

Abstract
The insulin/interleukin-4 (IL-4) receptor (I4R) motif mediates the association of insulin receptor substrate (IRS)-2 with the interleukin-4 (IL-4)Rα chain and transduces mitogenic signals in response to IL-4. Its physiological functions were analyzed in mice with a germline point mutation that changed the motif's effector tyrosine residue into phenylalanine (Y500F). The Y500F mutation abrogated IRS-2 phosphorylation and impaired IL-4–induced CD4+ T lymphocyte proliferation but left unperturbed Stat6 activation, up-regulation of IL-4-responsive gene products, and Th cell differentiation under Th2 polarizing conditions. However, in vivo the Y500F mutation was associated with increased allergen-induced IgE production, airway responsiveness, tissue eosinophilia, and mucus production. These results define an important role for the I4R motif in regulating allergic inflammation.
Keywords: interleukin 4 receptor, targeted mutagenesis, immunoglobulin E, insulin receptor substrate, asthma
Introduction
Interleukin-4 (IL-4) plays a key role in the development of Th2 responses and the evolution of immediate hypersensitivity reactions. IL-4 promotes the proliferation and survival of lymphocytes, Th2 lineage commitment of CD4+ T helper cells, and immunoglobulin isotype switching to IgE. It also exerts direct effects on antigen-presenting cells and on nonhematopoietic cells such as airway epithelial and smooth muscle cells. The actions of IL-4 are mediated by heterodimeric receptor complexes that have in common a 140-kD α subunit (IL-4Rα) that binds to and transduces growth-promoting and transcription-activating functions of IL-4 (1–3). In hematopoietic cells, IL-4Rα associates with the γc chain, a subunit common to several cytokine receptors (4–6; for review see reference 7). IL-4Rα also pairs with the IL-13Rα1, which is expressed on hematopoietic and nonhematopoietic cells to mediate signaling by both IL-4 and IL-13 (8). Targeted disruption of the IL-4Rα subunit in mice abrogates the IgE response, consistent with its central role in mediating allergic inflammation (9).
IL-4Rα, γc, and IL-13Rα1 physically associate with specific members of the Janus family of protein kinases, Jak1, Jak3, and Jak2 orTyK2, respectively, that serve to couple ligand binding to intracellular activation events (10). Binding of IL-4 to IL-4Rα chain is followed by receptor heterodimerization and activation of Jak kinases by transphosphorylation (11, 12). Activated Jak kinases mediate the phosphorylation of the cytoplasmic tail of IL-4Rα on conserved tyrosine residues that serve as docking sites for downstream Src homology domain (SH)2 and phosphotyrosine-binding domain (PTB) signaling proteins. Three closely clustered tyrosine residues serve as docking sites for Stat6, an SH2 domain–containing transcription factor selectively coupled to the IL-4R, which is indispensable for the induction of gene expression by IL-4, including the γ1 and ɛ heavy chain germline transcription (13). A distal tyrosine residue at the COOH terminus of IL-4Rα defines an immunorecptor tyrosine-based inhibitory motif (ITIM) that binds SH2 domains of several phosphatases, including the SH2-containing tyrosine phosphatase (SHP)-1 and SHP-2 and inositol 5′ phosphatase (14, 15).
The sequence surrounding the proximal tyrosine 500 (Y500) residue in the cytoplasmic tail of the murine IL-4α chain defines a motif that is highly homologous with sequences in the insulin and insulin-like growth factor 1 receptors (16). This motif, referred to as the insulin/IL-4 receptor (I4R) motif, interacts upon its phosphorylation at Y500 with several PTB domain–containing adaptor proteins that couple the IL-4Rα to downstream signaling cascades. In hematopoietic cells, the insulin receptor substrate (IRS)-2 is the most prominent phospho-I4R substrate (17, 18). However, phospho-I4R also interacts with other PTB-containing adaptors including IRS-1 (16), FRIP/Dok-R (19), and Shc (20). Recruitment of IRS-2 to IL-4Rα leads to IRS-2 phosphorylation, the subsequent recruitment and activation of PI3-K, and the activation of the down-stream protein kinase Akt (17, 21–23). Studies using mutant IL-4Rα chains transfected into cells lines and IRS-2–deficient lymphocytes have implicated the I4R motif and the IRS-2 signaling pathway in transmitting mitogenic signals in response to IL-4 (16, 18, 23, 24). Furthermore, a role for this motif in human allergic disorders has been suggested by a polymorphism in the human receptor, S503P, that strongly associates with atopy, asthma, and airway hyperresponsiveness (AHR) (25–27). In this study, we addressed the role of the I4R motif in IL-4 receptor signaling in vivo by introducing a germline mutation in the murine IL-4Rα gene that results in replacement of Y500 with a phenylalanine.
Materials and Methods
Construction of Targeting Vector and Generation of Mutant Mice.
Genomic DNA used to construct the targeting vector were isolated from a bacterial artificial chromosome clone containing the murine IL-4Rα gene (Genome Systems) (28). The replacement-type targeting vector (pKO; Lexicon) contained 13.5 kb of IL4RA sequences that extended from a XhoI site proximal to exon 9 to a KpnI site distal to exon 12 of the gene. A single A→T substitution was introduced by site-directed mutagenesis (QuickChange; Stratagene) at the second position of the tyrosine 500 codon (TAC), corresponding to position 1735 of murine IL-4Rα cDNA (1). The mutation changed the codon specificity from tyrosine to phenylalanine (TTC). Except for the Y500F mutation, the exonic sequences in the targeting vector were confirmed by DNA sequencing to be identical to those of the native allele. Positive selection of targeted clones was provided by a floxed PGK-neo cassette that was subcloned into an EcoRI site engineered by site-directed mutagenesis within intron 11 73 bp upstream of exon 12. The targeting vector also included a Diphtheria toxin gene (pKO SelectDT V840; Lexicon) to select against random integration.
Targeting plasmids were introduced by electroporation into RW4 embryonic stem (ES) cells and subjected to G418 selection. Homologous recombination was ascertained by Southern blotting using a probe corresponding to exon 4 of IL4RA, which lies 5′ to the homology sequence. Neomycin gene insertion is associated with the introduction of a new EcoRI site, leading to a decrease in the size of the genomic EcoRI fragment that normally hybridizes with the probe from 12.5 kb to 8.5 kb (see Fig. 1). Successfully targeted clones were transiently transfected with a Cre recombinase to remove the inserted neomycin gene, leaving in place one loxP sequence flanked by EcoRI sites.
Figure 1.
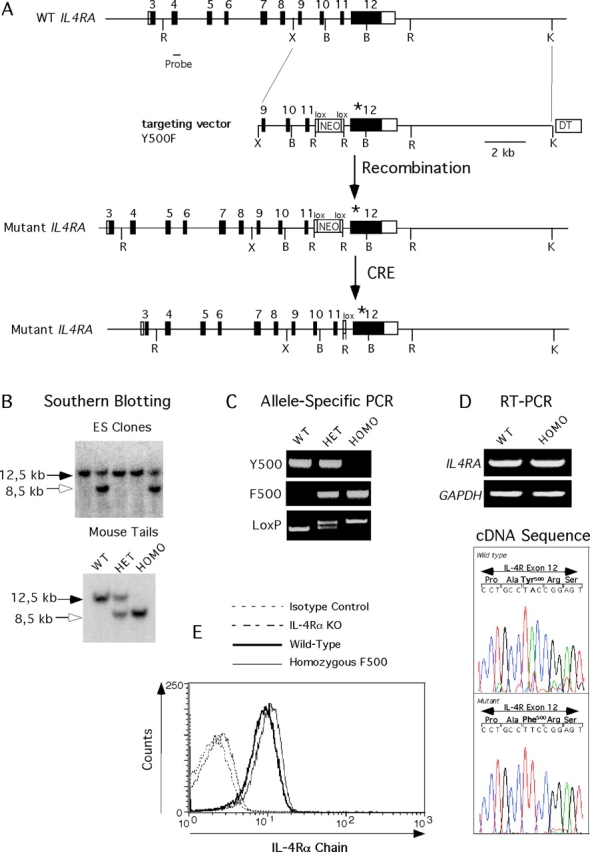
Targeted knock-in mutagenesis of murine IL-4Rα gene. (A) Targeting strategy. WT exon 12 was replaced with one carrying an A to T substitution in the second position of codon 500 in exon 12 of IL4RA, changing its specificity from a tyrosine (Y) to a phenylalanine (F). The mutant exon is marked with an asterisk. Successful targeting introduced within intron 11 a floxed Neo cassette flanked by EcoRI sites. Subsequent treatment with Cre recombinase removed the neo cassette, leaving one loxP sequence and the EcoRI site in place. Restriction enzyme abbreviations: B, BamHI; R, EcoRI; and X, XhoI. □ and ▪ denote no-coding and coding exonic sequences, respectively. (B) Southern blot analysis of EcoRI-digested mouse tail genomic DNA of WT, heterozygous (HET), and homozygous (HOMO) mutant mice. The introduction of a novel EcoRI site in intron 11 of targeted IL4A reduces the size of an EcoRI genomic fragment detected with an exon 4 probe from 12.5 kb in the WT allele to 8.5 kb in the mutant allele. (C) PCR analysis of mouse tail DNA using Y500 and F500 allele-specific primers (top and middle, respectively) or primers spanning the residual 34-bp loxP site left in intron 11 after Cre-mediated excision of the floxed Neo cassette. (D, top) RT-PCR analysis of IL-4Rα mRNA expression in splenocytes of WT and Y500F homozygous mutant mice. Amplified GAPDH transcripts were used as controls. (Bottom) Sequence analysis of RT-PCR products of IL-4Rα transcripts of WT and Y500F mutant mice. (E) Flow cytometric analysis of IL-4Rα expression in IL-4RαY500F lymphocytes. Splenic B cells of WT, IL-4Rα knock-out, and IL-4Rα Y500F mice were stained with PE-conjugated rat antimurine IL-4Rα mAb (mIL4R-M1). Control staining was performed on WT splenic B cells using a PE-conjugated, isotype-matched rat IgG2a antibody.
Heterozygous ES cells were injected into C57BL/6 blastocysts, and resultant male chimeras were mated with BALB/c females. Offspring were screened for heterozygotes by Southern blotting and PCR analysis. Heterozygotes were further bred for 8–10 generations on BALB/c background. Homozygous mutant mice and WT littermate controls were generated by mating Het parents. WT BALB/c and IL-4Rα knock-out mice (BALB/c-Il4ra tm 1Sz) were obtained from the Jackson laboratory. All protocols were in accordance with NIH guidelines and approved by the Animal Care and Use Committee of Washington University School of Medicine.
PCR Analysis.
Screening of WT and mutant IL4RA alleles was performed by PCR amplification using genomic DNA and the following allele-specific forward (F) primers: 5′-TTGCAGACAATCCTGCCTA-3′ (WT-specific) and 5′-TTGCAGACAATCCTGCCTT-3′ (mutant–specific), and a common reverse (R) primer: 5′-ACTGCCTGCACAAACTCCT-3′. Primers used for PCR screening of the residual LoxP-containing allele were: F, 5′-GGTGTCTATTTTAGGTGCC-3′ and R, 5′-TCTTCTCTCTTACTCTGTGCT-3′.
For RT-PCR analysis, total RNA was extracted from splenocytes of WT and IL-4Rα Y500F mutant mice using TRIzol (GIBCO BRL). The RNA was treated with Dnase I to remove residual genomic DNA contamination and then reverse transcribed. IL4RA transcripts were amplified by a two step process using the following pairs of nested primers: outer pair F (exon 11), 5′-CAGACCCGAAGCCAGGAGTCAACC-3′ and R (exon 12), 5′-CCCTGCTTCACTGCCTGCACAAAC-3′; inner pair F (both exon 12), 5′-GAGCAGCCTTCACACCAG-3′ and R, 5′-ACTGCCTGCACAAACTCCT. For GAPDH transcripts, the primers were F, 5′-ACCACAGTCCATGCCATCAC-3′ and R, 5′-TCCACCACCCTGTTGCTGTA-3′. For class switch recombination (CSR) studies, RNA was isolated from splenocytes that have been either left untreated or treated for 48 h with IL-4 at 50 ng/ml (R&D Systems) or with anti-CD40 mAb (clone HM40–3; BD Biosciences) plus IL-4. Primers used for RT-PCR amplification of germline, postswitch, and activation-induced cytidine deaminase (AID) transcripts were described previously (29). GAPDH transcripts were amplified as an internal control using the following primers: F, 5′-ACCACAGTCCATGCCATCAC-3′ and R, 5′-TCCACCACCCTGTTGCTGTA-3′. All PCR reactions were performed on various dilutions of cDNA to ensure that the products measured were in the linear range. Depending on the PCR product being measured, 5–30 ng of cDNA fell within this range.
Digestion-circularization (DC)–PCR.
Genomic DNA was isolated from cultured splenic B cells stimulated for 6 d with anti-CD40 mAb+ IL-4. DNA was digested with EcoR1, circularized, and used as template for PCR using primers as reported previously for Sμ-Sγ1 and nicotinic acetylcholine receptor unit (AChR) (30) and for Sμ-Sɛ (31).
Flow Cytometry Analysis.
Single cell suspensions were stained with the indicated antibodies and analyzed on a FACSCalibur® cytometer (Becton Dickinson). FITC- or PE-conjugated mAbs used were anti-TCRβ (H57), CD3 (145–2C11), CD4 (L3T4), CD8 (53–6.7), CD23 (B3B4), CD124 (mIL4R-M1), B220 (RA3–6B2), and anti I-Ad (AMS-32.1) (BD Biosciences).
Lymphocyte Cultures and In Vitro Th Cell Differentiation.
Splenocytes were fractionated into CD4+ T cells or B cells by magnetic-activated cell sorting using isolation kits for the respective lymphocyte population (Miltenyi Biotec). Purified B cells were >97% B220+ and <1% TCRβ+, and CD4+ T cells were >85% TCRβ+/CD4+ and <1% B220+. T and B lymphocyte cultures (5 × 105 cells/well in 96-well flat-bottomed plates) were treated with the indicated mitogens for 48 or 72 h, respectively. The following mitogens were used: anti-TCRβ mAb and anti-CD40 mAb (BD Biosciences), F(ab′)2 goat anti–mouse IgM antibodies (Jackson ImmunoResearch Laboratories), PMA and ionomycin (Sigma-Aldrich), recombinant IL-4 (Peprotech), and IL-2 (Biological Resources Branch, National Cancer Institute). The cultures were pulsed with 0.4 μCi/well of 3H-TdR for 18 h and then harvested. Proliferation was measured as counts per minute incorporated.
T cell blasts were generated by culturing splenocytes on plates coated with anti-TCRβ mAB (10 μg/ml) for 3 d followed by expansion of cell cultures with IL-2 (100 U/ml) for 7 d. Induction of in vitro IgE production was accomplished by culturing splenocytes for 7 d with anti-CD40 mAB in the presence of IL-4 at 100 ng/ml. For in vitro Th cell differentiation, splenocytes were cultured for 3 d on anti-TCRβ–coated plates in the presence of IL-4 (100 ng/ml) and anti-IFN-γ mAb (10 μg/ml) (Th2 conditions) or IL-12 (1 ng/ml) and anti–IL-4 mAb (10 μg/ml) (Th1 conditions) (R&D Systems). The cultures were expanded for an additional 4 d with IL-2 (100 U/ml) after which the cells were washed and restimulated with plate-coated anti-TCRβ mAb for 48 h. Culture supernatants were assayed for IL-4, IL-5, IL-13, and IFN-γ by ELISA (IL-4 and IFN-γ; PharMingen and IL-5 and IL-13; R&D Systems).
Immunoblotting and Immunoprecipitation.
Cells were stimulated with IL-4 (100 ng/ml) for the indicated periods of time and then lysed with either 2× Laemmli gel loading buffer or 0.5% NP-40 detergent buffer (32). Whole cell lysates or immunoprecipitates derived from NP-40 detergent extracts were resolved by SDS/PAGE, transferred to nitrocellulose filters, and immunoblotted with the respective antibodies: anti-Dok1, Dok-R, IL-4Rα, Shc and Stat6, (Santa Cruz Biotechnology, Inc.), anti–phospho-Akt (serine 473), Akt, phospho-Shc (tyrosine 239/240), phospho–Stat-6 (tyrosine 641), and phosphotyrosine (Cell Signal), anti–phospho–Jak-1 (tyrosine 1022/1023) and Jak-1 (Sigma-Aldrich), and anti–IRS-2 (Upstate Biotechnology). The blots were developed using horseradish peroxidase–conjugated secondary antibodies and enzyme-linked chemiluminescence (ECL; Amersham Biosciences).
Immunizations.
Mice were injected i.p. on day 0 with 100 μg OVA in alum together with 300 ng of pertussis toxin in a final volume of 100 μl. They were reimmunized on day 14 with 100 μg OVA in alum. Saline control groups of mice were similarly treated except for inclusion of saline in place of OVA.
Measurement of Immunoglobulin Levels.
Total immunoglobulin levels were determined by ELISA. The standard BD Biosciences protocol for sandwich ELISA was used to quantify OVA-specific IgE Abs (33). Results of OVA-specific IgE were expressed as ng per milliliter by comparison with a standard consisting of purified mouse OVA-specific IgE secreted by the hybridoma TOɛ, a gift from Mamoru Kiniwa (Immunology Research Laboratory, Hanno Research Center, Taiho Pharmaceutical Co. Ltd.). For OVA-specific antibodies of other isotypes, pooled sera from five mice that had been immunized i.p. with OVA were aliquoted and used as an ELISA reference. For each IgG subclass, a dilution of this standard serum was prepared and arbitrarily assigned a concentration of 1 U/ml. Standard curves constructed using this reference were used to calculate the amounts of OVA-specific antibody in test samples.
Analysis of Bronchoalveolar Lavage Fluid.
Cells in the airways were recovered by flushing 0.8 ml of bronchoalveolar lavage (BAL) fluid (1 mM EDTA, 10% FCS, PBS) into the lungs via the trachea. Total cell counts were determined, and 100 μl of fluid was cytospun onto glass slides. Differential cell counts were performed after staining with Diff-Quik Stain Set (Baxter Healthcare Corp.) (34).
Measurement of Airway Hyperresponsiveness.
OVA-immunized mice were exposed to aerosolized OVA (1% in saline for 20 min by ultrasonic nebulization) on days 28, 29, and 30, and their lung function was tested on day 31 after their initial immunization. For IL-4 and IL-13 instillation, 5 μg of the respective cytokine in 50 μl of PBS buffer were administered daily for three consecutive days. Control instillation was performed using 5 μg BSA in 50 μl PBS. Lung function was measured 24 h after the last cytokine or BSA instillation. Enhanced pause (Penh) was measured using whole body plethysmography (BUXCO) (34). Increases in airway resistance to aerosolized methacholine were determined as Penh values.
Statistical Analysis.
Student's t test was used to compare the groups of mice. Where appropriate, Penh results were analyzed by two-way ANOVA. A P value smaller than 0.05 was considered statistically significant.
Results
Generation of IL-4Rα Y500F Mutant Mice by Targeted Knock-in Mutagenesis.
A targeting construct was designed to replace exon 12 of IL4RA of ES cells with another bearing an A→T single base pair substitution at the second position of the Y500 codon that changed the codon specificity from tyrosine to phenylalanine (28) (Fig. 1 A). This mutation has previously been demonstrated to abrogate pY500-dependent binding of PTB domain–containing adaptor proteins to the I4R motif and consequently to uncouple downstream signaling pathways activated via pY500 (16, 18). Successfully targeted ES clones were used to derive murine male chimera that transmitted the Y500F mutation into the germline (Fig. 1 B). Matings of IL-4Rα Y500F heterozygotes resulted in generation of Y500F homozygous mutant mice in numbers consistent with autosomal recessive Mendelian inheritance. Homozygous mutant mice of both sexes were phenotypically indistinguishable from their WT and heterozygous littermates. The transmission and integrity of the mutant allele was verified by several methods including Southern blot analysis, allele-specific PCR amplification, and direct sequencing of genomic DNA (Fig. 1, B and C).
RT-PCR analysis revealed that the mutant allele was transcribed at levels equivalent to those of the WT allele (Fig. 1 D). The presence of the A→T substitution in mutant transcripts was directly confirmed by sequencing of the RT-PCR products. (Fig. 1 D). Expression of mutant receptor protein was verified by flow cytometry to be closely matched to WT controls (Fig. 1 E). Analysis revealed the T and B lymphocytes of the IL-4Rα Y500F mutant mice to be normal in number and phenotype (unpublished data).
The Y500F Mutation Abrogates IRS-2 Activation.
The impact of the Y500F mutation of IL-4Rα signaling events was examined in T and B lymphocyte populations treated with IL-4. Treatment of splenic T lymphoblasts with IL-4 revealed that Jak1 kinase activation was normally induced (Fig. 2 A). However, phosphorylation of IL-4Rα was markedly decreased in the Y500F mutant lymphocytes compared with WT controls, consistent with Y500 being a major target of phosphorylation by Jak kinases (Fig. 2 A). Significantly, the Y500F mutation abrogated the tyrosine phosphorylation of IRS-2 in response to IL-4 treatment, indicating failure to recruit IRS-2 to the mutant receptor (Fig. 2 B). In contrast, IL-4–induced phosphorylation of Dok-R (FRIP), a PTB domain protein reported previously to associate with phospho-Y500/I4R (19), was unaffected. This indicated that phosphorylation of Dok-R by IL-4 can proceed in a phospho-Y500–independent manner. IRS-2 phosphorylation results in the recruitment and activation of phosphatidylinositol 3′-kinase (PI3-K) and activation of the downstream kinase Akt, which play an important role in IL-4–induced proliferation (35). The latter event is associated with the phosphorylation of Akt on serine 473. In Y500F T lymphocytes, Akt activation by IL-4 was abrogated as evidenced by the failure of IL-4 treatment to induce Akt phosphorylation on serine 473 (Fig. 2 C). In contrast to the failure to activate the IRS-2/Akt pathway in Y500F, activation of Stat-6 was unaffected. Tyrosine phosphorylation of Stat-6 in response to IL-4 proceeded equally well in WT and mutant T lymphocytes (Fig. 2 D).
Figure 2.

The Y500F mutation selectively impairs activation of the IRS-2/AKT signaling cascade. (A–D) IL-4–induced signaling events in WT and Y500F T cells. (A) Jak1 and IL-4Rα tyrosine phosphorylation. (Top) Lysates of T cell lymphoblasts were directly blotted with an anti–phospho-Jak1 antibody (pJak1) then reprobed with an anti-Jak1 protein antibody. (Bottom) IL-4Rα immunoprecipitates were probed with an antiphosphotyrosine antibody (pY-IL-4Rα) then reprobed with an anti–IL-4Rα antibody. The IL-4Rα Y500 lysates were overloaded to reveal low level phosphorylation of mutant IL-4Rα in response to IL-4 treatment. (B) IRS-2 and Dok-R tyrosine phosphorylation. IRS-2 and Dok-R immunoprecipitates were probed with an antiphosphotyrosine antibody followed by anti–IRS-2 and anti–Dok-R antibodies, respectively. (C) Akt phosphorylation. Cell lysates were directly blotted with an anti–phospho-Akt serine 473 antibody (pAkt) and then reprobed with an anti-Akt protein antibody. (D) Time course of Stat6 phosphorylation. Stat6 immunoprecipitates were probed with an anti–phospho-Stat6 antibody (pY-Stat6) and then reprobed with an anti-Stat6 antibody. (E–H) IL-4–induced signaling events in WT and Y500F splenic B cells. (E) Tyrosine phosphorylation of IRS-2 (left) and Dok1 (right). (F) p46Shc phosphorylation. Lysates were probed with anti–phospho-Shc antibody and then reprobed with an anti-Shc antibody. (G) Akt phosphorylation on serine 473. (H) Time course of Stat6 phosphorylation.
Impairment of the IRS-2 signaling cascade was also noted in IL-4–treated Y500F primary splenic B cells in which induction of IRS-2 phosphorylation by IL-4 was abolished (Fig. 2 E). The specificity of this finding was demonstrated using another PTB domain adaptor protein, Dok1, which was found constitutively phosphorylated in B cells. When normalized for protein content, its phosphorylation level was marginally increased after IL-4 treatment but without appreciable difference between WT and mutant B cells (Fig. 2 E).
B cells express the p46/p52 isoforms of Shc, a PTB domain–containing adaptor that exhibits low affinity binding to IL-4Rα pY500 relative to IRS proteins (36). IL-4 treatment induces Shc phosphorylation in some tissues including B cells (37), prompting examination of Shc phosphorylation in response to IL-4 treatment of WT and Y500F B cells. In WT B cells, IL-4 treatment induced sustained phosphotyrosine phosphorylation of the p46 isoform (Fig. 2 F). IL-4 also induced p46 Shc phosphorylation in Y500F mutant B cells but with reduced intensity. In contrast, the p52 isoform was constitutively phosphorylated in both cell types and was not affected by IL-4 treatment. These results indicated partial dependence of p46 Shc phosphorylation by IL-4 on the Y500 residue.
Failure of IL-4 treatment to induce IRS-2 phosphorylation in Y500F mutant B cells was associated with abrogation of downstream Akt activation (Fig. 2 G). In contrast, induction by IL-4 of Stat-6 phosphotyrosine phosphorylation was not impaired (Fig. 2 H). These results established that in both T and B lymphocytes the Y500F mutation abrogated the activation by IL-4 of the IRS-2–dependent signaling cascade, whereas sparing other cascades activated via IL-4Rα.
The Y500F Mutation Impairs IL-4–induced CD4+ T Cell Proliferation.
The role of the Y500 residue in transducing mitogenic signals in lymphocytes was examined in splenic CD4+ T cells and B cells of WT and Y500F mutant mice. Fig. 3 A shows that in contrast to WT CD4+ T cells, which exhibited a modest proliferative response to IL-4, the response of mutant T cells was substantially decreased. Significantly, the Y500F mutation abrogated the comitogenic function of IL-4 in supporting T cell proliferation induced by anti-TCR antibodies. This effect was specific in that the response to stimulation with anti-TCR antibodies and IL-2 or with phorbol ester PMA and the calcium ionophore ionomycin was not affected.
Figure 3.

Effect of the Y500F mutation on IL-4–induced lymphocyte proliferation and gene expression. (A) T cell proliferation. (Left) CD4+ splenic T cells of WT and homozygous Y500F littermate mice were cultured with plate-bound anti-TCRβ mAb (100 ng/ml) and IL-4 as indicated. Proliferation was assessed by 3H-thymidine incorporation. Results are mean responses of triplicate cultures ± SE and are representative of three experiments. (Right) Purified CD4+ T cells were cultured with PMA (25 ng/ml) and ionomycin (1 μM) or with plate-bound anti-TCRβ and IL-2 (100 U/ml). (B) B cell proliferation. (Left) Splenic B cells were cultured with anti-CD40 mAb (1 μg/ml) and IL-4 as indicated. (Right) Splenic B cells were cultured with IL-4 (10 ng/ml), anti-IgM antibodies (10 μg/ml), or IL-4 and anti-IgM antibodies as indicated. (C) IL-4–induced up-regulation of CD23 and MHC class II antigens. Splenic B cells were cultured for 48 h in the absence (thin line) or in the presence (thick line) of IL-4 at 50 ng/ml and then examined for CD23 and I-Ad expression by flow cytometry. Results are representative of five experiments.
IL-4 also induced a modest proliferative response in purified B cells that, unlike the case in CD4+ T cells, was not compromised by the Y500F mutation (Fig. 3 B). Furthermore, the Y500F mutation did not impact the capacity of IL-4 to support B cell proliferation induced by B cell mitogens including anti-CD40 and anti-IgM antibodies (Fig. 3 B). These results established that the Y500 pathway is selectively required to support IL-4 mitogenic function in T but not B lymphocytes.
The impact of the Y500F mutation on the up-regulation of classical IL-4 responsive genes was examined by determining the expression levels of CD23 and the MHC class II antigen I-Ad in IL-4–treated WT and Y500F B cells. Fig. 3 C demonstrates that the Y500F mutation had no effect on the up-regulation by IL-4 of CD23 and I-Ad. This is consistent with previous observations that the up-regulation by IL-4 of CD23 and MHC class II antigens proceeds in a Stat-6–dependent manner (38, 39).
Impact of Y500F Mutation on Th Cytokine Production.
The impact of the Y500F mutation on Th cell differentiation was examined by comparing the in vitro differentiation of WT and Y500F T cells into Th1 or Th2 effector lymphocytes in the presence of IL-12 and IL-4, respectively. There was no significant difference in IL-4, IL-5, or IL-13 production between WT and Y500F T cells that have been differentiated under Th2 polarizing conditions and then stimulated by T cell receptor engagement (Fig. 4 A). Similarly, there was no significant difference in IFN-γ production between WT and Y500F T cells that have been differentiated under Th1 conditions. However, the residual production of all three Th2 cytokines was significantly decreased in Y500F T cell cultures that have been differentiated under Th1 polarizing conditions compared with their WT counterparts. This suggests that the Y500 pathway modulates Th cell differentiation by promoting the persistence of Th2 cells in the context of Th1 skewing conditions.
Figure 4.

Impact of the Y500F mutation on Th cytokine production. (A) In vitro Th cell polarization. Naive WT and Y500F splenic T cells were differentiated into Th1 or Th2 cells and then stimulated with anti-TCRβ mAB for 48 h. Culture supernatants were then harvested and examined for IL-4, IL-5, IL-13, and IFN-γ production by ELISA. (B) Cytokine production in response to antigenic stimulation. Splenocytes and mesenteric LN cells of OVA-immunized WT and Y500F mice were cultured for 4 d in the presence of 50 μg/ml of OVA. Culture supernatants were assayed for IL-4, IL-5, IL-13, and IFN-γ by ELISA. In both panels, results are means ± SE (n = 4–5 mice/group) and are representative of three experiments. *P < 0.001; **P = 0.013; ***P = 0.002.
The impact of Y500F mutation on Th cytokine production was further analyzed in mice immunized with OVA mixed with alum, a Th2-promoting adjuvant. Lymphocytes of WT and IL-4Rα Y500F mutant mice that have been immunized with OVA proliferated equally well upon in vitro stimulation with OVA (unpublished data). Furthermore, splenocytes and LN cells of OVA-immunized WT and IL-4Rα Y500F mutant mice produced similar amounts of IL-4, IL-5, IL-13, and IFN-γ upon stimulation with OVA in vitro (Fig. 4 B). These results indicated that the Y500F mutation did not compromise Th2 cell differentiation in the context of a Th2-biased in vivo immune response.
The Y500F Mutation Enhances Antibody Responses.
The impact of the Y500F mutation on humoral immunity was examined by determining the total and antigen-specific antibody responses after immunization with OVA mixed with alum adjuvant to promote Th2-type responses (40), or with saline/alum as immunization control. Fig. 5 A reveals that Y500F mutant mice exhibited substantially higher total IgE levels than WT controls both after immunization with OVA/alum and after control injection of saline/alum (2.5- and 7-fold, respectively). In contrast, the levels of IgM, IgG1, 2a, 2b, and 3 isotypes were similar. Significantly, OVA-specific antibody responses of several isotypes, including IgG1, IgG2a, and IgE, were three- to five-fold higher in the IL-4Rα Y500F mutant mice compared with control (Fig. 5 B). In contrast, levels of OVA-specific IgM, IgG2b, IgG3, and IgA were not significantly different in WT and mutant mice.
Figure 5.

The Y500F mutation up-regulates total IgE production and antigen-specific IgG1, IgG2a, and IgE antibody responses. Mice were immunized with OVA (100 μg) mixed with alum or sham immunized with saline/alum on day 0 and 14. Total (A) and OVA-specific Ig isotypes (B) were determined on day 28 postimmunizations by ELISA. Results are means ± SE (n = 6/group).
Enhanced IgE production was also observed upon in vitro stimulation of naive Y500F splenocytes with anti-CD40 mAb and IL-4, which induce isotype switching to IgE and IgG1 independent of T cell help. Under optimal stimulation conditions, the Y500F splenocytes produced about threefold higher amount of IgE compared with splenocytes of WT littermate controls. In contrast, both WT and Y500F cells produced similar amounts of IgG1 (Fig. 6 A). These results indicated that the enhanced IgE antibody responses in the IL-4Rα Y500F mutant mice reflected, at least in part, altered B cell responsiveness to IL-4.
Figure 6.
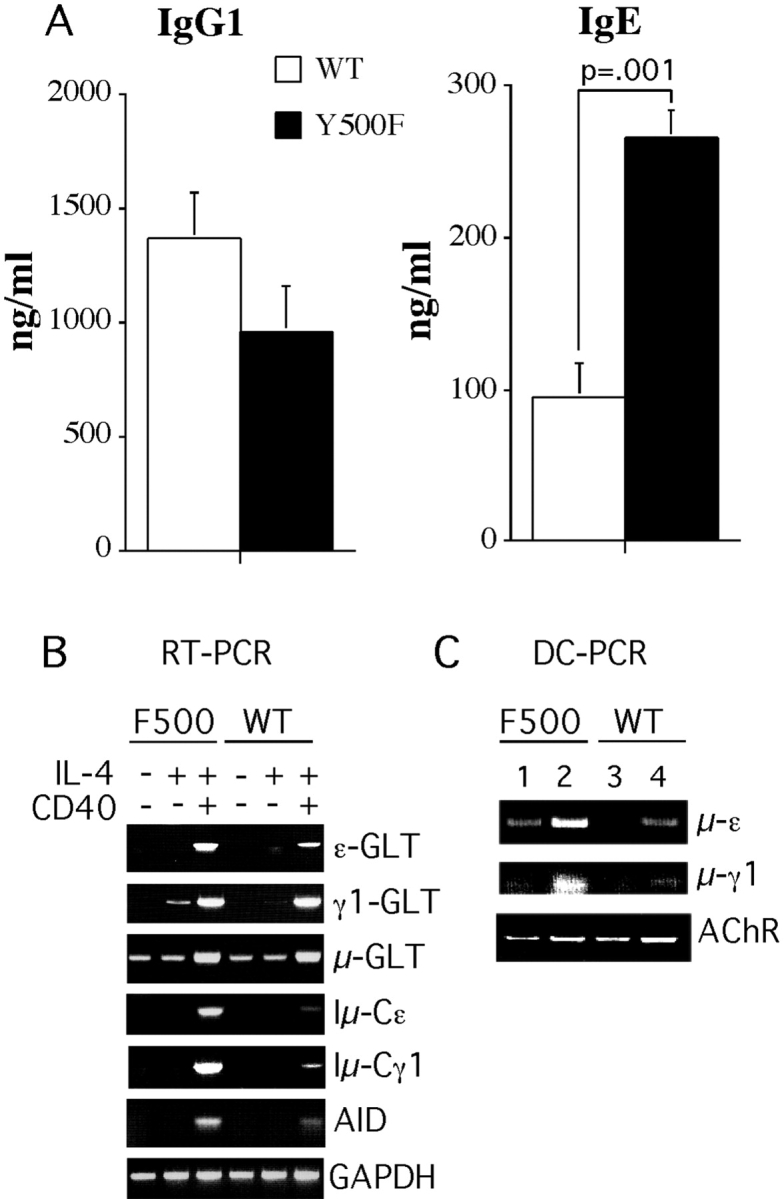
The Y500F mutation augments isotype switching. (A) Production of IgG1 and IgE by cultured splenocytes treated with an anti-CD40 mAb + IL-4. Levels of IgG1 and IgE antibodies present in IgG1 and IgE in supernatants of unstimulated cells were <8 ng/ml, respectively. Results are means ± SE antibody (4 mice/group). Similar results were found in three separate experiments. (B) Molecular events in isotype switching. RT-PCR analysis of μ, γ1, and ɛ germline (GLT) and γ1 and ɛ postswitch transcripts (Iμ→γ1 and Iμ→ɛ, respectively) in splenic B cells treated for 48 h with IL-4, anti-CD40 mAB, or both. Results are representative of four pairs of WT and mutant mice from two independent experiments. (C) Sμ-Sγ1 and Sμ-Sɛ deletional switch recombination measured by DC-PCR at day 6 after treatment with anti-CD40 mAb + IL-4. PCR was performed using 5 ng (lanes 1 and 3) or 20 ng (lanes 2 and 4) of template DNA.
To elucidate the mechanism of enhanced IgE production, we analyzed the induction of molecular events associated with CSR. These include expression of Cɛ and Cγ1 germline transcripts (GLT), expression of the gene for AID followed by Sμ→Sɛ and Sμ→Sγ1 deletional switch recombination, and expression of mature Iμ-Cɛ and Iμ-Cγ1 transcripts (41). Fig. 6 B demonstrates that the Y500F mutation was associated with enhanced induction of ɛ germline transcripts (GLT) in splenic B cells stimulated for 48 h with anti-CD40mAb + IL-4. γ1 GLT were only modestly increased with IL-4, whereas μ GLT were unaffected. In contrast, both ɛ and γ1 postswitch transcripts (Iμ→Cγ1 and Iμ→Cɛ, respectively) were markedly increased in Y500F splenic B cells stimulated with anti-CD40 mAb + IL-4 compared with WT control B cells. The increased levels of ɛ and γ1 postswitch transcripts in Y500F splenic B cells were associated with heightened expression of AID, an enzyme that plays an obligate role in CSR and somatic hypermutation (29). Treatment with IL-4 alone or with anti-CD40 + IL-4 induced higher levels of AID transcripts in mutant compared with WT B cells.
Further evidence of enhanced CSR to the γ1 and ɛ heavy chain loci in Y500F mutant B cells was obtained by DC-PCR amplification. The results showed enhanced Sμ-Sγ1 and Sμ-Sɛ deletional switch recombination in anti-CD40 mAB + IL-4–treated Y500F B cells compared with similarly treated WT B cells (Fig. 6 C). Neither stimulus induced these events by itself (unpublished data). Overall, these results indicated that the Y500F mutation potentiated molecular mechanisms involved in switching to both γ1 and ɛ loci. The observation that in vitro IgE but IgG1 synthesis was increased in Y500F B cells treated with anti-CD40 + IL-4 in the face of an early surge in both γ1 and ɛ postswitch transcripts is most likely due to subsequent sequential switching from γ1 to ɛ (42).
Potentiation of Allergen-induced Airway Inflammation by the Y500F Mutation.
The functional consequences of Y500F mutation were further analyzed in a model of antigen-induced airway inflammation. Mice immunized i.p. with OVA then challenged by OVA inhalation develop an IL-4Rα–dependent allergic inflammatory airway response characterized by AHR to methacholine, eosinophilic infiltration, and goblet cell hyperplasia. Accordingly, mice were immunized with OVA then subjected to inhalation challenge with OVA. Fig. 7 A reveals that at baseline, both WT and mutant mice immunized with OVA exhibited similar Penh. OVA-sensitized IL-4Rα Y500F mutant mice exposed to aerosolized OVA exhibited significantly enhanced bronchial responses to methacholine compared with similarly treated WT littermates (P = 0.03). In contrast, both WT and mutant mice sham immunized with saline/alum and then exposed to aerosolized OVA exhibited an equally modest increase in Penh upon methacholine challenge.
Figure 7.
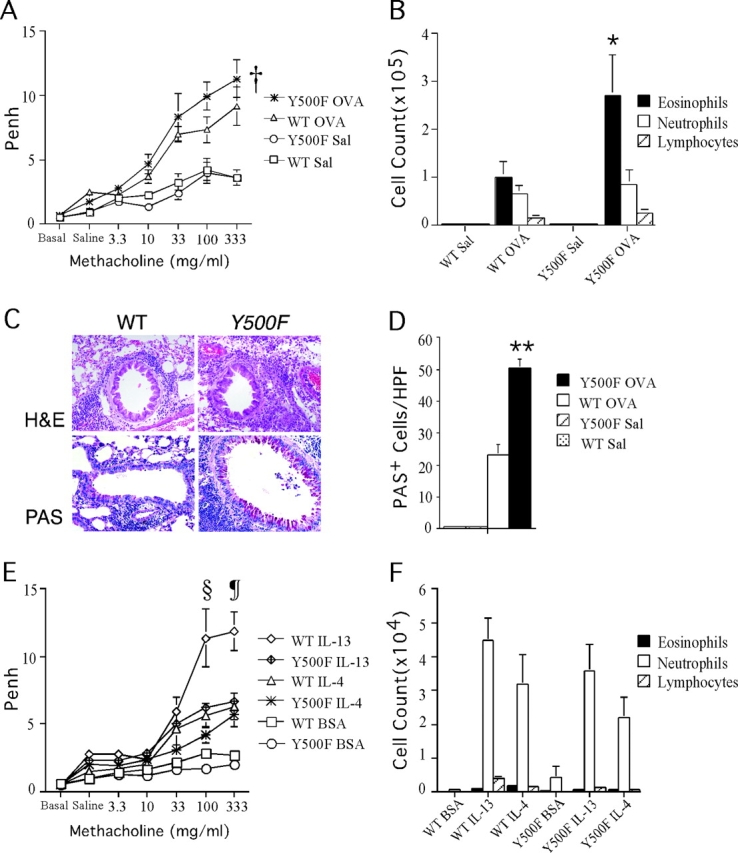
The Y500F mutation enhances AHR, tissue eosinophilia, and goblet cell metaplasia in an antigen-induced model of allergic airway inflammation. (A) AHR to aerosolized methacholine in WT and Y500F mutant mice (n = 5–6/group) immunized with OVA/alum or sham immunized with saline/alum mix and then subsequently challenged with aerosolized OVA. AHR was assessed by enhanced respiratory pause (Penh). Results are means ± SE; †P = 0.03 (versus IL-4Rα Y500F OVA). (B) Differential count of inflammatory cells found in BAL fluid of mice tested in A. *P = 0.04 (versus IL-4Rα Y500F OVA). (C) Lung histology of WT and IL-4Rα Y500F mutant mice sensitized and challenged with OVA. H&E, hematoxylin-eosin; PAS, periodic acid-schiff reagent. (D) The number of mucin-positive cells in the bronchi of OVA-treated WT and Y500F animals. PAS-stained lung sections were scored for the number of mucin-secreting cells per high power field (HPF, 400×) in central medium-sized bronchi. Three representative fields were counted for each of three mice in each group. **P < 0.0001 (versus IL-4Rα Y500F OVA). (E) AHR to aerosolized methacholine in WT and Y500F mutant mice (n = 14–15 mice/group) tested after intranasal instillation of IL-4, IL-13, or BSA at 5 μg each. (F) Differential count of inflammatory cells found in BAL fluid of mice tested in E. §P = 0.03; ¶P = 0.016 versus IL-4Rα Y500F IL-13 at 100 and 333 mg methacholine/ml, respectively.
The enhanced bronchial responsiveness observed in OVA-treated Y500F mutants was accompanied by an exaggerated allergic inflammatory response. Examination of lung tissue of WT and mutant mice stained with hematoxylin and eosin revealed patchy peribronchial inflammatory infiltrates, composed primarily of eosinophils and lymphocytes, that were substantially more intense in OVA-sensitized and challenged Y500F mutant mice compared with WT controls (Fig. 7 B). In contrast, sham-immunized mice exposed to aerosolized OVA showed normal lung histology (unpublished data). Analysis of BAL fluid of OVA-sensitized and challenged mice revealed that OVA-sensitized Y500F mutants exhibited greater recruitment of eosinophils into the airways compared with similarly treated WT mice (Fig. 7 C). In contrast, the two strains of mice exhibited similar numbers of eosinophils in the peripheral blood (unpublished data).
Allergen-driven inflammation of the airways is often accompanied by goblet cell metaplasia. IL-4 and IL-13 are mediators of this response, which proceeds in an IL-4Rα–dependent manner (43–46). Significantly, the IL-4Rα Y500F mutants exhibited markedly increased goblet cell metaplasia with mucus overproduction, revealed by periodic acid–Schiff staining, compared with controls (Fig. 7 D). These results indicated that the Y500F mutation exaggerated antigen-induced airway allergic inflammatory responses.
To determine whether the airways of Y500F mutant mice are intrinsically more responsive to signaling via IL-4Rα, we examined their response to direct IL-4 and IL-13 instillation compared with WT control mice. AHR measured after intranasal instillation of IL-4 was not significantly different between WT and the IL-4Rα Y500F mutant mice (Fig. 7 E). AHR induced by IL-13 instillation was significantly decreased at higher concentrations of methacholine challenge in Y500F mutant mice compared with WT controls. Treatment with both cytokines was associated with a modest, predominantly neutrophilic inflammatory cell population in the BAL fluid that was not significantly different between WT and IL-4Rα Y500F mutant mice (Fig. 7 F). These results indicated that the enhanced AHR observed in OVA-sensitized and challenged IL-4Rα Y500F mutant mice was likely due to the exaggerated allergic inflammatory response rather than to increased responsiveness of resident airway cells to IL-4 and IL-13.
Discussion
By using mice with a point mutation in the germline that targets the effector tyrosine residue of the I4R motif, an important role for this motif was revealed in the regulation of IL-4Rα–dependent immune responses in vivo. Mutagenesis of the Y500 residue resulted in the uncoupling of IL-4Rα from the IRS-2 pathway and its downstream components PI3-K/Akt. It also resulted in impaired CD4+ T cell proliferation in response to IL-4. However, rather than dampening IL-4Rα–dependent responses in vivo, the Y500F mutation enhanced antigen-specific antibody responses and allergic airway inflammation, consistent with a previously unrecognized negative regulatory function of the I4R motif in in vivo allergic responses.
The targeting strategy specifically inactivated Y500-dependent signaling while leaving unperturbed signaling via other tyrosine residues in the cytoplasmic domain of IL-4Rα. Of the adaptor molecules implicated previously in docking at the I4R motif, only IRS-2 was completely uncoupled from IL-4Rα by Y500 mutagenesis. IL-4–induced tyrosine phosphorylation of Dok-R was not impaired, suggesting a redundant function of Y500 in its recruitment. Shc phosphorylation was attenuated but not abrogated, indicating partial dependence on Y500 for IL-4–induced Shc activation. Other signaling events that are mediated by IL-4Rα in a Y500-independent manner proceeded unimpaired, including Jak kinase activation and Stat6 tyrosine phosphorylation,
Consistent with studies implicating signaling pathways downstream of IL-4Rα Y500 in cell proliferation, the mitogenic response of IL-4Rα Y500F CD4+ T cells to IL-4 was greatly decreased and comitogenesis with anti-TCR antibodies abolished. A previous observation that IRS-2–deficient lymphocytes exhibit impaired IL-4–induced proliferation implicates failure of IRS-2 activation in the defective IL-4 mitogenic response of IL-4Rα Y500F T cells (23). Surprisingly, however, a proliferative defect was not observed in B cells treated with IL-4 alone or in combination with anti-CD40 or anti-IgM antibodies. IL-4–induced IRS-2 and Akt phosphorylation was similarly abrogated in IL-4Rα Y500F T and B cells, ruling out both signaling intermediates as the immediate locus of this discrepancy. The rescue of IL-4 mitogenic function in IL-4Rα Y500 B cells suggests compensation by a currently unknown adaptor or signaling intermediate that is activated in a Y500-independent manner.
Given the critical function of Y500-coupled pathways in IL-4–induced T cell mitogenesis, the Y500F mutation would have been expected to impair evolution of a Th2-type, IL-4Rα– dependent immune response by restricting IL-4–dependent expansion of antigen-specific lymphocyte populations. However, differentiation into Th2 cells proceeded unimpaired both in vitro in response to Th2 polarizing signals and in vivo in response to a Th2-promoting antigenic stimulus (OVA mixed with alum) (40). This indicated that the Y500F mutation did not restrict Th2 cell differentiation under these experimental paradigms in agreement with previous studies on CD4+ T cells made to express I4R mutant IL-4Rα chains by retroviral transduction (47). However, decreased residual Th2 cytokine secretion was observed in Y500F that have been differentiated under Th1 polarizing conditions. This suggests a role for the Y500-coupled pathways in supporting Th2 cell differentiation and/or expansion that becomes limiting only under polarizing Th1 conditions. Further studies will be required to verify this proposition.
The Y500F mutation augmented the production of antigen-specific IgG1 and IgE antibodies associated with Th2 responses and antigen-specific IgG2a antibodies, which are normally associated with Th1-type immunity but are suppressed in Th2-type responses (40, 48). The up-regulation by the Y500F mutation of antigen-specific antibody responses was not associated with altered Th cytokine production, since the magnitude of the antigen-induced IL-4, IL-5, IL-13, and IFN-γ production was similar in WT and mutant lymphocytes. In contrast, the Y500F mutation up-regulated in vitro IgE production and augmented molecular events associated with CSR in response to treatment with anti-CD40 mAb and exogenously added IL-4. These results suggest negative regulatory function of IL-4Rα Y500–coupled signaling pathways in B cell antibody production. On the other hand, the enhanced antigen-specific IgG2a antibody responses in the context of a Th2-type immune response implicate the I4R motif in the suppression of Th1-associated antibody isotypes. Switching to IgG2a is dependent on the transcription factor T-bet (49). I4R signaling may alter the activity of this pathway and/or other pathways involved in switching to IgG2a.
The Y500F mutation enhanced allergen-induced AHR, tissue eosinophilia, and goblet cell metaplasia, consistent with a regulatory function of the I4R motif in allergic inflammation. Induction of AHR and inflammation is an IL-4Rα–dependent process involving hematopoietic and resident airway cells. In particular, induction of AHR, goblet cell metaplasia and mucin production is a direct attribute of IL-4Rα signaling in airway epithelial cells. Both IL-4 and IL-13 act directly on resident epithelial cells to induce goblet cell metaplasia in a Stat-6–dependent manner but independent of recruitment of hematopoietic inflammatory cells (46). AHR induced by cytokine instillation in the airways was either similar (IL-4) or decreased (IL-13) in IL-4Rα Y500F mutants compared with WT controls, indicating that the enhanced allergen-induced AHR and goblet cell metaplasia in the Y500F mutants was not due to heightened IL-4Rα responses in airway tissues. Rather, it incriminates the augmented allergic inflammatory response as the most likely mechanism. This may involve enhanced production by mast cells and basophils of IL-4 and IL-13 due to increased antigen-specific IgE antibody levels and/or altered recruitment to the airways of inflammatory cells including eosinophils and allergen-specific Th2 T cells.
In the human IL-4Rα, there exists a serine to proline polymorphism in the I4R motif (S503P), six amino acids down-stream of the effector tyrosine residue (Y497). The presence of the more common serine residue is associated with elevated IgE levels, atopy, and asthma in humans, a phenotype that strongly overlaps with that of mice with targeted inactivation of the I4R motif (25–27). An adverse effect of the S503 relative to the P503 substitution on the function of the I4R motif of the human IL-4Rα chain would provide a mechanism by which this and perhaps other IL-4Rα polymorphic amino acid residues promote human allergic disorders.
Acknowledgments
We thank Michael White for ES cell injection, Mendy Miller for expert technical assistance, and Traian Lupo for animal care.
This work was supported by National Institutes of Health grants HD35694 (to T.A. Chatila), AR47417 (to R.S. Geha), and AI054471 (to H.C. Oettgen) and a grant from the March of Dimes (to T.A. Chatila).
Abbreviations used in this paper: AHR, airway hyperresponsiveness; AID, activation-induced cytidine deaminase; BAL, bronchoalveolar lavage; CSR, class switch recombination; DC-PCR, digestion-circularization PCR; ES, embryonic stem; GLT, germline transcripts; I4R, insulin/IL-4 receptor; IL-4, interleukin-4; IRS, insulin receptor substrate; ITIM, immunoreceptor tyrosine-based inhibitory motif; PI3-K, phosphatidylinositol 3′-kinase; PTB, phosphotyrosine-binding domain; SH, Src homology domain; SHP, SH2-containing tyrosine phosphatase.
References
- 1.Mosely, B., M.P. Beckmann, C.J. March, R.L. Idzerda, S.D. Gimpel, T. VandenBos, D. Friend, A. Alpert, D. Anderson, J. Jackson, et al. 1989. The murine interleukin-4 receptor: molecular cloning and characterization of secreted and membrane bound forms. Cell. 59:335–348. [DOI] [PubMed] [Google Scholar]
- 2.Idzerda, R.L., C.J. March, B. Mosely, S.D. Lyman, T. Vanden Bos, S.D. Gimpel, W.S. Din, K.H. Grabstein, M.B. Widmer, L.S. Park, et al. 1990. Human interleukin-4 receptor confers biological responsiveness and defines a novel receptor superfamily. J. Exp. Med. 171:861–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Galizzi, J.P., C.E. Zuber, N. Harada, D.M. Gorman, O. Djossou, R. Kastelein, J. Banchereau, M. Howard, and A. Miyajima. 1990. Molecular cloning of a cDNA encoding the human interleukin 4 receptor. Int. Immunol. 2:669–675. [DOI] [PubMed] [Google Scholar]
- 4.Kondo, M., T. Takeshita, N. Ishii, M. Nakamura, S. Watanabe, K. Arai, and K. Sugamura. 1993. Sharing of the interleukin-2 (IL-2) receptor γ chain between receptors for IL-2 and IL-4. Science. 262:1874–1877. [DOI] [PubMed] [Google Scholar]
- 5.Russell, S.M., A.D. Keegan, N. Harada, Y. Nakamura, M. Nogushi, P. Leland, M.C. Friedmann, A. Miyajima, R.K. Puri, W.E. Paul, and W.J. Leonard. 1993. Interleukin-2 receptor γ chain: a functional component of the interleukin-4 receptor. Science. 262:1880–1883. [DOI] [PubMed] [Google Scholar]
- 6.Oakes, S.A., F. Candotti, J.A. Johnston, Y.-Q. Chen, J.J. Ryan, N. Taylor, X. Liu, L. Hennighausen, L.D. Notarangelo, W.E. Paul, et al. 1996. Signaling via IL-2 and IL-4 in JAK3-deficient severe combined immunodeficiency lymphocytes: JAK3-dependent and independent pathways. Immunity. 5:605–615. [DOI] [PubMed] [Google Scholar]
- 7.Nelms, K., A.D. Keegan, J. Zamorano, J.J. Ryan, and W.E. Paul. 1999. The IL-4 receptor: signaling mechanisms and biologic functions. Annu. Rev. Immunol. 17:701–738. [DOI] [PubMed] [Google Scholar]
- 8.Jensen, P.L. 2000. The interleukin 13 receptor complex. Stem Cells. 18:61–62. [DOI] [PubMed] [Google Scholar]
- 9.Noben-Trauth, N., L.D. Shultz, F. Brombacher, J.F. Urban, H. Gu, and W.E. Paul. 1997. An interleukin 4 (IL-4)-independent pathway for CD4+ T cell IL-4 production is revealed in ILα4 receptor-deficient mice. Proc. Natl. Acad. Sci. USA. 94:10838–10843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ihle, J.N. 1995. Cytokine receptor signalling. Nature. 377:591–594. [DOI] [PubMed] [Google Scholar]
- 11.Fujiwara, H., S.H. Hanissian, A. Tsytsykova, and R.S. Geha. 1997. Homodimerization of the human IL-4 receptor α chain induces Cɛ germline transcripts in B cells in the absence of the interleukin 2 receptor γ chain. Proc. Natl. Acad. Sci. USA. 94:5866–5871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reichel, M., B.H. Nelson, P.D. Greenberg, and P.B. Rothman. 1997. The IL-4 receptor alpha-chain cytoplasmic domain is sufficient for activation of JAK-1 and stat6 and the induction of IL-4-specific gene expression. J. Immunol. 158:5860–5867. [PubMed] [Google Scholar]
- 13.Hou, J., U. Schindler, W. Henzel, T. Ho, M. Brasseur, and S. McKnight. 1994. An interleukin-4-induced transcription factor: IL-4 Stat. Science. 265:1701–1706. [DOI] [PubMed] [Google Scholar]
- 14.Kashiwada, M., C.C. Giallourakis, P.Y. Pan, and P.B. Rothman. 2001. Immunoreceptor tyrosine-based inhibitory motif of the IL-4 receptor associates with SH2-containing phosphatases and regulates IL-4-induced proliferation. J. Immunol. 167:6382–6387. [DOI] [PubMed] [Google Scholar]
- 15.Giallourakis, C., M. Kashiwada, P.Y. Pan, N. Danial, H. Jiang, J. Cambier, K.M. Coggeshall, and P. Rothman. 2000. Positive regulation of interleukin-4-mediated proliferation by the SH2-containing inositol-5′-phosphatase. J. Biol. Chem. 275:29275–29282. [DOI] [PubMed] [Google Scholar]
- 16.Keegan, A.D., K. Nelms, M. White, L.M. Wang, J.H. Pierce, and W.E. Paul. 1994. An IL-4 receptor region containing an insulin receptor motif is important for IL-4-mediated IRS-1 phosphorylation and cell growth. Cell. 76:811–820. [DOI] [PubMed] [Google Scholar]
- 17.Sun, X.J., L.M. Wang, Y. Zhang, L. Yenush, M.G. Myers, Jr., E. Glasheen, W.S. Lane, J.H. Pierce, and M.F. White. 1995. Role of IRS-2 in insulin and cytokine signalling. Nature. 377:173–177. [DOI] [PubMed] [Google Scholar]
- 18.Wang, H.Y., W.E. Paul, and A.D. Keegan. 1996. IL-4 function can be transferred to the IL-2 receptor by tyrosine containing sequences found in the IL-4 receptor α chain. Immunity. 4:113–121. [DOI] [PubMed] [Google Scholar]
- 19.Nelms, K., A.L. Snow, J. Hu-Li, and W.E. Paul. 1998. FRIP, a hematopoietic cell-specific rasGAP-interacting protein phosphorylated in response to cytokine stimulation. Immunity. 9:13–24. [DOI] [PubMed] [Google Scholar]
- 20.Wery, S., M. Letourneur, J. Bertoglio, and J. Pierre. 1996. Interleukin-4 induces activation of mitogen-activated protein kinase and phosphorylation of shc in human keratinocytes. J. Biol. Chem. 271:8529–8532. [DOI] [PubMed] [Google Scholar]
- 21.Sun, X.J., D.L. Crimmins, M.G. Myers, Jr., M. Miralpeix, and M.F. White. 1993. Pleiotropic insulin signals are engaged by multisite phosphorylation of IRS-1. Mol. Cell. Biol. 13:7418–7428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cerezo, A., A.C. Martinez, D. Lanzarot, S. Fischer, T.F. Franke, and A. Rebollo. 1998. Role of Akt and c-Jun N-terminal kinase 2 in apoptosis induced by interleukin-4 deprivation. Mol. Biol. Cell. 9:3107–3118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wurster, A.L., D.J. Withers, T. Uchida, M.F. White, and M.J. Grusby. 2002. Stat6 and IRS-2 cooperate in interleukin 4 (IL-4)-induced proliferation and differentiation but are dispensable for IL-4-dependent rescue from apoptosis. Mol. Cell. Biol. 22:117–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ryan, J.J., L.J. McReynolds, A. Keegan, L.H. Wang, E. Garfein, P. Rothman, K. Nelms, and W.E. Paul. 1996. Growth and gene expression are predominantly controlled by distinct regions of the human IL-4 receptor. Immunity. 4:123–132. [DOI] [PubMed] [Google Scholar]
- 25.Kruse, S., T. Japha, M. Tedner, S.H. Sparholt, J. Forster, J. Kuehr, and K.A. Deichmann. 1999. The polymorphisms S503P and Q576R in the interleukin-4 receptor alpha gene are associated with atopy and influence the signal transduction. Immunology. 96:365–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ober, C., S.A. Leavitt, A. Tsalenko, T.D. Howard, D.M. Hoki, R. Daniel, D.L. Newman, X. Wu, R. Parry, L.A. Lester, et al. Variation in the interleukin 4-receptor alpha gene confers susceptibility to asthma and atopy in ethnically diverse populations. Am. J. Hum. Genet. 66:517–526. [DOI] [PMC free article] [PubMed]
- 27.Howard, T.D., G.H. Koppelman, J. Xu, S.L. Zheng, D.S. Postma, D.A. Meyers, and E.R. Bleecker. 2002. Gene-gene interaction in asthma: IL4RA and IL13 in a Dutch population with asthma. Am. J. Hum. Genet. 70:230–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wrighton, N., L.A. Campbell, N. Harada, A. Miyajima, and F. Lee. 1992. The murine interleukin-4 receptor gene: genomic structure, expression and potential for alternative splicing. Growth Factors. 6:103–118. [DOI] [PubMed] [Google Scholar]
- 29.Muramatsu, M., K. Kinoshita, S. Fagarasan, S. Yamada, Y. Shinkai, and T. Honjo. 2000. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell. 102:553–563. [DOI] [PubMed] [Google Scholar]
- 30.Chu, C.C., W.E. Paul, and E.E. Max. 1992. Quantitation of immunoglobulin mu-gamma 1 heavy chain switch region recombination by a digestion-circularization polymerase chain reaction method. Proc. Natl. Acad. Sci. USA. 89:6978–6982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xu, L., and P. Rothman. 1994. IFN-gamma represses epsilon germline transcription and subsequently down-regulates switch recombination to epsilon. Int. Immunol. 6:515–521. [DOI] [PubMed] [Google Scholar]
- 32.Blaeser, F., N. Ho, R. Prywes, and T.A. Chatila. 2000. Ca2+-dependent gene expression mediated by MEF2 transcription factors. J. Biol. Chem. 275:197–209. [DOI] [PubMed] [Google Scholar]
- 33.Spergel, J.M., E. Mizoguchi, J.P. Brewer, T.R. Martin, A.K. Bhan, and R.S. Geha. 1998. Epicutaneous sensitization with protein antigen induces localized allergic dermatitis and hyperresponsiveness to methacholine after single exposure to aerosolized antigen in mice. J. Clin. Invest. 101:1614–1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hamelmann, E., J. Schwarze, K. Takeda, A. Oshiba, G.L. Larsen, C.G. Irvin, and E.W. Gelfand. 1997. Noninvasive measurement of airway responsiveness in allergic mice using barometric plethysmography. Am. J. Respir. Crit. Care Med. 156:766–775. [DOI] [PubMed] [Google Scholar]
- 35.Xiao, H., T. Yin, X.Y. Wang, T. Uchida, J. Chung, M.F. White, and Y.C. Yang. 2002. Specificity of interleukin-2 receptor gamma chain superfamily cytokines is mediated by insulin receptor substrate-dependent pathway. J. Biol. Chem. 277:8091–8098. [DOI] [PubMed] [Google Scholar]
- 36.Wolf, G., T. Trub, E. Ottinger, L. Groninga, A. Lynch, M.F. White, M. Miyazaki, J. Lee, and S.E. Shoelson. 1995. PTB domains of IRS-1 and Shc have distinct but overlapping binding specificities. J. Biol. Chem. 270:27407–27410. [DOI] [PubMed] [Google Scholar]
- 37.Crowley, M.T., S.L. Harmer, and A.L. DeFranco. 1996. Activation-induced association of a 145-kDa tyrosine-phosphorylated protein with Shc and Syk in B lymphocytes and macrophages. J. Biol. Chem. 271:1145–1152. [DOI] [PubMed] [Google Scholar]
- 38.Kaplan, M.H., U. Schindler, S.T. Smiley, and M.J. Grusby. 1996. Stat6 is required for mediating responses to IL-4 and for the development of Th2 cells. Immunity. 4:313–319. [DOI] [PubMed] [Google Scholar]
- 39.Shimoda, K., J. Van Deursen, M.Y. Sangster, Sr., R.T. Sarawar, R.A. Carson, C. Tripp, Q.F.W. Chu, T. Nosaka, D.A. Vignali, P.C. Doherty, et al. 1996. Lack of IL-4 induced Th2 response and IgE class switching in mice with disrupted stat6 gene. Nature. 380:630–633. [DOI] [PubMed] [Google Scholar]
- 40.Brewer, J.M., M. Conacher, C.A. Hunter, M. Mohrs, F. Brombacher, and J. Alexander. 1999. Aluminium hydroxide adjuvant initiates strong antigen-specific Th2 responses in the absence of IL-4- or IL-13-mediated signaling. J. Immunol. 163:6448–6454. [PubMed] [Google Scholar]
- 41.Manis, J.P., M. Tian, and F.W. Alt. 2002. Mechanism and control of class-switch recombination. Trends Immunol. 23:31–39. [DOI] [PubMed] [Google Scholar]
- 42.Mandler, R., F.D. Finkelman, A.D. Levine, and C.M. Snapper. 1993. IL-4 induction of IgE class switching by lipopolysaccharide-activated murine B cells occurs predominantly through sequential switching. J. Immunol. 150:407–418. [PubMed] [Google Scholar]
- 43.Dabbagh, K., K. Takeyama, H.M. Lee, I.F. Ueki, J.A. Lausier, and J.A. Nadel. 1999. IL-4 induces mucin gene expression and goblet cell metaplasia in vitro and in vivo. J. Immunol. 162:6233–6237. [PubMed] [Google Scholar]
- 44.Wills-Karp, M., J. Luyimbazi, X. Xu, B. Schofield, T.Y. Neben, C.L. Karp, and D.D. Donaldson. 1998. Interleukin-13: central mediator of allergic asthma. Science. 282:2258–2261. [DOI] [PubMed] [Google Scholar]
- 45.Grunig, G., M. Warnock, A.E. Wakil, R. Venkayya, F. Brombacher, D.M. Rennick, D. Sheppard, M. Mohrs, D.D. Donaldson, R.M. Locksley, and D.B. Corry. 1998. Requirement for IL-13 independently of IL-4 in experimental asthma. Science. 282:2261–2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kuperman, D.A., X. Huang, L.L. Koth, G.H. Chang, G.M. Dolganov, Z. Zhu, J.A. Elias, D. Sheppard, and D.J. Erle. 2002. Direct effects of interleukin-13 on epithelial cells cause airway hyperreactivity and mucus overproduction in asthma. Nat. Med. 8:885–889. [DOI] [PubMed] [Google Scholar]
- 47.Zhu, J., L. Guo, C.J. Watson, J. Hu-Li, and W.E. Paul. 2001. Stat6 is necessary and sufficient for IL-4's role in Th2 differentiation and cell expansion. J. Immunol. 166:7276–7281. [DOI] [PubMed] [Google Scholar]
- 48.Snapper, C.M., and W.E. Paul. 1987. Interferon-gamma and B cell stimulatory factor-1 reciprocally regulate Ig isotype production. Science. 236:944–947. [DOI] [PubMed] [Google Scholar]
- 49.Peng, S.L., S.J. Szabo, and L.H. Glimcher. 2002. T-bet regulates IgG class switching and pathogenic autoantibody production. Proc. Natl. Acad. Sci. USA. 99:5545–5550. [DOI] [PMC free article] [PubMed] [Google Scholar]