

Abstract
B cells express complement receptors (CRs) that bind activated fragments of C3 and C4. Immunized CR knockout (KO) mice have lower antibody titers and smaller germinal centers (GCs), demonstrating the importance of CR signals for the humoral immune response. CR ligands were thought to be generated via complement fixation mediated by preexisting “natural” IgM or early Ab from inefficiently activated B cells. This concept was recently challenged by a transgenic (Tg) mouse model that lacks circulating antibody but still retains membrane IgM (mIgM) and mounts normal immune responses. To test whether CR ligands could be generated by the B cell receptor (BCR) itself, we generated similar mice carrying a mutated mIgM that was defective in C1q binding. We found that B cells from such mutant mice do not deposit C3 on B cells upon BCR ligation, in contrast to B cells from mIgM mice. This has implications for the immune response: the mutant mice have smaller GCs than mIgM mice, and they are particularly deficient in the maintenance of the GC response. These results demonstrate a new BCR-dependent pathway that is sufficient and perhaps necessary to provide a CR1/2 ligand that promotes efficient B cell activation.
Keywords: C1q, IgM, germinal center, CD19, CD35
Introduction
It has become clear that various signals from the innate immune system activate the adaptive immune system. Specifically, signals generated by the complement system can play an important role in B cell immune responses. B cells express complement receptors (CR1 [CD35] and CR2 [CD21]), which bind fragments of C3 and C4 (1–4) that can be deposited on Ags upon complement system activation. Mice lacking C3, C4, or CR1/2 had reduced Ab titers in serum and reduced size and number of germinal centers (GCs), particularly when the immunogenic stimulus was relatively weak (1–8). CR1/2 is widely expressed, notably on B cells and follicular dendritic cells (FDCs). Chimera studies showed that CR1/2 needs to be expressed on the B cell per se for optimal immune responses (6, 9). CR1/2 on B cells also enhances their entrance and survival in GCs, as shown by comparison of CR1/2 deficient and wild-type B cells in adoptive transfer experiments (10), thus implicating CR1/2 signaling in GC maintenance and presumably in memory B cell development.
Signaling via CR1/2 in B cells requires the formation of a complex with CD19/TAPA-1 (11–14), which transduces the intracellular signals. Coligation of this receptor complex with the B cell receptor (BCR) was shown to enhance BCR-mediated signaling and reduce the amount of Ag required for B cell activation (15–17). Moreover, coligation of BCR and CR1/2 prolongs BCR signaling via lipid rafts and enhances presentation of BCR bound Ag by class II MHC (18–24). These results provide a biochemical basis for the role of CR1/2 in B cell immune responses in vivo.
The fact that C3, C4, and C1q deficient mice all have defects in primary B cell responses suggests that complement fixation during the initial steps of an immune response is required to generate the CR1/2 ligand (25–29). In particular, the requirement for C4 and C1q implicates the classical pathway, which is primarily if not exclusively activated by immune complexes in vivo. Moreover, factor B–deficient mice (30), which are defective in the alternative pathway, appear to have normal immune responses.
It has been assumed that the requirement for early complement fixation to provide CR1/2 ligation during a physiologic B cell immune response was met by either preexisting (“natural”) IgM Ab or very early Ab that was secreted from a few inefficiently activated B cells (6, 31, 32). These preexisting Abs are thought to form immune complexes (ICs) with Ag, which could in principle activate complement via the classical pathway, leading to C3d deposition on ICs. A priori, a problem for this model for C3 deposition on Ag is that it would require natural Ab with affinity and concentration high enough to form immune complexes. It seems unlikely this would be generally available for all important Ags. Additionally any ICs formed with natural Ab would be subjected to very efficient clearance mechanism via CRs on phagocytes, B cells, and red blood cells, which will at the least attenuate their ability to stimulate an Ag-specific B cell.
We have generated a Tg mouse model that has provided an additional challenge to this classical model (33). This mouse expresses a mutant Vh IgM Tg that lacks the secreted exon of IgM, and thus lacks circulating Ab. Surface Ig is expressed normally, and B cell development is rescued when the Tg is crossed on the Jh knockout background (JhD). The Tg carries a Vh186.2 VDJ rearrangement that confers NP specificity when paired with λ light chain. Though these mice lack detectable Ab, they had normal immune responses to NP-human serum albumin (NP-HSA) and NP-chicken gamma globulin (NP-CGG), with normal numbers and sizes of GCs and normal kinetics (33). This was unexpected if soluble preformed Abs were important to create ICs that in turn coligate CR1/2 and the BCR. To reconcile our findings with the observations that CR1/2 coligation is critical for optimal GC formation, we hypothesized that membrane IgM fixation of complement is both sufficient and necessary to provide local C3b deposition on mIgM and/or IgM-bound Ag, which in turn can coligate the CR1/2/CD19/TAPA-1 complex.
We have now tested this hypothesis by generating a mouse model similar to the mIgM mice that has an additional mutation in the CH3 domain which inhibits C1q binding to the BCR. We report here that these latter mice, which have no circulating Ig and are deficient in complement activation via the classical pathway by mIgM, show decreased responses to immunizations with NP-HSA and NP-CGG, in contrast to mIgM mice. By demonstrating the phenotype of mice that cannot activate complement via the surface BCR, we show the importance of this novel pathway for providing a ligand that can cocrosslink CR1/2 and the BCR. Since the previously reported mIgM mice make normal responses and have this pathway intact, yet lack soluble Ab to perform these functions, it follows that the fixation of complement by mIgM must be sufficient, and possibly necessary, to start optimal immune responses.
Materials and Methods
Construct and Tg Mouse Generation.
The Vh186.2-mIgM Tg mice have been described (33, 34). They lack the secreted exon of the IgM region and thus have 1,000-fold less than normal standing IgM levels. Their B cells neither secrete nor shed IgM in response to LPS in vitro and in spite of having 4% of B cells specific for the NP hapten, have undetectable levels of serum anti-NP. To generate mice with disabled C1q binding but the same properties as Vh186.2-mIgM mice we altered the Vh186.2-mIgM construct using a restriction fragment of the IgM constant region containing a point mutation encoding a Pro-Ser exchange at residue 436 [Vh186.2-mIgM(P436S)]. This mutation was shown to lower the Ka for C1 over 50-fold and to be >200-fold less active in hemolysis assays (35, 36). The altered construct was tested for expression by transfection of CH1, a λ/IgMb-expressing cell line (37). CH1 transfectants expressed surface IgMa anti-NP. The tested construct was then used to generate Tg mice by microinjection of purified DNA. Tg positive founders were identified by PCR and bred directly to the CB.17 mice carrying a homozygous deletion at the Jh locus (JhD/JhD.CB17) strain to eliminate endogenous H chain production. F1 generation mice that matched the previously generated mIgM Tg mice (33) in terms of mIgMa expression and nitro-iodo-phenyl (NIP)-binding specificity assessed by FACS® were selected for further backcrossing with JhD/JhD.CB17 mice. The constant part of the Vh186.2 heavy chain construct was used to perform Southern blots on tail DNA from mating pairs and their litters. The blots showed the stability of the unique integration sites over several generations and confirmed that each line has a single Tg locus. The mice were housed under specific pathogen free conditions and received Sulfatrim in drinking water every second week. Mice were used at backcross four for preliminary immunizations and the majority of experiments were done on backcross five and higher.
Antigens and Immunization.
Chicken gamma globulin (CGG; Sigma-Aldrich), human serum albumin (HSA; Amour Pharmaceutical), and SRBCs (Colorado Serum) were haptenated with NP-hydroxysuccinimide ester (Cambridge Research Biochemicals). 6–12-wk-old mice were immunized intraperitoneally with 50 μg or 5 μg alum precipitated NP22-CGG, NP3-CGG, or NP15-HSA as described. Controls were immunized with the same amount of alum used for precipitation of the Ags.
Flow Cytometry.
FACS® was performed as described (38). In brief, spleens were mechanically disrupted and cells harvested. Red blood cells were lysed by 5 min incubation in ACT at room temperature. Purified splenocytes were counted and 106 cells were stained in 96-well plates on ice for 20 min. Because of the low frequency of λ+ (Ag specific) cells we collected 105 events per sample. PE haptenated with NIP-hydroxysuccinimide ester (Cambridge Research Biochemicals) was used to identify NP-specific B cells, as Vh186.2/λ recognizes NIP with high affinity. In addition splenocytes were stained with anti-λ-fluorescein (B374-P584D; Southern Biotechnology Associates, Inc.) and anti-CD86-biotin/Cy5 (GL-1; BD Biosciences). In complement deposition assays, goat anti-C3-FITC (Cappel) was used in combination with anti B220-PE (RA3–6B2; BD Biosciences) and anti-λ-Alexa 647.
Histology.
5 μm frozen spleen sections were cut in a cryostat microtome, thaw mounted onto poly-l-lysine (Sigma-Aldrich)-coated slides, fixed in cold acetone for 10 min, and stored at −80°C (39, 40). Rehydrated sections were blocked with 3% BSA (Gemini Bioproducts) in PBS/0.1% Tween 20 and stained with anti-λ-alkaline phosphatase (Southern Biotechnology Associates, Inc.) and peanut agglutinin-biotin (PNA-bi; Vector Laboratories). Overlapping pictures of the whole spleen were captured and the area (in square μm) of GCs measured using Image Pro software (Media Cybernetics). The GC areas were added up for the whole section and then calculated as GC area per picture (each of the same unit area), thus normalizing for spleen and section size.
C3 Deposition Assays.
Splenocytes were isolated from mIgM and mIgM(P436S) mice. 3 × 107 cells were incubated in 200 μl PBS/2% BSA on ice with 8 μg NP26-BSA or BSA alone for 30 min. After washing off excess NP, the cells were injected into the tail vein of B cell–deficient mice. Half an hour after the injection, blood was collected directly into Heparin (5U)/EDTA (10 mM) to prevent blood clotting and nonspecific complement activation. Lymphocytes were separated from the anticoagulated blood by gradient centrifugation (Lymphocyte Separation Medium; Cellgro) and stained with anti-B220, anti-λ, and polyclonal goat anti-C3 Abs (Cappel) and analyzed via flow cytometry. The anti-C3 detects intact C3 and its fragments, such as C3d.
Reconstitution of Soluble Antibody in mIgM(P436) Mice.
Blood was taken via eye bleeding or cardiac puncture, as indicated in the figure legend. Antibody-free serum was obtained from JhD mice and NP anti-serum was obtained from (m+s)IgM mice that had been immunized with NP22-CGG 2 wk before. CGG-specific anti-serum came from BALB/c mice immunized with 100 μg CGG 2 wk before. Serum from naive BALB/c mice was considered as “natural” antibody. The serum was tested for presence and absence of the expected specific antibodies by ELISA (not shown) and 200 μl were injected i.v. into the tail vein of mIgM(P436S) mice. 3 h later the mice were immunized i.p. with NP22-CGG in alum. At day 12 after immunization the immune response was analyzed as described above.
Results
Complement Is Activated and C3 Fragments Are Deposited on B Cells in mIgM Mice but Not mIgM(P436S) Mice.
In most species, complement is activated and leads to C3d deposition when whole blood or serum is kept ex vivo. Unfortunately, mouse complement is particularly susceptible to this: blood clotting at room temperature leads to substantial cleavage of C3 (41, 42). In preliminary experiments we found overwhelming deposition of C3d on B cells whether or not they were incubated with Ag, using either freshly prepared serum or plasma in a variety of tested buffers. This was predicted by results of several other groups using B cell lines and has been attributed to the ability of the CR itself to trigger C3d deposition on B cells in vitro but not in vivo (43–48). Therefore, to prevent too much nonspecific complement activation we performed a combined in vitro and in vivo experiment by separating the Ag binding step (in vitro) from the C3d deposition step (in vivo, by injecting the coated cells into B cell–deficient mice, see Materials and Methods). Note that we used a highly multivalent Ag (NP26-BSA) that should effectively aggregate BCRs on the surface of B cells. Furthermore, by using B cell–deficient recipients as the in vivo source of complement, we avoided any potential influence of natural IgM in the recipient.
After injection, donor B cells are only a small fraction of PBL, and only ∼3% of these are lambda positive, requiring us to collect 106 events per sample. mIgM cells showed complement deposition on their surface when incubated with NP-BSA, but no deposition was observed after incubation with BSA alone (Fig. 1 A). In contrast, B cells from mIgM(P436S) mice did not show complement deposition, no matter if they were treated with NP-BSA or BSA alone (Fig. 1 B). Although only λ+ cells are specific for the Ag, we saw deposition of C3d on κ+ cells as well, but only in the samples where we could find deposition on λ+ cells (i.e., only in the mIgM but not P436S mutant mice and only when the B cells had been coated with Ag). This surprising but consistent finding indicated that injection of the Ag-coated cells had induced widespread, strong complement activation intravascularly in the recipients. Indeed, it is well known that injections of immune complexes can even cause anaphylactic death of mice presumably due to massive complement activation. In any case, these data show that mIgM, but not P436S mutant B cells can activate complement deposition directly on the B cell surface when the BCR is cross-linked.
Figure 1.

C3 is deposited on λ+ mIgM but not mIgM(P436S) splenocytes. Splenocytes of mIgM (A) and mIgM(P436) (B) mice were prepared. After lysis of red blood cells they were loaded with NP-BSA (fine line) or incubated in BSA alone (dark line). After washing off unbound NP-BSA, cells were injected into B cell–deficient mice and after 30 min blood was collected into Heparin/EDTA. Lymphocytes were purified by gradient centrifugation and stained for B220, λ, and C3. Histograms represent C3 staining gated on live, λ+ B cells. Histograms show a representative result out of 6 experiments with 1–4 animals per group. mIgM(P436) λ cells (B) display no difference in C3 staining of cells incubated with NP-BSA or BSA control. On mIgM cells (A) incubated with NP-BSA (fine line) elevated anti-C3 staining was detected to BSA controls (dark line).
Proliferative Immune Responses.
Primary immune responses were initially evaluated at day 12, the known peak of the response in mIgM mice (33). To evaluate a range of different “strengths” of stimulation, two different carriers were chosen and haptenated at two different ratios. Each carrier and haptenation ratio was used for immunization at two different doses, 50 μg as used in all previous immunizations of mIgM mice, and 5 μg as a low Ag dose, for a total of 6 conditions. The B cell immune response was measured via the expansion of λ+/NIP+ cells as determined by FACS® analysis (Fig. 2) . As will be detailed below, in all experiments performed we observed that the immune response of mIgM(P436S) mice was significantly lower than in mIgM mice with especially noticeable differences from the mIgM mice at the low dose of Ag (Table I). Nevertheless, the mIgM(P436S) mice responded to all immunization regimes.
Figure 2.
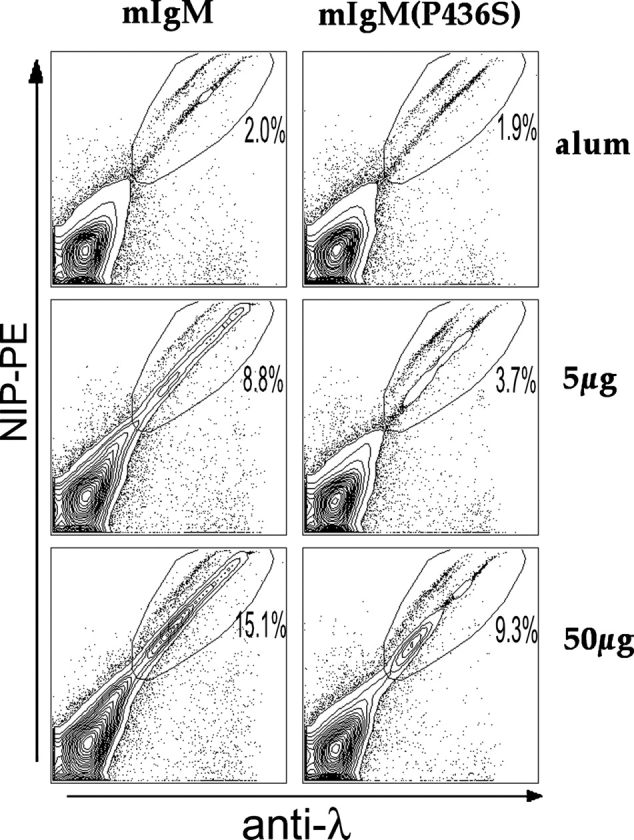
FACS® analysis of immune responses. Red cell–depleted splenocytes were stained as indicated and 105 events were collected per sample on a FACSCalibur™. Antigen-specific B cell expansion was measured by gating on λ+/NIP+, live splenocytes. Representative data of day 12 after immunization with NP22-CGG are shown here and summarized in Fig. 3 and Table I.
Table I.
Summary of the Percentages of Total λ+/NIP+ Splenic B Cells in Response to Various Ags and Doses at Day 12 After Immunization
NP22-CGG Immunization.
The strongest response in both types of mice was seen to immunization with NP22-CGG (Fig. 3 A). At 50 μg, both strains showed significant expansion of λ+/NIP+ cells. The λ+/NIP+ cell population of splenocytes in mIgM mice increased nearly eightfold whereas this population expanded only fivefold in mIgM (P436S) mice. We performed this experiment two times each with two different mIgM(P436S) strains obtained from different founders, always comparing to mIgM mice immunized at the same time with the same batch of Ag. No differences were found in comparing the two mIgM(P436S) strains to each other and only small variations were seen between immunizations as indicated by the error bars representing the pooled data of these four experiments. Importantly, both of the mIgM(P436S) strains showed lower responses than the wild type mIgM mice in all experiments, arguing against an effect of the Tg integration site. At the lower Ag dose (5 μg) the same pattern (threefold increase of λ+/NIP+ cells in mIgM mice in comparison to twofold in mIgM(P436S) strains) can be observed during a weaker but still significant response to the Ag. It is interesting that there are nearly identical percentages after immunization with 5 μg in mIgM mice and with 50 μg in mIgM(P436S) mice at day 12 (the peak response, see time course below). In this sense the inability of mIgM(P436S) to bind C1q results in a 10-fold decreased sensitivity in terms of Ag dose.
Figure 3.

mIgM(P436S) mice show diminished responses to immunization with different Ag carrier and haptenation ratios. Both mouse strains were immunized with NP22-CGG (A), NP3-CGG (B), and NP15-HSA (C) and the percentage of λ+/NIP+ splenocytes determined by FACS® analysis on day 12. Error bars indicate SEM of 3–4 mice per group in each of 2–3 separate experiments (i.e., at least 6 mice per group total). Mutant mice (solid bars) showed lower Ag-specific B cell expansion than mIgM mice (open bars) after all immunizations (P < 0.005 for NP22-CGG, NP3-CGG, and 50 μg of NP15-HSA) but responded to all of them except for 5 μg of NP3-CGG, in which case the response is not substantially different from the alum control.
Immune responses were also analyzed by immunohistology of cryosections of spleens. Staining with PNA to detect GCs was of special interest, as ligation of CR1/2 on B cells can enhance survival in GCs (10). To confirm Ag specificity of GCs we included an Ab to the λ light chain. At day 12 after a 50 μg NP22-CGG immunization, mIgM(P436S) spleens had GCs of only ∼2/3 the size of mIgM GCs (Figs. 4 and 6) and λ+ cells were visibly more abundant in mIgM spleens, confirming the flow cytometry data (Fig. 3 A). Nonetheless, spleens of both strains exhibited GCs homogeneously filled with λ+ cells. Distribution of λ+ cells in the white pulp was similar among the strains with most of the cells populating GCs and a smaller portion residing in the marginal zone. GC generation was also dose-dependent, as 50 μg immunizations always elicited bigger GCs than 5 μg immunizations (Fig. 4).
Figure 4.
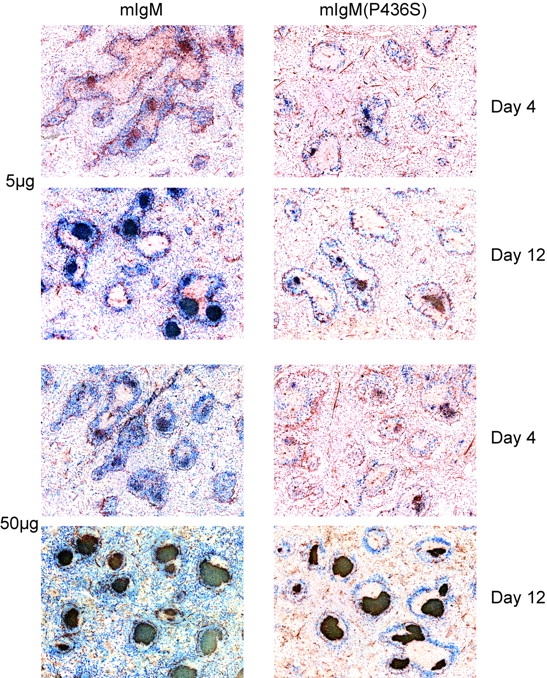
mIgM(P436S) mice generate smaller and fewer GCs throughout most of the primary immune response. Spleens were frozen from each experiment and 5 μm frozen sections were cut and stained with anti λ-AP (blue) and PNA-bi/SA-HRP (red). mIgM spleens consistently show more GCs of larger size than mIgM(P436S) spleens.
NP3-CGG Immunization.
Again mIgM(P436S) mice did not respond as well to NP3-CGG as mIgM mice when analyzed by flow cytometry (Fig. 3 B). In these immunizations all of the responses were weaker than in NP22-CGG experiments, as expected from the lower haptenation ratio. Still, the results paralleled the NP22-CGG immunizations, with the mutant mice having attenuated responses. 12 d after immunization with 50 μg NP3-CGG, mIgM mice show 5.5-fold and mIgM(P436S) mice 3.4-fold increase of λ+/NIP+ cells. The responses to 5 μg of NP3-CGG on the other hand resulted only in 2-fold increase for mIgM and 1.4-fold in the mutant mice.
Immunohistologic examination of spleens from mice immunized with NP3-CGG showed that GCs form in response to this sparsely-haptenated Ag at the higher Ag dose, but are much smaller than in NP22-CGG immunizations. In immunizations with 5 μg of NP3-CGG we could observe λ+ GCs only in mIgM but not in mIgM(P436S) spleens. Thus, a more absolute difference between the mutant and wild-type strains emerged at low doses of a lightly haptenated Ag.
NP15-HSA Immunization.
When HSA was used as a carrier for NP, B cell expansion was less robust than with CGG (Fig. 3 C), as we had observed before (33). Regardless, immunization with 50 μg NP15-HSA resulted in a weaker immune response in mutant mice (2.9% λ+/NIP+ cells) compared with mIgM mice (5.2% λ+/NIP+ cells). But after immunization with 5 μg NP15-HSA both mouse strains responded with only a ∼50% increase in the λ+/NIP+ cell population.
Stained sections of spleens immunized with NP15-HSA had smaller GCs in comparison with CGG as carrier. GCs were also not homogeneously packed with λ+ cells as seen at day 12 after NP22-CGG immunization. Nevertheless, the differences between the mouse strains remained, with smaller GCs containing few λ+ cells at the higher Ag dose for the mutant mice compared with larger GCs with more λ+ cells in the mIgM mice. Results for the lower Ag dose showed parallel differences between the strains, with small GCs in both strains, but very few λ+ cells only in the mIgM(P436S) mice.
Primary Immune Response Kinetics.
To better define differences in the primary immune response of the mice, we measured the increase of λ+/NIP+ cells and the size of GCs at days 0, 4, 8, 12, and 20 after immunization, studying responses to both doses of NP22-CGG.
After immunization with 50 μg NP22-CGG, mIgM mice doubled the frequency of λ+/NIP+ cells at day four in comparison to nearly no response in mutant mice (Fig. 5) . 8 d after immunization, mIgM mice displayed an even stronger increase of Ag specific cells. Interestingly, after overcoming the initial lag phase, the mutant mice show a growth rate between days 4 and 8 that is similar to mIgM mice. Given the selected time points for this time course experiment we observed the peak of the immune response of both mouse strains at day 12, as observed for mIgM mice previously. However, the shape of the growth curves of Ag-specific cells was quite different between the two strains. mIgM mice demonstrated a continuous increase in the fraction of λ+/NIP+ cells at each time point up to day 12 whereas in mutant mice, λ+/NIP+ cells increase little between days 8 and 12, a time when the GC response should be dominant (5, 33). This lower increase in Ag-specific B cells resulted in a plateau-shaped response in mIgM(P436S) mice, in contrast to the strong increase up to the peak in mIgM mice. Finally, at day 20, λ+/NIP+ cell frequencies are lower than on day 12 in both mouse strains.
Figure 5.

Time course analysis of the primary immune response to NP22-CGG. Splenocytes from mIgM (solid symbols) and mIgM(P436S) (open symbols) were analyzed by FACS® at several time points after immunization for the percentage of Ag specific B cells. Mice were given 5 μg (dotted lines, triangles) or 50 μg (solid lines, squares) of antigen. Error bars indicate SEM of 3–4 mice per group per experiment. Day 8 was done in two experiments, day 12 in three and the other time points are from 3–4 mice in one experiment (P < 0.005 at days 8, 12, and 20. At day 4 P < 0.05 for 5 μg and P < 0.01 for 50 μg). Diminished Ag-specific B cell expansion in mIgM(P436S) mice was seen throughout the primary immune response. Shown are the percentages of λ+/NIP+ live splenocytes minus the average percentage of λ+/NIP+ cells in the relevant preimmune Tg mouse.
Immunizations with 5 μg NP22-CGG resulted again in lower responses of mIgM(P436S) mice during the whole response than in mIgM mice (Fig. 5). 4 d after immunization we observed, similar to the higher Ag dose, a much stronger response in mIgM mice than in the mutant mice. Between days 4 and 8 the mutant and mIgM mice show parallel but still weaker increases in Ag specific cell numbers, again similar to the higher Ag dose. Between days 8 and 12, λ+ cells in mIgM(P436S) mice continue to expand, in contrast to the high Ag dose, almost matching the mIgM response to the same Ag. As will be seen below, in this case the percentages do not match the GC size data, indicating that most of the proliferation measured by FACS® in this interval must have come from non-GC cells in the mutant mice, in contrast to the mIgM mice. The decrease in cell numbers between day 12 and 20 is similar in both mouse strains at 5 μg NP22-CGG. On days 12 and 20 the percentages of λ+ cells from 5 μg NP22-CGG immunization in mIgM mice resemble those elicited by 50 μg immunizations in mIgM(P436S) mice.
Overall the mutant mice show a later onset of the response and less expansion of λ+/NIP+ cells. At the higher Ag dose the mutant mice seem to catch up between day 4 and 8 but are not able to sustain the increase in their response like mIgM mice up to day 12. At the low Ag dose mutant mice respond in a similar fashion to mIgM mice but with lower cell numbers. The rate of disappearance of Ag-specific cells between day 12 and day 20 is similar in both mouse strains. The observation that the mutant mice cannot sustain their response could be explained by the importance of complement for survival of B cells in GCs (10). Faster clearance within GCs could result in a lower λ+/NIP+ cell number that flattens out the response as seen with the 50 μg dose. To obtain more information about the influence of this mutation on the number and size of GCs, we followed the generation and disappearance of GCs during this 20-d time course.
GC Reactions.
Immunohistologic analysis was performed on spleens from mice used for the time course of the primary immune response to NP22-CGG. Similar to the flow cytometry data, GCs of mIgM(P436S) mice were smaller than in mIgM mice in nearly all groups. The peak of GC size in both strains occurred around day 12 (Fig. 6) , in accord with the FACS® analysis above.
Figure 6.

Quantitation of the GC response. Spleens obtained from mice immunized with 50 μg (A) and 5 μg NP22-CGG (B) were frozen at the several time points during the primary immune response and cryosections stained for GCs (PNA) and the λ light chain. The area of GCs was measured as described in Materials and Methods. Bars show the total area of λ+ GCs in square mm per 40× field. Error bars indicate standard error of 5 to 8 pictures per mouse and 3 mice per group (P < 0.005 for days 4, 12, and 20. On day 8, P > 0.05). Solid bars represent mIgM(P436S) mice and open bars mIgM mice.
After immunization with 50 μg NP22-CGG we saw small GCs in both strains at day 4 that were only partly filled with λ+ cells (Figs. 4 and 6 A). By day 8, GCs had increased in size in both strains and were more densely filled with λ+ cells. The size of GCs of both mouse strains was similar after 8 d, a point at which the number of λ+ cells by FACS® most closely matched between the two strains. Most importantly, however, the size of GCs continued to increase through day 12 in mIgM mice whereas GCs in mIgM(P436S) mice showed only a minimal increase after day 8. The halt in the growth of the GCs in the mutant mice also demonstrates that the differences between the strains overall could not just be due to the slightly different numbers of λ+ cells before immunization, as the size of GCs in both strains is nearly the same at day 8 but differs substantially at day 12. At day 20, the sizes of GCs were decreased in both strains. Still, at day 20, mIgM mice retained substantially bigger GCs (Fig. 6 A).
In responses to 5 μg of Ag, only mIgM mice had measurable λ+ GCs 4 d after immunization (Figs. 4 and 6 B). At day 8, mIgM(P436S) mice caught up with mIgM mice and both strains had about the same size of λ+ GCs which were also more densely filled with λ+ cells. After 12 d we observed not only a stagnation in GC size in mIgM(P436S) mice that received 5 μg of Ag, as with 50 μg of Ag, but actually a decrease in GC size compared with day 8. mIgM mice on the other hand still showed growth in GCs up to day 12, reaching an average area four times greater than in the mutant mice. At day 20 both strains had smaller GC sizes, as was observed in the 50 μg Ag dose. Nonetheless, the mIgM mice retained GCs with areas ∼5 times greater than in mIgM(P436S) mice.
Reconstitution of Soluble Antibody Restores Responses in mIgM(P436S) Mice.
Natural autoantibody is thought to play a role in generating the CR1/2 ligand under normal circumstances (6, 31, 32). In fact, there is little direct evidence for this, with the best support for the idea coming from the phenotype of mice that lack secreted IgM (but not IgG) and have delayed kinetics of primary immune responses (49, 50). In contrast to the natural situation in nonimmune mice, it is very clear that if soluble Ab, particularly of IgM and certain IgG isotypes is added to Ag, this will potentiate the primary immune response (8, 51–54). One would predict that such Ab, which is absent in the mIgM(P436S) mice, might restore the responses of these B cells by bypassing the defect in the mutant mIgM. We therefore did two types of experiments (Fig. 7) . In one case, we used polyclonal anti-carrier Ab raised by immunizing normal mice. Infusion of 0.2 ml of serum from such immune mice i.v. 3 h before i.p. immunization with NP22-CGG did indeed result in the restoration of proliferative responses to levels typically seen in mIgM mice (Fig. 7, A and B). Serum from nonimmune mice or from JhD mice had no significant effect. In a second approach, we immunized (m+s)IgM Tg mice with NP-CGG (33) to generate secreted IgM anti-NP and used this serum. This also resulted in restoration of the response of mIgM(P436S) mice to nearly the levels of mIgM mice (Fig. 7 C). Thus, in the case of NP-CGG, immune but not nonimmune sera restored responses of mIgM(P436S) mice.
Figure 7.

Specific antiserum rescues the primary response in mIgM(P436S) mice. Serum was obtained from cardiac puncture of JhD and BALB/c (naive and 2 wk after immunization with CGG) mice. Tail vein injection of 200 μl serum into mIgM(P436S) mice was followed by i.p. immunization with 5 μg of NP22-CGG three hours later. At day 12 the percentage of λ+/NIP+ (A) and λ+/PNA+ (B) splenocytes were determined by FACS® analysis. Administration of immune BALB/c serum (black) led to reconstitution of the primary immune response in comparison to mIgM(P436S) mice that received no serum (white, P < 0.005), JhD serum (horizontal stripes, P < 0.005), naive BALB/c serum (diagonal stripes, P < 0.005), or immune BALB/c serum (black, P < 0.005). The influence of NP specific IgM on the response of mIgM(P436S) mice (gray) was compared with the response in mIgM (black) and mIgM(P436S) (white) mice at 5 μg and 50 μg NP22-CGG (C) (P < 0.01 for 5 μg and P < 0.05 for 50 μg between mIgM or mIgM(P436S)+Ab and mIgM(P436S)).
Discussion
In this paper we show the activation of complement on the B cell surface in vitro and demonstrate its importance for the immune response in vivo. We did this by genetically eliminating two sources of C1q-dependent complement activation: soluble Ab and membrane-bound IgM. Only the inactivation of the BCR-intrinsic (mIgM) source gave a phenotype similar to the CR1/2 KO mice. Thus, we found that activation of complement by the BCR is sufficient to start the classical pathway of complement, leading to deposition of C3 fragments that in turn provide costimulation of B cells for an optimal immune response. Whereas, in the system we studied, soluble Ab was not necessary for these processes.
Though it is widely accepted that complement participates in B cell signaling and subsequent B cell responses, its relative importance and exact role are still debatable. It was first found that cocrosslinking of CD19 with the BCR leads to increased signaling (11, 15). Later, this process was found to be mediated by C3b that links the BCR to CR2, which is associated with CD19 and TAPA-1 (14). Therefore, in vivo–activated complement is necessary for increased signaling by cross-linking CD19 and the BCR. Several groups have investigated the primary immune response in CR knockout mice with different T cell–dependent Ags (5–7). Lower titers of specific Abs were found in all studies, although details varied. Also the results on the impairment of GCs in these mice were controversial. Nevertheless it was shown that CR deficiency on the B cell is important for this phenotype, though CR1/2 could also play a role on FDC's. More recent results indicate that high doses of Ag could at least in part compensate the effect of deficient CR1/2-mediated signaling (5).
Thus, complete deficiency in the ability to cocrosslink CR1/2 with the BCR, as in the CR KO mice, only results in a partial immune response defect. This limits the phenotype that we could expect to see in disabling classical pathway activation by both eliminating soluble Ab and preventing C1q binding by membrane IgM, as in our mIgM(P436S) Tg. In this regard, the phenotype we did see is completely consistent with the CR1/2 KO: (a) a partial but reproducible reduction in the size of the total proliferative response; (b) a more pronounced reduction in the GC response; and (c) altered kinetics consistent with the inability to sustain the GC response normally. One potential concern with our system is the unusually high precursor frequency of Ag-specific cells. As the fixation of complement is thought to be local at the B cell membrane and not via secreted IgM, the effects of nearby Ag-specific B cells should be minimized. More importantly, the strongest phenotype occurs at the GC cell stage; even in mice with normal precursor frequencies, GCs are typically composed of a few clones of Ag-specific B cells in close proximity and by this stage of the response, the GCs in our Ig-Tg mice are similar to the normal situation.
Complement deposition was tested after binding Ag to B cells in vitro, using both mIgM and mIgM(P436S) splenocytes either bound to multivalent Ag or not. As expected, mIgM(P436S) splenocytes showed no increase in C3 deposition on their surface after incubation with the Ag and exposure to complement. Cells from mIgM mice on the other hand displayed substantially higher anti-C3 staining when incubated with the Ag before exposure to a source of complement in comparison to cells that never saw the Ag. Only in samples of mIgM splenocytes did we also observe C3 deposition on κ+ cells, which might be explained by amplification of C3b via the alternative pathway and thereby spreading to nonspecific cells. The deposition of C3 on κ1 cells only in the mIgM mice cannot be explained by shedding of the BCR from the mIgM mice, as the mice have virtually undetectable Ab levels and do not shed BCR even in an LPS culture in vivo (33). Rather, this spreading of C3 deposition to other B lymphocytes could be mainly due to our assay system with the sudden injection of high numbers of opsonized lymphocytes (3 × 107 cells) in the peripheral blood. This massive complement activation on the B cell surface likely overwhelms soluble regulators of complement present in the blood which usually prevent spreading of C3. A similar situation probably occurs when immune complexes are injected into mice i.v., which can cause enough complement fixation to kill the animal. The intention of this assay was not to mimic physiologic complement activation but purely to show the ability of mIgM to activate complement in the absence of serum Ig. Nevertheless this assay also shows the lack of complement activation of Ag loaded mIgM(P436S) B cells, demonstrating the inability of this mutant mIgM to fix complement even in this setting where evidently activation can be amplified. These results demonstrated the plausibility of our hypothesis, directly showing that there is Ag-dependent complement activation even in the absence of secreted Ab as seen on mIgM B cells. Additionally, this experiment showed that the complement cascade can be started by the BCR on the target cells themselves when they are stimulated with Ag. Presumably complement on the surface of the B cell is activated by C1q binding that is catalyzed by the close proximity of BCRs after ligation by a multivalent Ag, much as IgG fixes complement only when several molecules are bound to Ag in close proximity.
As there are differences in deposition of complement on mIgM versus P436S Tg B cells, our hypothesis predicts differences in immune responses between the two types of Tg mice, given the known effects of complete inability to signal via CR1/2. To test this, we characterized the immune responses to different Ag carrier conjugations and different haptenation ratios. In all our experiments we found lower responses in the mutant than in mIgM mice. In the 50 μg group on the peak of the response of B cell expansion (day 12), we found only about half the frequency of Ag-specific cells in the mutant mice as in mIgM's, varying somewhat depending on the Ag-carrier combinations used (Fig. 2). Consistently, the mutant mice showed the same extent of B cell expansion in response to 50 μg as mIgM mice had to 5 μg, leading to the conclusion that the mutation leads to a 10-fold decrease in sensitivity to the Ags at the peak of the response. This finding is similar, perhaps coincidentally, to the ∼10-fold decrease in Ab titers observed in CR KO mice in comparison to wild-type mice (5–7), although in our system we are not able to measure Ab titers and therefore only looked at the percentage of Ag-specific B cell expansion. Moreover, the response to 5 μg of sparsely haptenated Ag (see NP3-CGG) or early in the response (NP22-CGG at day 4) was very weak in the mutant mice and not much different from controls that were immunized with alum alone. These observations are in concert with earlier observations that complement coactivation boosts and prolongs the immune response particularly to weak or lower dose immunization. Nonetheless, we did see responses to lower dose Ag in P436S mice, albeit smaller ones than in mIgM mice, which we believe are again consistent with the notion that signaling via CR1/2 is not absolutely necessary for B cell responses, as CR KO mice also show at least a weak response.
Perhaps the most interesting defect in the mIgM(P436S) mice was found during the analysis of GCs. Initially, studying day 12 after immunization, we saw a difference in GC size between the mutant mice and mIgM mice, paralleling the data on B cell expansion. Data on the GC area during the time course of the primary immune response showed two major differences between the strains. First, GC responses (and to some degree proliferative responses) were slow to start in P436S mice, which was seen at both doses of Ag. This could indicate a rather critical role in the initial generation of GC precursor cells that may depend on deposition of C3 fragments catalyzed at the B cell surface. Nonetheless, it seems likely that there is a bypass of this pathway at least for a minority of GC precursor cells. This could be due to alternative methods to activate complement or simply that it is not absolutely required. Once GC precursors are selected, evidently the early part of the GC response proceeds similarly in the two mouse strains. At this point, we would suggest that CR1/2 signals generated in this way may be less important; this would also be before Ag concentrations would become limiting, as indeed the GCs are manifestly growing at this stage.
Significantly, by day 12 the GC responses stagnate (50 μg dose) or involute (5 μg dose) in the mutant P436S mice, but continue expanding in the mIgM Tg mice. This could reflect an increased requirement for CR-mediated cosignals during the late GC response, when Ag concentrations are waning. This generally agrees with prior results in CR1/2 KO mice (5–7, 10). It also fits with a relatively greater dependence of the GC response and memory response on CR signaling that was previously reported (10). However, there are conflicting results regarding GCs. One group (6) found reduced numbers and size of GCs whereas a second group (7) did not find differences in GC morphology after immunization with SRBCs (though it was unclear how this was quantitated). In their discussion, Molina et al. (7) note that this is surprising because CD19 KO mice fail to generate GCs. A recent paper by Chen et al. (5) also shows lower numbers of GCs in CR1/2 KO mice at low doses but diminished differences to controls at a high Ag dose. The latter finding is due to both an increase of GC numbers in CR1/2 KO mice and also a decrease in GC numbers in C57BL/6 controls. These findings are similar to ours at the low Ag dose but differ at the high Ag dose as both mIgM and mIgM(P436S) mice display larger GCs at the peak of the response, yet a difference remains between them. It could be that the higher precursor frequency of Ag-specific B cells in our mice accounts for these minor differences between our observations and those of Chen (5).
One very interesting paper investigated more closely the GC response in CR1/2 KO B cells (10) by transferring them into mice with ongoing GC responses. It was found that with low affinity Ag, CR-deficient B cells were quickly cleared from follicles and they also did not enter GCs. After stimulation with high affinity Ag CR-deficient B cells were retained in follicles but only ∼1/10 of the GCs had specific B cells in comparison to ∼1/2 in CR-intact cells. It was also shown that CR KO cells did not divide, leading to the conclusion that CR-deficient B cells were only poorly sustained in GCs and only when the BCR cross-linking was relatively strong. Remarkably, it seems that our system has reproduced a similar phenotype by blocking both soluble IgM-mediated as well as membrane IgM-mediated C1q activation. We saw the most severe deficiencies of mIgM(P436S) mice in GC formation and especially with the low Ag dose. Moreover, the kinetic analysis showed that the GC response could not be sustained at a time when Ag should become limiting.
Lastly, we were able to test the role of immune and nonimmune serum to restore the immune response of mIgM(P436S) mice to levels seen in the wild-type mIgM Tg mice. We found that either anti-carrier or anti-hapten Ab could do this, but nonimmune serum had no effect. The anti-carrier serum would have been predominantly IgG, with some IgM, as it was raised by hyperimmunization of normal BALB/c mice. The anti-hapten serum would have been virtually all IgM, having been raised in the (m+s)IgM Tg mice (33). Thus, both IgG and IgM can have this effect, as expected from the ability of both isotypes to promote complement fixation and in accord with previous results (8, 51–54). Interestingly, at the doses used, nonimmune serum had no effect. This could be because we did not add enough serum, but in view of the positive results with immune serum, raises the point that for some antigens preimmune serum may not contain sufficient natural Ab to potentiate immune responses, further underscoring the relevance of the membrane-bound BCR-mediated pathway.
In summary, using a genetic approach and in vitro/in vivo assays as well as in vivo immunization, we found that complement can be activated, most likely via the classical pathway, on the surface of B cells as a consequence of cross-linking the BCR itself. We call this the BCR-intrinsic pathway of complement activation. This complement activation is sufficient for costimulation of B cells via CR1/2/CD19/TAPA-1 leading to a relatively enhanced immune response. We observed that in mice mutant for this pathway, 10 times the amount of Ag was necessary to induce the same peak B cell expansion as seen in mIgM mice. Differences in the kinetics and in particular the ability to sustain GC responses proved that the effect was independent of any differences in precursor frequency between the strains. Thus the mutant mice have revealed a novel pathway that is important in the initiation and development of B cell immune responses and links the complement pathway to adaptive B cell immunity. These results suggest a revision in the thinking on how the CR ligand is generated at the start of the B cell immune response. Although natural Ab could play a role if plentiful, it is fundamentally dispensable, whereas the BCR-intrinsic pathway is required, at least when sufficient preformed Ab to fix complement is absent. Physiologically, it seems likely that appropriate natural Ab will often be lacking and that the BCR-intrinsic pathway will play an important role.
Acknowledgments
We thank Ann Haberman, Mike Carroll, and Shannon Anderson for critical reading and useful discussions. We thank Wendy Hamelin and the Yale Animal Resource Center for excellent animal husbandry, and we thank Ashraf Khalil for expert technical assistance.
Supported by NIH Grant R01-AI43603.
Abbreviations used in this paper: BCR, B cell receptor; CGG, chicken gamma globulin; GC, germinal center; HSA, human serum albumin; IC, immune complex.
References
- 1.Carter, R.H., M.O. Spycher, Y.C. Ng, R. Hoffman, and D.T. Fearon. 1988. Synergistic interaction between complement receptor type 2 and membrane IgM on B lymphocytes. J. Immunol. 141:457–463. [PubMed] [Google Scholar]
- 2.Eden, A., G.W. Miller, and V. Nussenzweig. 1973. Human lymphocytes bear membrane receptors for C3b and C3d. J. Clin. Invest. 52:3239–3242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ross, G.D., M.J. Polley, E.M. Rabellino, and H.M. Grey. 1973. Two different complement receptors on human lymphocytes. One specific for C3b and one specific for C3b inactivator-cleaved C3b. J. Exp. Med. 138:798–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pepys, M.B. 1972. Role of complement in induction of the allergic response. Nat. New Biol. 237:157–159. [DOI] [PubMed] [Google Scholar]
- 5.Chen, Z., S.B. Koralov, M. Gendelman, M.C. Carroll, and G. Kelsoe. 2000. Humoral immune responses in Cr2−/− mice: enhanced affinity maturation but impaired antibody persistence. J. Immunol. 164:4522–4532. [DOI] [PubMed] [Google Scholar]
- 6.Ahearn, J.M., M.B. Fischer, D. Croix, S. Goerg, M. Ma, J. Xia, X. Zhou, R.G. Howard, T.L. Rothstein, and M.C. Carroll. 1996. Disruption of the Cr2 locus results in a reduction in B-1a cells and in an impaired B cell response to T-dependent antigen. Immunity. 4:251–262. [DOI] [PubMed] [Google Scholar]
- 7.Molina, H., V.M. Holers, B. Li, Y.-F. Fang, S. Mariathasan, J. Goellner, J. Strauss-Schoenberger, R.W. Karr, and D.D. Chaplin. 1996. Markedly impaired humoral immune response in mice deficient in complement receptors 1 and 2. Proc. Natl. Acad. Sci. USA. 93:3357–3361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Applequist, S.E., J. Dahlstrom, N. Jiang, H. Molina, and B. Heyman. 2000. Antibody production in mice deficient for complement receptors 1 and 2 can Be induced by IgG/Ag and IgE/Ag, but not IgM/Ag complexes. J. Immunol. 165:2398–2403. [DOI] [PubMed] [Google Scholar]
- 9.Croix, D.A., J.M. Ahearn, A.M. Rosengard, S. Han, G. Kelsoe, M. Ma, and M.C. Carroll. 1996. Antibody response to a T-dependent antigen requires B cell expression of complement receptors. J. Exp. Med. 183:1857–1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fischer, M.B., S. Goerg, L. Shen, A.P. Prodeus, C.C. Goodnow, G. Kelsoe, and M.C. Carroll. 1998. Dependence of germinal center B cells on expression of CD21/CD35 for survival. Science. 280:582–585. [DOI] [PubMed] [Google Scholar]
- 11.Carter, R.H., D.A. Tuveson, D.J. Park, S.G. Rhee, and D.T. Fearon. 1991. The CD19 complex of B lymphocytes. Activation of phospholipase C by a protein tyrosine kinase-dependent pathway that can be enhanced by the membrane IgM complex. J. Immunol. 147:3663–3671. [PubMed] [Google Scholar]
- 12.Bradbury, L.E., G.S. Kansas, S. Levy, R.L. Evans, and T.F. Tedder. 1992. The CD19/CD21 signal transducing complex of human B lymphocytes includes the target of antiproliferative antibody-1 and Leu-13 molecules. J. Immunol. 149:2841–2850. [PubMed] [Google Scholar]
- 13.Kansas, G.S., and T.F. Tedder. 1991. Transmembrane signals generated through MHC class II, CD19, CD20, CD39, and CD40 antigens induce LFA-1-dependent and independent adhesion in human B cells through a tyrosine kinase-dependent pathway. J. Immunol. 147:4094–4102. [PubMed] [Google Scholar]
- 14.Fearon, D.T., and R.H. Carter. 1995. The CD19/CR2/TAPA-1 complex of B lymphocytes: Linking natural to acquired immunity. Annu. Rev. Immunol. 13:127–149. [DOI] [PubMed] [Google Scholar]
- 15.Carter, R.H., and D.T. Fearon. 1992. CD19: lowering the threshold for antigen receptor stimulation of B lymphocytes. Science. 256:105–107. [DOI] [PubMed] [Google Scholar]
- 16.Weiss, A., and D.R. Littman. 1994. Signal transduction by lymphocyte antigen receptors. Cell. 76:263–274. [DOI] [PubMed] [Google Scholar]
- 17.Tedder, T.F., L.J. Zhou, and P. Engel. 1994. The CD19/CD21 signal transduction complex of B lymphocytes. Immunol. Today. 15:437–442. [DOI] [PubMed] [Google Scholar]
- 18.Dykstra, M., A. Cherukuri, and S.K. Pierce. 2001. Rafts and synapses in the spatial organization of immune cell signaling receptors. J. Leukoc. Biol. 70:699–707. [PubMed] [Google Scholar]
- 19.Dykstra, M.L., R. Longnecker, and S.K. Pierce. 2001. Epstein-Barr virus coopts lipid rafts to block the signaling and antigen transport functions of the BCR. Immunity. 14:57–67. [DOI] [PubMed] [Google Scholar]
- 20.Stoddart, A., M.L. Dykstra, B.K. Brown, W. Song, S.K. Pierce, and F.M. Brodsky. 2002. Lipid rafts unite signaling cascades with Clathrin to regulate BCR internalization. Immunity. 17:451–462. [DOI] [PubMed] [Google Scholar]
- 21.Pierce, S.K. 2002. Lipid rafts and B-cell activation. Nat. Rev. Immunol. 2:96–105. [DOI] [PubMed] [Google Scholar]
- 22.Cheng, P.C., M.L. Dykstra, R.N. Mitchell, and S.K. Pierce. 1999. A role for lipid rafts in B cell antigen receptor signaling and antigen targeting. J. Exp. Med. 190:1549–1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cheng, P.C., B.K. Brown, W. Song, and S.K. Pierce. 2001. Translocation of the B cell antigen receptor into lipid rafts reveals a novel step in signaling. J. Immunol. 166:3693–3701. [DOI] [PubMed] [Google Scholar]
- 24.Cherukuri, A., P.C. Cheng, H.W. Sohn, and S.K. Pierce. 2001. The CD19/CD21 complex functions to prolong B cell antigen receptor signaling from lipid rafts. Immunity. 14:169–179. [DOI] [PubMed] [Google Scholar]
- 25.Bottger, E.C., T. Hoffmann, U. Hadding, and D. Bitter-Suermann. 1985. Influence of genetically inherited complement deficiencies on humoral immune response in guinea pigs. J. Immunol. 135:4100–4107. [PubMed] [Google Scholar]
- 26.Fischer, M.B., M. Ma, S. Goerg, X. Zhou, J. Xia, O. Finco, S. Han, G. Kelsoe, R.G. Howard, T.L. Rothstein, et al. 1996. Regulation of the B cell response to T-dependent antigens by classical pathway complement. J. Immunol. 157:549–556. [PubMed] [Google Scholar]
- 27.Petry, F., M. Botto, R. Holtappels, M.J. Walport, and M. Loos. 2001. Reconstitution of the complement function in C1q-deficient (C1qa−/−) mice with wild-type bone marrow cells. J. Immunol. 167:4033–4037. [DOI] [PubMed] [Google Scholar]
- 28.Botto, M., C. Dell'Agnola, A.E. Bygrave, E.M. Thompson, H.T. Cook, F. Petry, M. Loos, P.P. Pandolfi, and M.J. Walport. 1998. Homozygous C1q deficiency causes glomerulonephritis associated with multiple apoptotic bodies. Nat. Genet. 19:56–59. [DOI] [PubMed] [Google Scholar]
- 29.Cutler, A.J., M. Botto, D. van Essen, R. Rivi, K.A. Davies, D. Gray, and M.J. Walport. 1998. T cell-dependent immune response in C1q-deficient mice: defective interferon gamma production by antigen-specific T cells. J. Exp. Med. 187:1789–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Matsumoto, M., W. Fukuda, A. Circolo, J. Goellner, J. Strauss-Schoenberger, X. Wang, S. Fujita, T. Hidvegi, D.D. Chaplin, and H.R. Colten. 1997. Abrogation of the alternative complement pathway by targeted deletion of murine factor B. Proc. Natl. Acad. Sci. USA. 94:8720–8725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Klaus, G.G.B., J.H. Humphrey, A. Kunkl, and D.W. Dongworth. 1980. The follicular dendritic cell: Its role in antigen presentation in the generation of immunological memory. Immunol. Rev. 53:3–28. [DOI] [PubMed] [Google Scholar]
- 32.Thornton, B.P., V. Vetvicka, and G.D. Ross. 1994. Natural antibody and complement-mediated antigen processing and presentation by B lymphocytes. J. Immunol. 152:1727–1737. [PubMed] [Google Scholar]
- 33.Hannum, L.G., A.M. Haberman, S.M. Anderson, and M.J. Shlomchik. 2000. Germinal center initiation, variable gene region hypermutation, and mutant B cell selection without detectable immune complexes on follicular dendritic cells. J. Exp. Med. 192:931–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chan, O.T., L.G. Hannum, A.M. Haberman, M.P. Madaio, and M.J. Shlomchik. 1999. A novel mouse with B cells but lacking serum antibody reveals an antibody-independent role for B cells in murine lupus. J. Exp. Med. 189:1639–1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Arya, S., F. Chen, S. Spycher, D.E. Isenman, M.J. Shulman, and R.H. Painter. 1994. Mapping of amino acid residues in the C mu 3 domain of mouse IgM important in macromolecular assembly and complement-dependent cytolysis. J. Immunol. 152:1206–1212. [PubMed] [Google Scholar]
- 36.Wright, J.F., M.J. Shulman, D.E. Isenman, and R.H. Painter. 1988. C1 binding by murine IgM. The effect of a Pro-to-Ser exchange at residue 436 of the mu-chain. J. Biol. Chem. 263:11221–11226. [PubMed] [Google Scholar]
- 37.Lynes, M.A., L.L. Lanier, G.F. Babcock, P.J. Wettstein, and G. Haughton. 1978. Antigen-induced murine B cell lymphomas. I. Induction and characterization of CH1 and CH2. J. Immunol. 121:2352–2357. [PubMed] [Google Scholar]
- 38.Shlomchik, M.J., D. Zharhary, T. Saunders, S.A. Camper, and M.G. Weigert. 1993. A rheumatoid factor transgenic mouse model of autoantibody regulation. Int. Immunol. 5:1329–1341. [DOI] [PubMed] [Google Scholar]
- 39.Jacob, J., R. Kassir, and G. Kelsoe. 1991. In situ studies of the primary immune response to (4-hydroxy-3-nitrophenyl)acetyl. I. The architecture and dynamics of responding cell populations. J. Exp. Med. 173:1165–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jacob, J., and G. Kelsoe. 1992. In situ studies of the primary immune response to (4-hydroxy-3-nitrophenyl)acetyl. II. A common clonal origin for periarteriolar lymphoid sheath-associated foci and germinal centers. J. Exp. Med. 176:679–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Borso, T., and M. Cooper. 1961. On the hemolytic activity of mouse complement. Prc. Soc. Exp. Biol. Med. 107:227–237. [Google Scholar]
- 42.Klaus, G.G., M.B. Pepys, K. Kitajima, and B.A. Askonas. 1979. Activation of mouse complement by different classes of mouse antibody. Immunology. 38:687–695. [PMC free article] [PubMed] [Google Scholar]
- 43.Johnson, A.A., A. Mirowski Rosengard, K. Skjodt, J.M. Ahearn, and R.G. Leslie. 1999. The structural basis for complement receptor type 2 (CR2, CD21)-mediated alternative pathway activation of complement: studies with CR2 deletion mutants and vaccinia virus complement-control protein-CR2 chimeras. Eur. J. Immunol. 29:3837–3844. [DOI] [PubMed] [Google Scholar]
- 44.Marquart, H.V., S.E. Svehag, and R.G. Leslie. 1994. CR2 is the primary acceptor site for C3 during alternative pathway activation of complement on human peripheral B lymphocytes. J. Immunol. 153:307–315. [PubMed] [Google Scholar]
- 45.Nielsen, C.H., H.V. Marquart, W.M. Prodinger, and R.G. Leslie. 2001. CR2-mediated activation of the complement alternative pathway results in formation of membrane attack complexes on human B lymphocytes. Immunology. 104:418–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nielsen, C.H., M.L. Pedersen, H.V. Marquart, W.M. Prodinger, and R.G. Leslie. 2002. The role of complement receptors type 1 (CR1, CD35) and 2 (CR2, CD21) in promoting C3 fragment deposition and membrane attack complex formation on normal peripheral human B cells. Eur. J. Immunol. 32:1359–1367. [DOI] [PubMed] [Google Scholar]
- 47.Nielsen, C.H., and R.G. Leslie. 2002. Complement's participation in acquired immunity. J. Leukoc. Biol. 72:249–261. [PubMed] [Google Scholar]
- 48.Olesen, E.H., A.A. Johnson, G. Damgaard, and R.G. Leslie. 1998. The requirement of localized, CR2-mediated, alternative pathway activation of complement for covalent deposition of C3 fragments on normal B cells. Immunology. 93:177–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ehrenstein, M.R., T.L. O'Keefe, S.L. Davies, and M.S. Neuberger. 1998. Targeted gene disruption reveals a role for natural secretory IgM in the maturation of the primary immune response. Proc. Natl. Acad. Sci. USA. 95:10089–10093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Boes, M., C. Esau, M.B. Fischer, T. Schmidt, M. Carroll, and J. Chen. 1998. Enhanced B-1 cell development, but impaired IgG antibody responses in mice deficient in secreted IgM. J. Immunol. 160:4776–4787. [PubMed] [Google Scholar]
- 51.Heyman, B. 1990. The immune complex: possible ways of regulating the antibody response. Immunol. Today. 11:310–313. [DOI] [PubMed] [Google Scholar]
- 52.Heyman, B., L. Pilstrom, and M.J. Shulman. 1988. Complement activation is required for IgM-mediated enhancement of the antibody response. J. Exp. Med. 167:1999–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Klaus, G.G.B. 1978. The generation of memory cells II. Generation of B memory cells with preformed antigen-antibody complexes. Immunology. 34:643–652. [PMC free article] [PubMed] [Google Scholar]
- 54.Klaus, G.G.B. 1979. The generation of memory cells III. Antibody class requirements for the generation of B-memory cells by antigen-antibody complexes. Immunology. 37:345–351. [PMC free article] [PubMed] [Google Scholar]