

Abstract
Little is known about the biochemical environment in phagosomes harboring an infectious agent. To assess the state of this organelle we captured the transcriptional responses of Mycobacterium tuberculosis (MTB) in macrophages from wild-type and nitric oxide (NO) synthase 2–deficient mice before and after immunologic activation. The intraphagosomal transcriptome was compared with the transcriptome of MTB in standard broth culture and during growth in diverse conditions designed to simulate features of the phagosomal environment. Genes expressed differentially as a consequence of intraphagosomal residence included an interferon γ– and NO-induced response that intensifies an iron-scavenging program, converts the microbe from aerobic to anaerobic respiration, and induces a dormancy regulon. Induction of genes involved in the activation and β-oxidation of fatty acids indicated that fatty acids furnish carbon and energy. Induction of σE-dependent, sodium dodecyl sulfate–regulated genes and genes involved in mycolic acid modification pointed to damage and repair of the cell envelope. Sentinel genes within the intraphagosomal transcriptome were induced similarly by MTB in the lungs of mice. The microbial transcriptome thus served as a bioprobe of the MTB phagosomal environment, showing it to be nitrosative, oxidative, functionally hypoxic, carbohydrate poor, and capable of perturbing the pathogen's cell envelope.
Keywords: microarray gene expression analysis, macrophage activation, inducible nitric oxide synthase, innate immunity, pathogenicity
Introduction
The phagosome is a difficult organelle to study because profound biochemical shifts accompany the host cell's effort to kill and degrade microbial pathogens. These shifts affect the levels of ions, radicals, lipids, and metabolites whose nature is not completely known, whose concentrations will change during subcellular fractionation, and whose characterization thus remains refractory to experimental assessment. We reasoned that a powerful route around this technical obstacle is provided by the ability of a select group of pathogens to adapt to these harsh conditions. Thus, in theory, the transcriptional response of surviving facultative intracellular pathogens should serve as a mirror reflecting conditions prevalent in the organelle in which they reside.
Macrophages are central to the effector arm of immune defense against most long-lived, nonviral intracellular pathogens. Through a variety of cell surface receptors, macrophages internalize microbes into phagosomes that undergo maturational events that expose the microbes to acid, lytic enzymes, oxygenated lipids, fatty acids, and reactive oxygen and nitrogen intermediates (1, 2). Despite this battery of antimicrobial molecules, some pathogens are able to survive and replicate within macrophage phagosomes (3, 4). The mechanisms that allow some pathogens to survive within phagosomes include inhibition of phagosomal maturation, resistance against antimicrobial molecules, and adaptation to host-induced metabolic constraints (3).
Mycobacterium tuberculosis (MTB) is among the microorganisms most successful at adapting to long-term residence in macrophage phagosomes. Inhalation of MTB leads to phagocytosis by alveolar macrophages. Before macrophages are immunologically activated, MTB blocks maturation of phagosomes into phagolysosomes (5). Replication of the pathogen follows until the onset of cell-mediated immunity, critically involving IFN-γ, which capacitates the macrophage to proceed with phagosomal maturation (6) and produce microbicidal molecules, including nitric oxide (NO; 7). Surviving bacteria are believed to enter a period of nonreplicating persistence in the phagosome until waning host immunity leads to reactivation from the latent state and the onset of disease (8). Thus, adaptation of MTB to the phagosomal compartment of the macrophage is an essential component of its pathogenesis, transmission, and continued survival as a life form (5, 8, 9).
In this study we asked the MTB transcriptome to serve as bioprobe to disclose biochemical features of MTB-infected phagosomes within primary bone marrow–derived murine macrophages. Global expression profiling identified the genes that are differentially expressed by intraphagosomal MTB in naive and IFN-γ–activated macrophages, compared with MTB grown in standard broth culture. Expression profiles of MTB exposed to ten different in vitro conditions of growth were then used to identify conditions that mimic the phagosomal environment.
Materials and Methods
Isolation of RNA from Intraphagosomal MTB.
Murine bone marrow macrophages and MTB clinical isolate 1254 were prepared as previously described (10). For each time point, 6–8 × 107 macrophages were infected with a multiplicity of 2–5. Uptake of bacteria by macrophages was analyzed by microscopy and serial dilutions of culture medium on 7H11 agar plates. If necessary, extracellular bacteria were removed by washing with cell culture medium prewarmed to 37°C before harvesting of MTB. 4, 24, or 48 h after infection macrophages were lysed with guanidine thiocyanate and bacteria were harvested by centrifugation (11, 12). Pellets were frozen on dry ice and stored at −80°C. 2–4 μg bacterial RNA were isolated from each sample using previously described procedures (11, 12, 13).
Experiments with H2O2 and Palmitic Acid.
H2O2 (5 mM final concentration) was added to early log-phase cultures (7H9 medium supplemented with bovine serum albumin, NaCl, glucose, glycerol, and Tween 80). RNA was isolated 40 min after the addition of H2O2. Experiments with palmitic acid were done in minimal medium (0.5 g/liter asparagine, 1 g/liter KH2PO4, 2.5 g/liter Na2PO4, 10 mg/l MgSO4·7H2O, 50 mg/l ferric ammonium citrate, 0.5 mg/l CaCl2, 0.1 mg/l ZnSO4, 0.05% Triton WR1339, 10 mM glucose, pH 6.6). Palmitic acid was dissolved in water at 50°C (0.2%) and added to early log-phase cultures at a final concentration of 0.05 mM. RNA was isolated 1 h after the addition of palmitic acid.
Microarray Experiments.
Labeling of RNA and hybridizations to amplicon arrays were performed as previously described (13). Oligonucleotide microarrays were first prehybridized for 1 h in 5X SSC, 1% BSA, and 0.1% SDS and washed with H2O and isopropanol. After the prehybridization, 10 μl hybridization solution (labeled cDNA, 5 μg tRNA, 2X SSC, 25% formamide, 0.1% SDS) was hybridized at 54°C for 16–20 h.
Microarray Data Analysis.
Microarrays were scanned using a GenePix 4000A (Axon Instruments, Inc.). Fluorescence intensities of the two dyes at each spot were quantified using the ScanAlyze software (http://rana.lbl.gov/EisenSoftware.htm). Amplicon microarrays were used to analyze RNA isolated from intraphagosomal MTB 4, 24, and 48 h after infection. Amplicon microarray-determined ratios were calculated from three biological replicates and two microarrays for each biological replicate (4-h time point) and four biological replicates and one to two microarrays for each biological replicate (24- and 48-h time points). Oligonucleotide microarrays were used to analyze RNA isolated from intraphagosomal MTB 24 h after infection. Oligonucleotide microarray-determined ratios were calculated from three biological replicates and two microarrays for each biological replicate. Genes were classified as intraphagosomally regulated if they met both of the following criteria: (a) They were regulated in intraphagosomal MTB with a false discovery rate of <1% in both the amplicon and the oligonucleotide datasets and (b) they were regulated at least twofold in one and at least 1.8-fold in the other dataset. False discovery rates were determined using the significance analysis of microarrays program (14). The fold-change criteria were used because congruence in the fraction of genes regulated according to both datasets increased with the magnitude of regulation (see Fig. S3, available at http://www.jem.org/cgi/content/full/jem.20030846/DC1) .
Identification of Significant Clusters of Regulated Genes in the Chromosome of MTB.
We determined the number of regulated genes within ±20 positions of each regulated gene and divided it by 40 (the number of genes in the segment of the chromosome being analyzed). Each resulting number was divided by the ratio of the number of regulated genes minus one to the number of genes in the chromosome minus one. The resulting quotients are the relative densities and were calculated independently for induced and repressed genes. Values for repressed genes were multiplied by −1. To derive the 5% null hypothesis cut-off value, 200 datasets were generated in which the positions of regulated genes were randomly selected from 3,924 possible positions. The maximum relative density was calculated for each of the 200 datasets and the 10th largest of these numbers was used as the 5% cut-off value. Relative densities greater than the 5% cut-off value define “clusters.”
Analysis of Mycobacterial RNA with Quantitative Real Time Reverse Transcription-PCR (qrtPCR).
8 × 106 macrophages were infected as described above and lysed with TRIZOL (Invitrogen). Bacteria were harvested by centrifugation and disrupted by bead beating (13). C57BL/6 mice were i.v. infected with 106 colony-forming units of MTB strain H37Rv. Lungs were isolated 21 and 56 d after infection and partly used to determine the number of viable bacteria. The remainder was homogenized in 4 ml TRIZOL. RNA was prepared as previously described (13). cDNA was prepared as for microarray experiments, except that gene-specific primers were used. Real time PCR was performed using specific TaqMan probes. A reaction lacking reverse transcriptase was performed for every RNA sample. The major housekeeping sigma factor gene sigA was used as an internal control for the normalization of mRNA levels (15). Gene induction ratios were calculated from the comparison with RNA from bacteria grown to mid-log phase in 7H9. RNA was isolated from two independent macrophage infections and from three mice killed 21 d after infection and three mice killed 56 d after infection.
Online Supplemental Material.
Gene regulation data for all analyzed genes are available at http://www.jem.org/cgi/content/full/jem.20030846/DC1. Regulated genes were selected as described in Results.
Results
Expression Profiling of Intraphagosomal MTB.
Microbicidal capacity of the primary bone marrow–derived murine macrophages in these experiments required IFN-γ, was NO synthase 2 (NOS2)-dependent, and was accompanied by the production of NO, as reported (10). Activated wild-type macrophages killed MTB by 120 h after infection. However, the number of viable intracellular bacteria was stable over the first 72 h and was similar in naive and activated macrophages during that time (Fig. 1 A). Quantitative cultures of the tissue culture medium of this infection experiment showed that ≥99% of the MTB were macrophage associated 24 and 48 h after infection, and our prior observations and those of other investigators show that most of these bacteria are intraphagosomal (16, 17, 18).
Figure 1.
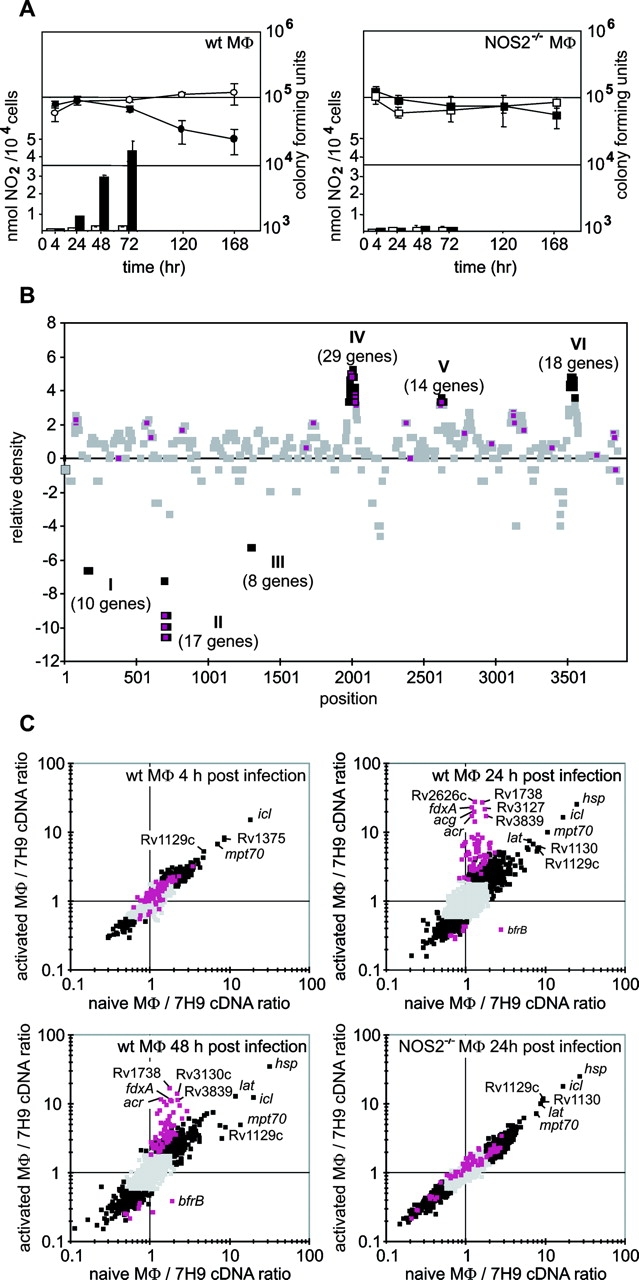
Impact of macrophage activation on gene expression by intraphagosomal MTB. (A) Survival of MTB in macrophages (CFU) and generation of NO2 −, an accumulating oxidation product of NO. Left, wild-type (wt) macrophages; right, NOS2-deficient macrophages. Open symbols and bars are for macrophages not treated with IFN-γ (“naive”) and solid symbols and bars are for IFN-γ–stimulated macrophages. NO2 − secretion was measured as described 4, 24, 48, and 72 h after infection (reference 10). Means ± SD of triplicates in one experiment representative of three. (B) Distribution of differentially intraphagosomally regulated genes on the chromosome of MTB. Clusters were identified as described in Materials and Methods. Regulated genes outside of clusters are shown as gray squares, whereas genes within clusters are indicated by black squares. Centers of squares representing activation-specific genes are purple. (C) Comparison of gene regulation in MTB residing in naive or IFN-γ–stimulated macrophages. Data are averages of six (4 h after infection) or seven (24 and 48 h after infection) amplicon microarray experiments. Gray squares indicate genes regulated less than twofold in naive and activated macrophages. Black squares indicate genes regulated at least twofold in naive or activated macrophages. Purple squares indicate genes whose regulation was activation specific in wild-type macrophages. Activation-specific genes were defined as genes for which regulation in activated versus naive macrophages was significantly different (FDR <1% 24 or 48 h after infection in the amplicon array dataset and 24 h after infection in the oligonucleotide array dataset) and at least twofold different 24 h after infection. wt MΦ, wild-type macrophages; NOS2−/− MΦ, NOS2-deficient macrophages.
RNA was prepared from intracellular MTB at 4, 24, and 48 h, times when the intracellular pathogen was fully viable, using a method that lyses the macrophage, but not the bacteria, while rapidly blocking bacterial mRNA degradation and preventing further transcription. Separation of bacteria from the macrophage lysate by centrifugation and the extraction of RNA from the pellet yields intact mycobacterial RNA free from macrophage RNA and provides a faithful representation of bacterial transcripts present within the phagosome. This method for differential lysis and bacterial RNA stabilization has been validated both by whole genome analysis using microarrays and slot-blot hybridization analysis of individual genes (11, 12). A “two-color” microarray hybridization method was used to compare each RNA sample of intracellular bacteria with RNA from bacteria grown to mid-log phase in 7H9, a standard MTB culture medium (19). Six DNA microarrays that had been fabricated with PCR-derived amplicons were used for the 4-h experiments and seven amplicon microarrays were used for the 24- and 48-h experiments. This analysis provides a genome-wide inventory of genes that are differentially expressed (regulated) in the two environments. Genes showing similar expression levels in liquid culture and in the phagosome of macrophages were excluded from further investigation, but are listed in Table S1, available at http://www.jem.org/cgi/content/full/jem.20030846/DC1.
A comparison of the 4, 24, and 48 h postinfection time points disclosed significant changes in MTB gene expression between 4 and 24 h, for both the resting and IFN-γ–activated macrophages. By contrast, the expression profiles of MTB 24 and 48 h after infection were very similar (see Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20030846/DC1). We concluded that at 24 h after infection the transcriptome of MTB reaches a state that is stable for at least the next 24 h. Because the half-lives of most bacterial transcripts are <8 min (20), these MTB expression profiles must represent de novo transcription in response to specific intracellular cues. As such, they form a part of the adaptive survival strategy of the organism immediately after phagocytosis and in response to the reshaping of the internal macrophage environment with time.
The MTB Differential Intraphagosomal Transcriptome.
The differential intraphagosomal transcriptome was operationally defined as all MTB genes significantly regulated 24 or 48 h after infection in either naive or IFN-γ–stimulated wild-type macrophages. To reduce crosshybridization artifacts, six additional experimental replicates of the same 24-h postinfection RNA samples were analyzed with oligonucleotide microarrays. These results and those from the six to seven amplicon microarrays were analyzed by the significance analysis of microarrays program using a false discovery rate of <1% (14). By these criteria, 601 MTB genes, representing ∼15% of MTB open reading frames, composed the differential intraphagosomal transcriptome. Of these, 454 were induced and 147 repressed (see Tables S2 and S3, available at http://www.jem.org/cgi/content/full/jem.20030846/DC1). The 454 induced genes identified here include 22 genes that were previously found by other methods to be expressed by intraphagosomal MTB (see Table S4, available at http://www.jem.org/cgi/content/full/jem.20030846/DC1).
qrtPCR was used to determine if 21 genes within the microarray-determined differential intraphagosomal transcriptome could be shown to be differentially expressed by a more quantitative measure of transcript abundance (Table I). These genes were selected based on their predicted function as well as their specificity of regulation. 12 genes were selected because they are predicted or known to be involved in transcriptional regulation (nadR, Rv0792c, Rv1129c), intermediary metabolism (icl, gltA1), lipid metabolism (fadE5, fadB2, fadD19, echA19, fadA6, desA1) or iron acquisition (mbtB). Regulation of these genes suggests specific metabolic adaptations occurring in intraphagosomal MTB. The genes fdxA, acr, mbtB, Rv1738, and Rv2626c were selected because they were expressed more strongly in activated than naive macrophages. Five genes of unknown function that were regulated similarly in naive and activated macrophages (mpt83, pe11, ppe37, and Rv1462) were also analyzed by qrtPCR. Icl was analyzed because it has been reported as specifically regulated in response to macrophage activation (21) but was strongly induced in activated and naive macrophages in the present array experiments. As a negative control, fadD28, which was not intraphagosomally regulated according to the microarray experiments, was also analyzed. Each of the 21 intraphagosomal transcriptome genes was shown by qrtPCR to be induced 3–200-fold by MTB residing in the macrophage phagosome compared with bacteria growing in 7H9 medium (Table I). Genes that were more strongly regulated in activated macrophages according to our microarray experiments were also more strongly regulated in the qrtPCR analysis. Expression of icl was intraphagosomally induced but not affected by macrophage activation. Similar amounts of fadD28 RNA were detected in both in vitro–grown and intraphagosomal MTB.
Table I.
qrtPCR Analysis of MTB Gene Regulation after Cultivation in Macrophages (MΦ) and Mice
| Gene induction ratios
|
|||||
|---|---|---|---|---|---|
| Genea | Function of encoded protein | Naive MΦ | Activated MΦ | Mice | |
| 24 hours | 24 hours | 21 days | 56 days | ||
| nadR | Transcriptional regulation | 5.0 ± 0.9 | 5.3 ± 1.1 | not detected | 0.6 ± 1.4 |
| Rv0792c | Transcriptional regulation | 12 ± 2.4 | 11 ± 1.4 | 10 ± 2.6 | 6.3 ± 1.4 |
| Rv1129c | Transcriptional regulation | 16 ± 1.8 | 17 ± 0.4 | 14 ± 2.8 | 7.0 ± 1.6 |
| icl | Glyoxylate cycle | 98 ± 12 | 103 ± 7.5 | 44 ± 13 | 37 ± 9.0 |
| gltA1 | Alternative citrate synthase | 4.9 ± 0.3 | 5.3 ± 0.2 | 15 ± 4.1 | 7.6 ± 2.1 |
| fadE5 | Fatty acid degradation | 10 ± 0.2 | 9.7 ± 0.6 | 14 ± 3.3 | 27 ± 4.8 |
| fadB2 | Fatty acid degradation | 13 ± 0.7 | 15 ± 0.8 | 10 ± 1.4 | 7.4 ± 1.9 |
| fadD19 | Fatty acid degradation | 3.8 ± 0.5 | 4.4 ± 0.5 | 16 ± 3.3 | 14 ± 5.2 |
| echA19 | Fatty acid degradation | 3.0 ± 0.4 | 3.0 ± 0.3 | 8.4 ± 1.5 | 5.9 ± 2.1 |
| fadA6 | Fatty acid degradation | 3.2 ± 0.4 | 2.9 ± 0.4 | not detected | 3.1 ± 1.1 |
| desA1 | Fatty acid desaturation | 3.2 ± 0.1 | 3.9 ± 0.2 | 11 ± 3.0 | 6.3 ± 1.5 |
| fdxA | Electron transfer | 2.5 ± 0.9 | 70 ± 17 | 604 ± 165 | 759 ± 276 |
| acr | α-crystallin | 1.5 ± 0.5 | 201 ± 57 | 798 ± 269 | 695 ± 224 |
| mbtB | Mycobactin biosynthesis | 2.5 ± 0.4 | 4.6 ± 0.5 | 26 ± 5.1 | 26 ± 5 |
| acg | Unknown | 1.9 ± 0.9 | 26 ± 5.0 | 277 ± 68 | 94 ± 28 |
| mpt83 | Unknown | 4.9 ± 0.3 | 10 ± 1.0 | 29 ± 2.3 | 16 ± 4.0 |
| pe11 | Unknown | 5.9 ± 1.1 | 5.3 ± 1.0 | 5.4 ± 1.4 | 5.4 ± 1.9 |
| ppe37 | Unknown | 3.7 ± 0.5 | 5.5 ± 1.0 | 59 ± 12 | 68 ± 17 |
| Rv1462 | Unknown | 3.0 ± 0.2 | 3.2 ± 0.3 | 6.8 ± 1.5 | 5.3 ± 1.5 |
| Rv1738 | Unknown | 1.6 ± 0.5 | 66 ± 16 | 851 ± 403 | 346 ± 124 |
| Rv2626c | Unknown | 3.8 ± 2.0 | 40 ± 8.4 | 420 ± 170 | 264 ± 91 |
| fadD28 b | Lipid synthesis | 0.9 ± 0.1 | 0.9 ± 0.0 | 1.4 ± 0.6 | 2.2 ± 0.7 |
Gene induction ratios were calculated from the comparison with RNA from bacteria grown to mid-log phase in 7H9. Data are means (±SEM) from at least six independent measurements analyzing RNA from at least two (MΦ) or three different infections (mice).
According to Cole et al. (reference 28) and http://genolist.pasteur.fr/TubercuList/.
Not regulated in intraphagosomal MTB according to microarray experiments.
To correlate the microarray-determined differential intraphagosomal transcriptome with MTB gene expression in infected tissues, RNA was isolated from mouse lungs 21 and 56 d after infection with MTB. By qrtPCR, RNA levels for 19 of the 21 assayed intraphagosomally induced genes were increased between 5- and 800-fold compared with in vitro grown bacteria. By contrast, RNA levels for the negative control gene, fadD28, increased only slightly (1.4-fold, 2.2-fold) in vivo (Table I). Thus, transcriptional adaptation of MTB to the phagosomal environment of activated bone marrow–derived macrophages reflects adaptations that also occur in murine tuberculosis. This conclusion is supported by previous observations that some of the genes found to be intraphagosomally induced by our microarray experiments are important for MTB virulence: fadD26 (22, 23), mbtABCDEFGH (24), icl (21), furA (25), umaA, alkB (26), and fadD33 (27).
A statistical analysis revealed that 96 of the 601 genes were localized in six separate clusters of contiguous genes on the chromosome (Fig. 1 B). Clusters I–III contain four putative operons of repressed genes, including the mycobacterial cell entry operon 1 (see Fig. S2, available at http://www.jem.org/cgi/content/full/jem.20030846/DC1, Cluster I; 28, 29). Clusters II and III encode ribosomal proteins and the ATP-synthase, respectively. Clusters IV–VI contain 61 induced intraphagosomal genes that are distributed across at least 27 different transcriptional units. 20 of these genes are macrophage activation specific, as defined below. Several genes in cluster VI are predicted by their annotated functions (28) to be involved in lipid metabolism, whereas most genes in clusters IV and V encode proteins of unknown function.
Macrophage Activation-specific Genes.
To reveal the effect of macrophage activation on the differential intraphagosomal transcriptome we compared the expression profiles of MTB residing in naive macrophages with those of bacteria in IFN-γ–stimulated macrophages (Fig. 1 C). 68 genes were identified whose expression changed significantly in response to IFN-γ stimulation 24 and 48 h after infection (Fig. 1 C, purple squares). These were operationally defined as “activation-specific” (see Table S5, available at http://www.jem.org/cgi/content/full/jem.20030846/DC1). 60 of the activation-specific genes were induced and 8 were repressed. IFN-γ–specific regulation was not observed 4 h after infection. We conclude that ∼11% of the genes comprising the differential intraphagosomal transcriptome are regulated by IFN-γ activation of the macrophage between 4 and 24 h after infection, coincident with the production of NO by IFN-γ–stimulated macrophages from wild-type mice (Fig. 1 A). To further explore the relationship between NO production and the activation-specific gene set, we compared the expression levels of genes in IFN-γ–activated macrophages from wild-type and NOS2−/− mice. Stimulation with IFN-γ failed to significantly change the expression levels of any of the 68 activation-specific genes in NOS2−/− macrophages (Fig. 1 C). Thus, NOS2 activity is necessary for the IFN-γ–dependent induction of the activation-specific gene set.
Condition-specific Regulation of the Intraphagosomal Transcriptome.
To deduce physiochemical conditions encountered by MTB in the macrophage, we compared the differential intraphagosomal transcriptome with expression profiles previously obtained for MTB in response to in vitro conditions thought to simulate features of the phagosome: exposure to low and high concentrations of NO (the NO donor DETA/NO, 0.05 mM, and 0.5 mM for 40 min; 30), low oxygen (0.2% O2 for 2 h; 31), low iron (2 μM Fe3+ through mid-log phase; 32), low pH (pH 5.5 for 15 min; 33), nutrient starvation (PBS for 4, 24, and 96 h; 34), heat shock (45°C for 30 min; 35), thiol-specific oxidative stress (5 mM diamide for 1 h; 36), and a cell wall–perturbing detergent (0.05% SDS for 90 min; 37). In addition, we obtained expression profiles of MTB to oxidative stress (5 mM H2O2 for 40 min) and during growth in a medium containing a free fatty acid (50 μM palmitic acid for 1 h) as a source of carbon and energy. Taken together, 8 of the 10 in vitro conditions induced regulation of ∼60% of the genes that comprise the differential intraphagosomal transcriptome (Fig. 2, A and B) .
Figure 2.

Comparison of gene regulation in IFN-γ–stimulated macrophages with gene regulation in response to various stresses in broth culture. (A) Stresses mimicking those imposed selectively by activated macrophages. Each scatter plot contains all genes induced in activated macrophages 24 h after infection (FDR <1% in oligonucleotide and amplicon array datasets, magnitude of regulation at least twofold in amplicon array dataset), most of which were also induced in resting macrophages. The x axes indicate regulation in intraphagosomal MTB and the y axes indicate regulation in culture. In each panel, squares located in the horizontal gray areas indicate genes regulated less than twofold in vitro. Red squares indicate activation-specific genes that were also induced with 0.05 mM DETA/NO and blue squares indicate activation-specific genes that were not induced with 0.05 mM DETA/NO but were induced with 5 mM H2O2. Most of the latter were also induced with 0.5 mM DETA/NO. All other genes were indicated by black squares if regulated more than twofold in culture under the conditions indicated in a given panel and by gray squares if regulated less than twofold in the same culture conditions. Thus, genes regulated similarly in naive and activated macrophages are marked by neither red nor blue. They are depicted by black or gray symbols according to their behavior under the in vitro conditions indicated. The numbers of coregulated genes give the number of all intraphagosomally induced genes that were also induced more than twofold by culture under the conditions indicated on the y axis. Data for DETA/NO, 0.2% oxygen, and low iron are from Voskuil et al. (reference 30), Sherman et al. (reference 31), and Rodriguez et al. (reference 32), respectively. (B) Stresses mimicking those imposed by both naive and activated macrophages. Data for PBS, heat shock (45°C), and SDS were from Betts et al. (reference 34), Stewart et al. (reference 35), and Manganelli et al. (reference 36), respectively. Squares are colored as described in A. Regulation for genes not reported as significantly regulated during incubation in PBS was set to 1 because primary data for these genes were not available.
To identify a physical or chemical instigator of the activation-specific gene response, the expression profiles for each of these in vitro conditions were compared with the transcriptional response of MTB in IFN-γ–activated macrophages. Based on this analysis, the activation-specific gene set was divided into two groups (Figs. 2 A and 3): (a) those induced in response to low concentrations of NO (0.05 mM DETA/NO) or hypoxia (Fig. 2 A, red squares) and (b) those regulated by H2O2 (Fig. 2 A, blue squares). Regulation of genes of the first group did not change in response to stimulation of NOS2−/− macrophages with IFN-γ. This observation and their induction by NO in vitro shows that their expression in IFN-γ–activated wild-type macrophages is due to the direct effect of NO on MTB transcription rather than the indirect effect of IFN-γ on the regulation of diverse macrophage genes (10). Most genes in the second, H2O2 group, were also induced by exposure of MTB to high NO (Fig. 2 A, 0.5 mM DETA/NO), but not to low NO (0.05 mM DETA/NO) concentrations. Remarkably, the H2O2/high dose NO response was also not induced by MTB in IFN-γ–stimulated NOS2−/− macrophages. Therefore, the expression of this gene set is also NOS2 dependent and NO regulated. The NO-dependent induction of the H2O2 genes in the phagosomal compartment of activated, wild-type macrophages suggests that NO concentrations reach levels high enough to induce or contribute to an oxidative stress response. However, the NO-mediated oxidative stress response induced remarkably few potential antioxidant genes in the phagosome. Most prominent among those induced were ahpC and ahpD, components of a peroxynitrite reductase/peroxidase (Fig. 4 ; 38).
Figure 4.
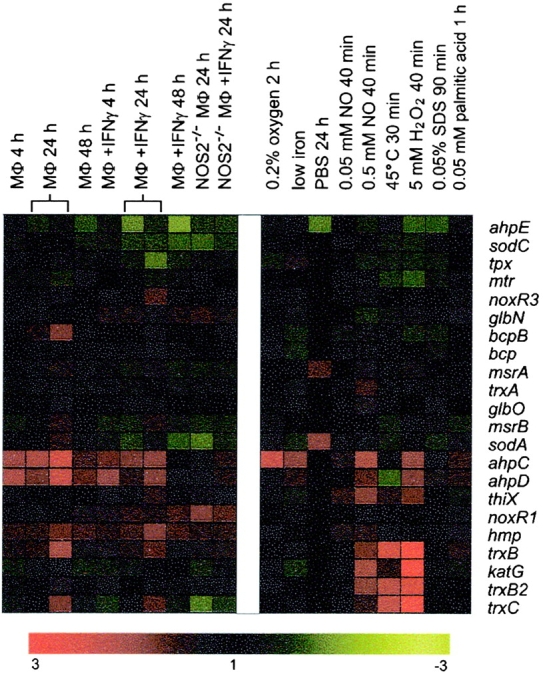
Relative expression levels of antioxidant genes. Genes were selected based on a demonstrated or presumed antioxidant function of the encoded proteins. cDNA ratios were averaged, log2 transformed, and displayed according to the color code at the bottom of the display. The order of genes was determined by hierarchical clustering.
A common set of 35 MTB genes was induced in IFN-γ–stimulated macrophages and during growth in a medium containing low concentrations of iron (Figs. 2 A and 3). Many of the low iron response genes were also induced in resting macrophages and NOS2−/− macrophages (Fig. 3) , indicating that the phagosome is perceived by MTB to be a low iron environment. However, most of the low iron response genes were more strongly induced in IFN-γ–activated wild-type macrophages than resting macrophages (Fig. 3), including the mbt operon, which directs the synthesis of mycobacterial siderophores (24, 39). Most of these genes were also induced in vitro by 0.5 mM DETA/NO and 5 mM H2O2, presumably due to oxidative damage to IdeR, a negative regulator of the MTB iron scavenging response (32). Thus, the increased induction of low iron response genes in activated, wild-type macrophages might be caused by NO rather than a decrease in the availability of iron in this environment. If so, then NO-mediated induction of the iron uptake system could aid in the repair or replacement of damaged iron-containing proteins, which often result from exposure to oxidative and nitrosative stress (40). At the same time, the increase in iron uptake may lead to high free iron concentrations within the bacteria and increased oxidative stress (41).
Figure 3.
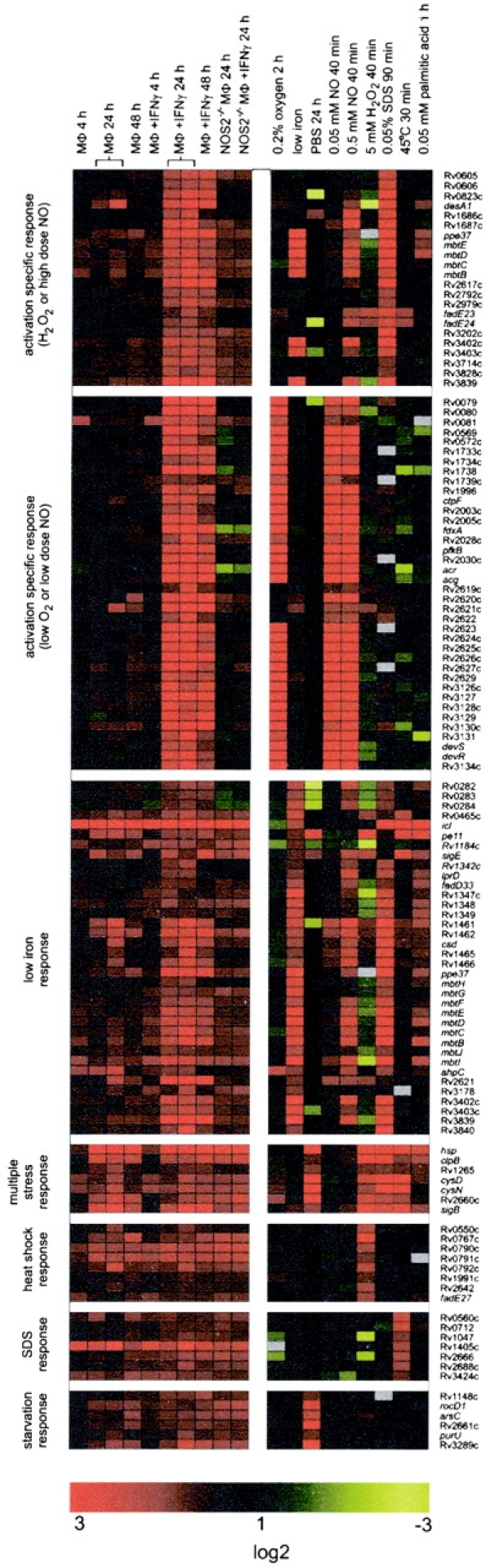
Red-green display summarizing the regulation of selected intraphagosomally induced genes. cDNA ratios were averaged, log2 transformed, and displayed according to the color code at the bottom of the display. Gray fields indicate missing data. Experimental conditions were as indicated at the top of the display. The two columns shown for intraphagosomal regulation 24 h after infection indicate data from amplicon arrays and oligonucleotide arrays, respectively. Activation-specific genes were selected as described (Figs. 1 C and 2 A). The low iron response genes are all the genes induced in IFN-γ–stimulated macrophages that were induced at least twofold during growth with low iron. The multiple stress response genes are the genes that most strongly reacted to all three of the following conditions: PBS, heat shock, and SDS. Genes labeled as heat shock, SDS, and starvation responses were genes induced more than twofold during heat shock, SDS treatment, or starvation, respectively, and induced in activated macrophages but induced less than twofold in response to all other in vitro stresses.
Nutrient starvation, heat shock, and exposure to SDS in vitro also induced subsets of genes in the differential intraphagosomal transcriptome (Fig. 2 B). However, in contrast to the NO, low oxygen, and iron scavenging responses described above, most of these genes were not affected by macrophage activation. Many of the starvation-, heat shock–, and SDS-induced genes were induced in vitro by more than one of these stimuli. The most strongly regulated genes in this group are shown in Fig. 3 as the multiple stress response gene set. However, other genes of the differential intraphagosomal transcriptome were only induced in response to one of these in vitro stimuli (Fig. 3). The latter result suggests that the macrophage, independent of its stimulation by IFN-γ, exerts several different types of stress. One of these, simulated in vitro by exposure of the organism to SDS, is particularly notable because the transcriptional response to SDS requires σE, an extracellular function sigma factor that seems to protect the organism from treatments that disrupt the cell envelope (37). Mutants of MTB deficient in σE are unable to proliferate in macrophage-like cell lines. The lipid-rich nature of the MTB surface, the release of lipids derived from intraphagosomal MTB (42), the cell envelope–perturbing effect of SDS, and the capacity of SDS to mimic a stress within the phagosome suggest that the macrophage damages a lipoidal component of the mycobacterial cell surface.
The survival of cell envelope–damaged MTB in the macrophage phagosome suggests that part of the response to this stress entails remodeling of the mycobacterial surface. FadD26 and umaA, which might direct this kind of remodeling, were identified within the differential intraphagosomal transcriptome and also induced in vitro by exposure of the microbe to SDS (37). FadD26 is involved in the synthesis of phthiocerol dimycocerosate, a complex lipid in the cell envelope of pathogenic mycobacteria that serves as a mycobacterial virulence factor. MTB strains that are unable to produce phthiocerol dimycocerosate show increased cell wall permeability and are more sensitive to detergent (43). UmaA, a protein of unknown function, belongs to a highly conserved family of mycolic acid methyl transferases that modify lipids of the mycobacterial cell wall (44). UmaA also seems important for MTB virulence (26). Remodeling of the cell envelope appears to be accompanied by a DNA recombination and repair response because alkA, recX, recG, dinF, and radA were induced in the phagosome. These genes were similarly regulated in wild-type and NOS2-deficient macrophages. Thus, the macrophage appears to induce DNA damage independently of NO.
Metabolic Adaptations.
MTB can survive and even replicate in naive macrophages and thus must assimilate carbon and produce energy in the phagosome (45). Analysis of genes within the differential intraphagosomal transcriptome that code for functions of intermediary metabolism suggests that MTB switches its carbon source from glucose and glycerol during in vitro growth to fatty acids in the phagosome (Fig. 5 A). This interpretation is based on the induction of 18 genes that collectively are predicted to encode all enzymes necessary for the biochemical activation and β-oxidation of fatty acids, including fatty acid-coenzyme A (CoA) synthase (fadD3,9,10,19), acyl-CoA dehydrogenase (fadE5,14,22–24,27–29,31), enoyl-CoA hydratase (echA19), hydroxybutyryl-CoA dehydrogenase (fadB2,3), and acetyl-CoA transferase (fadA5,6). The induction of multiple genes for four of the five transformations suggests that different isoenzymes are required to catabolize structurally diverse fatty acids (Fig. 5 A). Six of these genes were also induced in vitro during degradation of palmitic acid by MTB (Fig. 5 A). Fatty acids might be derived from triglycerides by PE30, a putative lipase encoded by an intraphagosomally induced gene. Breakdown products of fatty acids (i.e., acetyl-CoA and propionyl-CoA) are likely metabolized via the citric acid and glyoxylate cycles as suggested by the induction of the genes gltA1, Rv1130, and icl (Fig. 5 A). Proteins encoded by gltA1 and Rv1130 are similar to PrpC and PrpD of Salmonella, which participate in the catabolism of propionyl-CoA (46). The gene icl encodes isocitrate lyase, an enzyme necessary for the glyoxylate cycle and for the persistence of MTB in a murine model of latent tuberculosis (21). Induction of pckA, which encodes the gluconeogenetic rate–limiting enzyme phosphoenolpyruvate carboxykinase, suggests that fatty acids are in part converted into sugars via gluconeogenesis. We used qrtPCR to demonstrate that a subset of these genes (icl, gltA1, fadE5, fadD19, echA19, and fadA6) was also induced in the MTB-infected mouse lung (Table I) and other investigators using different methods have shown that fadE5, echA19, icl, and pckA (47, 48) and the protein product of Rv1130 (Schaible, U.E., and Kaufmann, S.H.E., personal communication) are induced in human macrophages or macrophage-like cell lines. Taken together, these data support the view that MTB uses fatty acids as a carbon source during in vivo growth (21, 28, 45).
Figure 5.

Regulation of genes encoding proteins predicted to be involved in intermediary metabolism and energy metabolism. (A) Catabolism of fatty acids. cDNA ratios were averaged, log2 transformed, and are shown in the red green display according to the color code shown in B. Experimental conditions were as indicated at the top of the display (see Fig. 2 for details). The two columns shown for intraphagosomal regulation 24 h after infection indicate data from amplicon arrays and oligonucleotide arrays, respectively. Genes were selected based on their annotation (reference 15) and grouped into those that were either regulated in response to at least one of the in vitro stresses or by none of them. Red arrows in the graphical representation of the β-oxidation and glyoxylate cycles indicate reactions that we infer to be induced in intraphagosomal MTB based on the pattern of gene expression. MΦ, macrophages. (B) Energy generation and NAD+ regeneration. Genes were selected based on their annotation and ordered based on their location in the chromosome. Gray field indicates missing data. On the amplicon arrays, narX was only represented by a DNA that also contained unspecific PCR products and is therefore not included in the red-green display. Regulation of narX in the oligonucleotide array dataset was activation specific (1.3 ± 0.4 [mean ± SEM] in naive macrophages vs. 5.2 ± 1.2 in activated macrophages 24 h after infection). Red and green arrows in the graphical representation indicate reactions that we infer to be induced (red) and repressed (green) in intraphagosomal MTB based on the pattern of gene expression.
Energy Production During Anaerobic Respiration.
Changes in energy metabolism (Fig. 5 B) accompanied changes in intermediary metabolism. Genes that encode subunits of NADH dehydrogenase (NDH)1 (nuoABDFIL), the ubiquinol–cytochrome C complex (qcrCA), and ATP synthase (atpABDEFGH) were repressed. Because RNA from intraphagosomal MTB was compared with RNA from logarithmically growing bacteria, repression of genes encoding NDH-1, the cytochrome C complex, and the ATP synthase presumably reflects the reduced need for energy generation during bacteriostasis, the growth state of intraphagosomal MTB (Fig. 1), compared with logarithmic growth. Consistent with this, operons encoding NDH-1 and the ATP synthase were repressed during starvation (Fig. 5 B; 34).
By contrast, frdA, narX, and ndh, encoding fumarate and nitrate reductases and NDH-2, respectively, were induced. The modulation of the respiratory chain that likely occurs as a consequence of the induction of frdA, narX, and ndh may not serve to increase energy production. Indeed, the induction of ndh by intraphagosomal MTB could reduce the bioenergetic efficiency of the respiratory chain, as shown for the Escherichia coli homologue of NDH-2, whose oxidation of NADH does not contribute to the generation of proton motive force (49). Alternatively, the induction of these genes could signal an increased need for NAD+ regeneration, the second major function of the respiratory chain besides energy production. β-oxidation of fatty acids consumes NAD+ and FADH in every cycle of fatty acid oxidation (Fig. 5 A). Induction of NDH-2 thus might represent an adaptation necessary to ensure NAD+ levels that are sufficient to allow increased β-oxidation of fatty acids during bacteriostasis. The induction of ald, which encodes an l-alanine dehydrogenase, might also contribute to the regeneration of NAD+ because the reductive amination of pyruvate is the kinetically preferred reaction at physiological pH (Fig. 5 B; 8, 50).
narX, encoding a nitrate reductase, was only induced in activated macrophages and is located in a region of the chromosome containing four other activation-specific genes. That many of the genes specifically induced in activated macrophages were also induced with low oxygen in vitro (Figs. 2 A and 3) suggests that intraphagosomal MTB generates energy by anaerobic respiration. Remarkably, this switch in energy generation is most likely imposed on the pathogen by inhibition of aerobic respiration by NO (51, 52, and unpublished data) and is also an activation-specific component of the MTB metabolic response in IFN-γ–stimulated wild-type macrophages.
Discussion
There is no one phagosome. Phagosomes differ depending on the cell in which they arise, their point along the developmental cycle of the organelle, and the nature of the microbe resident within them. Intraphagosomal microbes can profoundly alter host cell physiology, sometimes to the point of inducing apoptosis. In striking contrast to phagosomes containing most pyogens, which evolve and resolve over a few hours, the MTB-containing phagosomes of macrophages are notable for their longevity and relative stability over several days in vitro and years in vivo. MTB may achieve this effect by controlling phagosomal development, preventing its fusion with the lysosome, and reducing maximal acidification of phagosomal contents (5). Here we show that MTB perceives the phagosome of naive and activated macrophages as an environment that provides fatty acids (but relatively few carbohydrates), is DNA and cell envelope damaging, and of reduced iron availability. Changes in the transcriptome suggest that MTB adapts to this environment by the induction of fatty acid–degrading enzymes and DNA repair proteins, the remodeling of its envelope, and the production of secreted siderophores to facilitate the acquisition of iron. Examination of the expression profile also showed that the generation of NO by IFN-γ–activated macrophages induces additional responses, yielding a phagosomal environment that is nitrosative, oxidative, inhibitory to aerobic respiration, and conducive to increased uptake of iron by the pathogen.
The manipulation of host cellular processes by bacterial pathogens can vary significantly according to the organism and the intracellular target of microbial effector molecules (53). A comparison of the intraphagosomal expression profile of MTB with those of E. coli (54) and Salmonella enterica (55) points to differences in the environments encountered by these microbes within host cells. The oxidative stress response of MTB in the macrophage phagosome is induced by NO and thus differs markedly from the phagocyte oxidase–driven response of E. coli in the neutrophil (54). The carbon and energy metabolism of intraphagosomal MTB seems to depend on the β-oxidation of fatty acids. This is fundamentally different from the proposed use of gluconate by intraphagosomal S. enterica. S. enterica furthermore does not perceive its macrophage phagosome as a low iron environment (55). These differences suggest that the kinds of nutrients found in the phagosome are also subject to pathogen manipulation. Collectively, these differences likely account in part for the dramatic differences in the natural histories and pathogenesis of these infectious agents.
Acknowledgments
We thank Kevin Visconti, Kimberly Chong, and Sangeeta Balakrishnan for assistance with microarray work and Drs. Ken Duncan and Graham Stewart for sharing microarray data.
This work was supported by the Action TB Program (G.K. Schoolnik), National Institutes of Health grants AI 44826 (to G.K. Schoolnik), HL61241 (to C. Nathan), and HL68525 (to S. Ehrt), grant BAA-00-33 from the Defense Advanced Research Projects Agency (to G.K. Schoolnik), the British Medical Research Council Cooperative Group grant G9800300 (to P.D. Butcher), and a postdoctoral fellowship from the Deutsche Forschungsgemeinschaft (to D. Schnappinger). The Department of Microbiology & Immunology acknowledges the support of the William Randolph Hearst Foundation.
Abbreviations used in this paper: CoA, coenzyme A; MTB, Mycobacterium tuberculosis; NDH, NADH dehydrogenase; NO, nitric oxide; NOS2, NO synthase 2; qrtPCR, quantitative real time reverse transcription-PCR.
The online version of this article contains supplemental material.
References
- 1.Aderem, A., and D.M. Underhill. 1999. Mechanisms of phagocytosis in macrophages. Annu. Rev. Immunol. 17:593–623. [DOI] [PubMed] [Google Scholar]
- 2.Nathan, C., and M.U. Shiloh. 2000. Reactive oxygen and nitrogen intermediates in the relationship between mammalian hosts and microbial pathogens. Proc. Natl. Acad. Sci. USA. 97:8841–8848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Finlay, B.B., and S. Falkow. 1997. Common themes in microbial pathogenicity revisited. Microbiol. Mol. Biol. Rev. 61:136–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Amer, A.O., and M.S. Swanson. 2002. A phagosome of one's own: a microbial guide to life in the macrophage. Curr. Opin. Microbiol. 5:56–61. [DOI] [PubMed] [Google Scholar]
- 5.Russell, D.G. 2001. Mycobacterium tuberculosis: here today, and here tomorrow. Nat. Rev. Mol. Cell Biol. 2:569–577. [DOI] [PubMed] [Google Scholar]
- 6.Schaible, U.E., S. Sturgill-Koszycki, P.H. Schlesinger, and D.G. Russell. 1998. Cytokine activation leads to acidification and increases maturation of Mycobacterium avium-containing phagosomes in murine macrophages. J. Immunol. 160:1290–1296. [PubMed] [Google Scholar]
- 7.Flynn, J.L., and J. Chan. 2001. Immunology of tuberculosis. Annu. Rev. Immunol. 19:93–129. [DOI] [PubMed] [Google Scholar]
- 8.Wayne, L.G., and C.D. Sohaskey. 2001. Nonreplicating persistence of Mycobacterium tuberculosis. Annu. Rev. Microbiol. 55:139–163. [DOI] [PubMed] [Google Scholar]
- 9.Glickman, M.S., and W.R. Jacobs, Jr. 2001. Microbial pathogenesis of Mycobacterium tuberculosis: dawn of a discipline. Cell. 104:477–485. [DOI] [PubMed] [Google Scholar]
- 10.Ehrt, S., D. Schnappinger, S. Bekiranov, J. Drenkow, S. Shi, T.R. Gingeras, T. Gaasterland, G. Schoolnik, and C. Nathan. 2001. Reprogramming of the macrophage transcriptome in response to interferon γ and Mycobacterium tuberculosis: signaling roles of nitric oxide synthase-2 and phagocyte oxidase. J. Exp. Med. 194:1123–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Butcher, P.D., J. Mangan, and I.M. Monahan. 1998. Intracellular gene expression: analysis of RNA from mycobacteria in macrophages using RT-PCR. Mycobacteria Protocols. T. Parish and N.G. Stoker, editors. Humana Press, Totowa. 285–306. [DOI] [PubMed]
- 12.Mangan, J.A., I.M. Monahan, and P.D. Butcher. 2002. Gene expression during host-pathogen interactions: approaches to bacterial mRNA extraction and labeling for microarray analysis. Functional Microbial Genomics. B.W. Wren and N. Dorrell, editors. Academic Press, London. 137–151.
- 13.Ehrt, S., M. Voskuil, G.K. Schoolnik, and D. Schnappinger. 2002. Genome-wide expression profiling of intracellular bacteria: the interaction of Mycobacterium tuberculosis with macrophages. Immunology of Infection. S.H.E. Kaufmann and D. Kabelitz, editors. Academic Press, London. 169–180.
- 14.Tusher, V.G., R. Tibshirani, and G. Chu. 2001. Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl. Acad. Sci. USA. 98:5116–5121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Manganelli, R., E. Dubnau, S. Tyagi, F.R. Kramer, and I. Smith. 1999. Differential expression of 10 sigma factor genes in Mycobacterium tuberculosis. Mol. Microbiol. 31:715–724. [DOI] [PubMed] [Google Scholar]
- 16.Hu, C., T. Mayadas-Norton, K. Tanaka, J. Chan, and P. Salgame. 2000. Mycobacterium tuberculosis infection in complement receptor 3-deficient mice. J. Immunol. 165:2596–2602. [DOI] [PubMed] [Google Scholar]
- 17.Bekker, L.G., S. Freeman, P.J. Murray, B. Ryffel, and G. Kaplan. 2001. TNF-alpha controls intracellular mycobacterial growth by both inducible nitric oxide synthase-dependent and inducible nitric oxide synthase-independent pathways. J. Immunol. 166:6728–6734. [DOI] [PubMed] [Google Scholar]
- 18.Clemens, D.L., B.Y. Lee, and M.A. Horwitz. 2002. The Mycobacterium tuberculosis phagosome in human macrophages is isolated from the host cell cytoplasm. Infect. Immun. 70:5800–5807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eisen, M.B., and P.O. Brown. 1999. DNA arrays for analysis of gene expression. Methods Enzymol. 303:179–205. [DOI] [PubMed] [Google Scholar]
- 20.Bernstein, J.A., A.B. Khodursky, P.H. Lin, S. Lin-Chao, and S.N. Cohen. 2002. Global analysis of mRNA decay and abundance in Escherichia coli at single-gene resolution using two-color fluorescent DNA microarrays. Proc. Natl. Acad. Sci. USA. 99:9697–9702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McKinney, J.D., K. Honer zu Bentrup, E.J. Munoz-Elias, A. Miczak, B. Chen, W.T. Chan, D. Swenson, J.C. Sacchettini, W.R. Jacobs, Jr., and D.G. Russell. 2000. Persistence of Mycobacterium tuberculosis in macrophages and mice requires the glyoxylate shunt enzyme isocitrate lyase. Nature. 406:735–738. [DOI] [PubMed] [Google Scholar]
- 22.Cox, J.S., B. Chen, M. McNeil, and W.R. Jacobs, Jr. 1999. Complex lipid determines tissue-specific replication of Mycobacterium tuberculosis in mice. Nature. 402:79–83. [DOI] [PubMed] [Google Scholar]
- 23.Camacho, L.R., D. Ensergueix, E. Perez, B. Gicquel, and C. Guilhot. 1999. Identification of a virulence gene cluster of Mycobacterium tuberculosis by signature-tagged transposon mutagenesis. Mol. Microbiol. 34:257–267. [DOI] [PubMed] [Google Scholar]
- 24.De Voss, J.J., K. Rutter, B.G. Schroeder, H. Su, Y. Zhu, and C.E. Barry, III. 2000. The salicylate-derived mycobactin siderophores of Mycobacterium tuberculosis are essential for growth in macrophages. Proc. Natl. Acad. Sci. USA. 97:1252–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pym, A.S., P. Domenech, N. Honore, J. Song, V. Deretic, and S.T. Cole. 2001. Regulation of catalase-peroxidase (KatG) expression, isoniazid sensitivity and virulence by furA of Mycobacterium tuberculosis. Mol. Microbiol. 40:879–889. [DOI] [PubMed] [Google Scholar]
- 26.McAdam, R.A., S. Quan, D.A. Smith, S. Bardarov, J.C. Betts, F.C. Cook, E.U. Hooker, A.P. Lewis, P. Woollard, M.J. Everett, et al. 2002. Characterization of a Mycobacterium tuberculosis H37Rv transposon library reveals insertions in 351 ORFs and mutants with altered virulence. Microbiology. 148:2975–2986. [DOI] [PubMed] [Google Scholar]
- 27.Rindi, L., L. Fattorini, D. Bonanni, E. Iona, G. Freer, D. Tan, G. Deho, G. Orefici, and C. Garzelli. 2002. Involvement of the fadD33 gene in the growth of Mycobacterium tuberculosis in the liver of BALB/c mice. Microbiology. 148:3873–3880. [DOI] [PubMed] [Google Scholar]
- 28.Cole, S.T., R. Brosch, J. Parkhill, T. Garnier, C. Churcher, D. Harris, S.V. Gordon, K. Eiglmeier, S. Gas, C.E. Barry, III, et al. 1998. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 393:537–544. [DOI] [PubMed] [Google Scholar]
- 29.Arruda, S., G. Bomfim, R. Knights, T. Huima-Byron, and L.W. Riley. 1993. Cloning of an M. tuberculosis DNA fragment associated with entry and survival inside cells. Science. 261:1454–1457. [DOI] [PubMed] [Google Scholar]
- 30.Voskuil, M.T., D. Schnappinger, K.C. Visconti, M.I. Harrell, G.M. Dolganov, D.R. Sherman, and G.K. Schoolnik. 2003. Inhibition of respiration by nitric oxide induces a Mycobacterium tuberculosis dormancy program. J. Exp. Med. 198:705–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sherman, D.R., M. Voskuil, D. Schnappinger, R. Liao, M.I. Harrell, and G.K. Schoolnik. 2001. Regulation of the Mycobacterium tuberculosis hypoxic response gene encoding alpha-crystallin. Proc. Natl. Acad. Sci. USA. 98:7534–7539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rodriguez, G.M., M.I. Voskuil, B. Gold, G.K. Schoolnik, and I. Smith. 2002. ideR, an essential gene in Mycobacterium tuberculosis: role of IdeR in iron-dependent gene expression, iron metabolism, and oxidative stress response. Infect. Immun. 70:3371–3381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fisher, M.A., B.B. Plikaytis, and T.M. Shinnick. 2002. Microarray analysis of the Mycobacterium tuberculosis transcriptional response to the acidic conditions found in phagosomes. J. Bacteriol. 184:4025–4032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Betts, J.C., P.T. Lukey, L.C. Robb, R.A. McAdam, and K. Duncan. 2002. Evaluation of a nutrient starvation model of Mycobacterium tuberculosis persistence by gene and protein expression profiling. Mol. Microbiol. 43:717–731. [DOI] [PubMed] [Google Scholar]
- 35.Stewart, G.R., L. Wernisch, R. Stabler, J.A. Mangan, J. Hinds, K.G. Laing, D.B. Young, and P.D. Butcher. 2002. Dissection of the heat-shock response in Mycobacterium tuberculosis using mutants and microarrays. Microbiology. 148:3129–3138. [DOI] [PubMed] [Google Scholar]
- 36.Manganelli, R., M.I. Voskuil, G.K. Schoolnik, E. Dubnau, M. Gomez, and I. Smith. 2002. Role of the extracytoplasmic-function sigma factor sigma(H) in Mycobacterium tuberculosis global gene expression. Mol. Microbiol. 45:365–374. [DOI] [PubMed] [Google Scholar]
- 37.Manganelli, R., M.I. Voskuil, G.K. Schoolnik, and I. Smith. 2001. The Mycobacterium tuberculosis ECF sigma factor sigmaE: role in global gene expression and survival in macrophages. Mol. Microbiol. 41:423–437. [DOI] [PubMed] [Google Scholar]
- 38.Bryk, R., P. Griffin, and C. Nathan. 2000. Peroxynitrite reductase activity of bacterial peroxiredoxins. Nature. 407:211–215. [DOI] [PubMed] [Google Scholar]
- 39.Quadri, L.E., J. Sello, T.A. Keating, P.H. Weinreb, and C.T. Walsh. 1998. Identification of a Mycobacterium tuberculosis gene cluster encoding the biosynthetic enzymes for assembly of the virulence-conferring siderophore mycobactin. Chem. Biol. 5:631–645. [DOI] [PubMed] [Google Scholar]
- 40.Ding, H., and B. Demple. 2000. Direct nitric oxide signal transduction via nitrosylation of iron-sulfur centers in the SoxR transcription activator. Proc. Natl. Acad. Sci. USA. 97:5146–5150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Touati, D. 2000. Iron and oxidative stress in bacteria. Arch. Biochem. Biophys. 373:1–6. [DOI] [PubMed] [Google Scholar]
- 42.Russell, D.G., H.C. Mwandumba, and E.E. Rhoades. 2002. Mycobacterium and the coat of many lipids. J. Cell Biol. 158:421–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Camacho, L.R., P. Constant, C. Raynaud, M.A. Laneelle, J.A. Triccas, B. Gicquel, M. Daffe, and C. Guilhot. 2001. Analysis of the phthiocerol dimycocerosate locus of Mycobacterium tuberculosis. Evidence that this lipid is involved in the cell wall permeability barrier. J. Biol. Chem. 276:19845–19854. [DOI] [PubMed] [Google Scholar]
- 44.Glickman, M.S., S.M. Cahill, and W.R. Jacobs, Jr. 2001. The Mycobacterium tuberculosis cmaA2 gene encodes a mycolic acid trans-cyclopropane synthetase. J. Biol. Chem. 276:2228–2233. [DOI] [PubMed] [Google Scholar]
- 45.Honer zu Bentrup, K., and D.G. Russell. 2001. Mycobacterial persistence: adaptation to a changing environment. Trends Microbiol. 9:597–605. [DOI] [PubMed] [Google Scholar]
- 46.Horswill, A.R., and J.C. Escalante-Semerena. 2001. In vitro conversion of propionate to pyruvate by Salmonella enterica enzymes: 2-methylcitrate dehydratase (PrpD) and aconitase enzymes catalyze the conversion of 2-methylcitrate to 2-methylisocitrate. Biochemistry. 40:4703–4713. [DOI] [PubMed] [Google Scholar]
- 47.Graham, J.E., and J.E. Clark-Curtiss. 1999. Identification of Mycobacterium tuberculosis RNAs synthesized in response to phagocytosis by human macrophages by selective capture of transcribed sequences (SCOTS). Proc. Natl. Acad. Sci. USA. 96:11554–11559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dubnau, E., P. Fontan, R. Manganelli, S. Soares-Appel, and I. Smith. 2002. Mycobacterium tuberculosis genes induced during infection of human macrophages. Infect. Immun. 70:2787–2795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gennis, R.B., and V. Stewart. 1996. Respiration. Escherichia coli and Salmonella. Cellular and Molecular Biology. F.C. Neidhardt, R. Curtiss, J.L. Ingraham, E.C.C. Lin, K.B. Low, B. Magasanik, W.S. Reznikoff, M. Riley, M. Schaechter, and H.E. Umbarger, editors. ASM Press, Washington, D.C. 217–261.
- 50.Hutter, B., and M. Singh. 1999. Properties of the 40 kDa antigen of Mycobacterium tuberculosis, a functional L-alanine dehydrogenase. Biochem. J. 343:669–672. [PMC free article] [PubMed] [Google Scholar]
- 51.Pathania, R., N.K. Navani, A.M. Gardner, P.R. Gardner, and K.L. Dikshit. 2002. Nitric oxide scavenging and detoxification by the Mycobacterium tuberculosis haemoglobin, HbN in Escherichia coli. Mol. Microbiol. 45:1303–1314. [DOI] [PubMed] [Google Scholar]
- 52.Boon, C., and T. Dick. 2002. Mycobacterium bovis BCG response regulator essential for hypoxic dormancy. J. Bacteriol. 184:6760–6767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Knodler, L.A., J. Celli, and B.B. Finlay. 2001. Pathogenic trickery: deception of host cell processes. Nat. Rev. Mol. Cell Biol. 2:578–588. [DOI] [PubMed] [Google Scholar]
- 54.Staudinger, B.J., M.A. Oberdoerster, P.J. Lewis, and H. Rosen. 2002. mRNA expression profiles for Escherichia coli ingested by normal and phagocyte oxidase-deficient human neutrophils. J. Clin. Invest. 110:1151–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Eriksson, S., S. Lucchini, A. Thompson, M. Rhen, and J.C. Hinton. 2003. Unravelling the biology of macrophage infection by gene expression profiling of intracellular Salmonella enterica. Mol. Microbiol. 47:103–118. [DOI] [PubMed] [Google Scholar]