. Agents Chemother. 46:1336–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nagashima, K.A., D.A. Thompson, S.I. Rosenfield, P.J. Maddon, T. Dragic, and W.C. Olson. 2001. Human immunodeficiency virus type 1 entry inhibitors PRO 542 and T-20 are potently synergistic in blocking virus-cell and cell-cell fusion. J. Infect. Dis. 183:1121–1125. [DOI] [PubMed] [Google Scholar]
- 61.Shattock, R.J., G.E. Griffin, and G.I. Gorodeski. 2000. In vitro models of mucosal HIV transmission. Nat. Med. 6:607–608. [DOI] [PubMed] [Google Scholar]
- 62.Greenhead, P., P. Hayes, P.S. Watts, K.G. Laing, G.E. Griffin, and R.J. Shattock. 2000. Parameters of human immunodeficiency virus infection of human cervical tissue and inhibition by vaginal virucides. J. Virol. 74:5577–5586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Frank, I., and M. Pope. 2002. The enigma of dendritic cell-immunodeficiency virus interplay. Curr. Mol. Med. 2:229–248. [DOI] [PubMed] [Google Scholar]
- 64.Pope, M., and A.T. Haase. 2003. Transmission, acute HIV-1 infection and the quest for strategies to prevent infection. Nat. Med. 9:847–852. [DOI] [PubMed] [Google Scholar]
- 65.Kawamura, T., F.O. Gulden, M. Sugaya, D.T. McNamara, D.L. Borris, M.M. Lederman, J.M. Orenstein, P.A. Zimmerman, and A. Blauvelt. 2003. R5 HIV productively infects Langerhans cells, and infection levels are regulated by compound CCR5 polymorphisms. Proc. Natl. Acad. Sci. USA. 100:8401–8406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kwon, D.S., G. Gregorio, N. Bitton, W.A. Hendrickson, and D.R. Littman. 2002. DC-SIGN-mediated internalization of HIV is required for trans-enhancement of T cell infection. Immunity. 16:135–144. [DOI] [PubMed] [Google Scholar]
- 67.Pohlmann, S., F. Baribaud, and R.W. Doms. 2001. DC-SIGN and DC-SIGNR: helping hands for HIV. Trends Immunol. 22:643–646. [DOI] [PubMed] [Google Scholar]
- 68.Kawamura, T., S.S. Cohen, D.L. Borris, E.A. Aquilino, S. Glushakova, L.B. Margolis, J.M. Orenstein, R.E. Offord, A.R. Neurath, and A. Blauvelt. 2000. Candidate microbicides block HIV-1 infection of human immature Langerhans cells within epithelial tissue explants. J. Exp. Med. 192:1491–1500. [DOI] [PMC free article] [PubMed] [Google Scholar]