

Abstract
Besides orthologs of other eukaryotic mismatch-repair (MMR) proteins, plants encode MSH7, a paralog of MSH6. The Arabidopsis thaliana recognition heterodimers AtMSH2·MSH6 (AtMutSα) and AtMSH2·MSH3 (AtMutSβ) were previously found to bind the same subsets of mismatches as their counterparts in other eukaryotes—respectively, base–base mismatches and single extra nucleotides, loopouts of extra nucleotides (one or more) only—but AtMSH2·MSH7 (AtMutSγ) bound well only to a G/T mismatch. To test hypotheses that MSH7 might be specialized for G/T, or for base mismatches in 5-methylcytosine contexts, we compared binding of AtMutSα and AtMutSγ to a series of mismatched DNA oligoduplexes, relative to their (roughly similar) binding to G/T DNA. AtMutSγ bound G/G, G/A, A/A and especially C/A mispairs as well or better than G/T, in contrast to MutSα, for which G/T was clearly the best base mismatch. The presence of 5-methylcytosine adjacent to or in a mispair generally lowered binding by both heterodimers, with no systematic difference between the two. Alignment of protein sequences reveals the absence in MSH7 of the clamp domains that in bacterial MutS proteins—and by inference MSH6 proteins—non-specifically bind the backbone of mismatched DNA, raising new questions as to how clamp domains enhance mismatch recogni tion. Plants must rigorously suppress mutation during mitotic division of meristematic cells that eventually give rise to gametes and may also use MMR proteins to antagonize homeologous recombination. The MSH6 versus MSH7 divergence may reflect specializations for particular mismatches and/or sequence contexts, so as to increase both DNA-replication and meiotic-recombination fidelity, or dedication of MSH6 to the former and MSH7 to the latter, consistent with genetic evidence from wheat.
INTRODUCTION
DNA mismatch-repair (MMR) systems promote genomic stability in most prokaryotic and eukaryotic organisms (reviewed in 1–3). MMR initiates with recognition of base mismatches, or some DNA lesions, by evolutionarily conserved MutS-homolog (MSH) proteins: MutS homodimers in bacteria, two or three MSH heterodimers in eukaryotes. Recognition of DNA replication errors initiates excision-replacement correction pathways that reduce spontaneous mutation rates by as much as two to three orders of magnitude. Similarly, recognition of mismatches in heteroduplex recombination joints is thought to provoke processes that antagonize homeologous recombination between partially diverged DNA sequences. Suppression of homeologous crossovers would prevent chromosome rearrangement via ectopic recombination between similar DNA sequences during mitosis, and reduce meiotic recombination in progeny of inter-species crosses. Recognition of a variety of DNA base lesions in replication-template strands, including UV photoproducts, either ‘matched’ with their canonical Watson–Crick complements or ‘mismatched,’ may trigger excisive correction that averts lesion-induced mutation, or signaling to cell-cycle-arrest and programmed-cell-death pathways, depending on the circumstances (4–9).
Prokaryotic MutS homodimers recognize both base–base mismatches and one to four extra ‘looped out’ nucleotides (thought to arise during DNA replication by template-primer slip-mispairings). In most eukaryotes, MSH2·MSH6 heterodimers recognize base-mispairs and very short loopouts, but MSH2·MSH3 exclusively recognizes a broad range of loopout sizes. A mismatched-DNA-binding motif near the N-terminus is highly conserved in MutS, MSH3 and MSH6, but not MSH2 polypeptides (10).
Genomic stabilities of plants are threatened not only by sustained exposure to solar UV-B radiation and oxyradical by-products of vigorous oxygen metabolism, and by their inability to escape localized environmental genotoxins, but also by the lack of reserved germ lines: gametes arise only at ends of life cycles, from meristematic cells whose previous divisions provide opportunities for spontaneous and environmental mutagenesis. Thus, plants might be expected to promote genomic fidelity in meristematic cells, and their direct floral descendants, as scrupulously as do other eukaryotic cells. Indeed, the genomes of Arabidopsis thaliana and other plants reveal orthologs of most mammalian and yeast genomic-stability proteins, including those comprising excision-repair and DNA-break-repair pathways, DNA translesion polymerases and DNA-damage surveillance/signaling proteins (11).
In particular, Arabidopsis and other plants encode a complete suite of MMR proteins orthologous to those in other eukaryotes: the recognition heterodimers MSH2·MSH3 and MSH2·MSH6, and the recognition–excision coupling component MLH1PMS2 (10,12,13). In vitro AtMSH3 and AtMSH6 form specific heterodimers with the constant AtMSH2 component; their mismatch-recognition specificities resemble those of eukaryotic counterparts (14). Progeny of parental Arabidopsis plants in which AtMSH2 is inactivated by gene disruption or RNA interference display nucleotide-repeat-sequence (microsatellite) instabilities similar to those seen in MMR-deficient mice (15).
We previously discovered in Arabidopsis a third mismatch-recognition paralog, apparently unique to plants, designated AtMSH7 (14). This polypeptide retains the conserved N-terminal F-Y-E mismatch-recognition motif, and a putative proliferating-cell nuclear antigen (PCNA)-interaction domain in its extreme N-terminus. AtMSH7 also formed a heterodimer with AtMSH2 in vitro. However, despite phylogenetic evidence that MSH7 arose from MSH6, perhaps early in plant evolution, the mismatch-recognition properties of the corresponding heterodimers AtMSH2·MSH6 (AtMutSα) and AtMSH2·MSH7 (AtMutSγ) appeared to differ sharply (14). In particular, MutSγ bound poorly to DNA containing a single extra ‘looped out’ (+T) nucleotide, a substrate typically recognized well by MutS and by both eukaryotic MSH2·MSH6 and MSH2·MSH3 (MutSβ) proteins, including those in Arabidopsis. However, AtMutSγ recognized a G/T mismatch nearly as well as did AtMutSα; most other base mispairs were not tested.
A second striking difference between AtMSH7 and AtMSH6 is the absence from the former of an extended internal stretch of amino acids (AtMSH6 884–984) fairly well conserved among eukaryotic MSH6 proteins. This difference, and the possibility that MutSγ was specific for G/T, suggested that its role in plant genomic stability might be other than the correction of DNA replication errors, as mediated by MutSα and MutSβ in other eukaryotes. MutSγ might be dedicated to correction of T/G mispairs that arise in non-replicating DNA via deamination of 5-methylcytosine (mC) at the mCpG and mCpNpG motifs that are particularly frequent in plant DNA (16). Alternatively, AtMutSγ might generally recognize replication errors in mC-containing contexts. A different special role for plant MutSγ protein is suggested by recent genetic evidence that links the wheat MSH7 gene to the Ph2 locus, where mutations increase recombination between homeologous chromosomes (17). Respectively, MutSγ would be predicted to recognize mostly G/T arising from 5-methylcytosine deamination, presumably in non-replicating as well as replicating DNA, or recognize a broad set of mismatches containing or adjacent to 5-methylcytosine, or recognize in recombining chromosomes a range of base–base mismatches. To semi-quantitatively test these alternatives, we synthesized AtMutSα and AtMutSγ in vitro, and used electrophoretic mobility shift assays to compare binding to a range of mismatches, in some cases in contexts that included 5-methylcytosine. AtMutSγ proved to bind all base-mispairs tested, in most cases as well or better than AtMutSα. Mispairs containing or adjacent to 5-methylcytosine were generally bound slightly less well by both heterodimers.
The recent availability of MSH7 sequences from maize and wheat (17) made it possible to confirm the absence in MSH7 of roughly 100 central amino acids (relative to MSH6), and to identify the deleted residues with the clamp domain that, in the recently published bacterial MutS structures, makes non-specific DNA contacts (18,19).
MATERIALS AND METHODS
In vitro protein synthesis
Derivatives of plasmids pGEM-3Z (Promega, Madison, WI), containing cDNAs encoding AtMSH2, AtMSH6 and AtMSH7, each modified to include a Kozak consensus sequence 5′ to the respective coding sequences, were described previously (14). These are designated here as pGEMatMSH2, pGEMatMSH6 and pGEMatMSH7. Synthesis of AtMSH polypeptides in rabbit reticulocyte lysates (TnT Quick, Promega) was essentially as described (14). Co-synthesis yields more heterodimers than mixing separately synthesized MSH peptides (14). To optimize yields, we systematically varied amounts of each plasmid in 50-µl lysate mixtures, from 0.5 to 2.0 µg. We subsequently used mixtures in which amounts of the two polypeptide products were approximately balanced, judged by incorporation of [35S]methionine (taking account of respective methionine contents), as measured by electrophoresis on 10% polyacrylamide gels in 0.1% SDS buffer (SDS–PAGE) and autoradiography. Figure 1 shows SDS–PAGE analyses of typical balanced-yield synthesis reactions. As recommended by the manufacturer, Mg-acetate and KCl concentrations were varied to optimize polypeptide yields. Typically, lysates were made 60 and 6 mM in Mg-acetate. After incubation at 30°C for 90 min, mixtures were diluted 20-fold with buffer A (50 mM potassium phosphate, pH 7.5, 50 mM NaCl, 0.1 M Na2EDTA, 1 mM dithiothreitol, 5% glycerol) and centrifuged, until back to the original volume, at 1000 g, in a Microcon 100 filter cartridge (nominal cut-off 105 kDa; Millipore, New Bedford, MA). This removes considerable lysate protein, as well as ATP, which inhibits binding of MSH proteins to mismatched DNA (20,21). Aliquots were flash-frozen in liquid N2 and stored at –80°C.
Figure 1.
Analysis of co-synthesized Arabidopsis MSH proteins. Balanced synthesis of AtMSH7 plus AtMSH2 (lane 1) and AtMSH6 plus AtMSH2 in rabbit-reticulocyte lysates in the presence of [35S]methionine, Micron 100 filtration/centrifugation, electrophoresis of denatured proteins in 10%-acrylamide 0.1% SDS gels and autoradiography were as described in Materials and Methods.
Because the amounts of cDNA-encoded protein synthesized are very low relative to the amount of protein in reticulocyte lysates, and it is difficult to determine the absolute specific activities of [35S]methionine incorporated into proteins, no quantitative measurements of amounts of protein synthesized in these lysates were possible. However, we previously compared synthesis of AtMutSα and AtMutSγ to synthesis of luciferase programmed by a control cDNA supplied by the manufacturer, who stated 150–500 ng yields per 50 µl lysate to be typical. Based on relative 35S radioactivities of luciferase, AtMutSα and AtMutSγ in parallel reactions (and taking account of methionine contents), a very approximate estimate of AtMutSα and AtMutSγ yields would be 30–100 ng per 50 µl (K.Culligan and J.Hays, unpublished data). Previous gel filtration analyses showed heterodimer yields to be 84% and 76%, respectively (14).
Preparation of duplex oligomers for binding assays
DNA substrates were prepared essentially as described previously (14). Briefly, gel-purified synthetic DNA ‘top strand’ 51mers were 5′-end-labeled by incubating 50-µl reaction mixtures containing 20 pmol of DNA oligomer, 40 pmol of [γ-32P]ATP, and 10 U of T4 polynucleotide kinase, in kinase buffer (Gibco BRL, Gaithersburg, MD), for 30 min at 37°C; reactions were terminated by heating at 70°C for 10 min. To produce desired mismatched-DNA substrates, 20 pmol purified synthetic 51mer bottom strands (Table 1) were annealed to 20 pmol 32P-end-labeled top strands, by heating mixtures at 85°C for 5 min and slowly cooling down to 4°C. To purify DNA duplexes away from single-stranded DNA (ssDNA) and unincorporated [32P]ATP, 0.2 vol of benzoyl-napthoyl-DEAE (BND) cellulose slurry (Sigma; washed and resuspended in 0.3 M KCl such that settled resin occupies half the volume) was added to the reaction mixtures, which were then made up to 1 M NaCl using 5 M NaCl, incubated at room temperature for 5 min, and layered on a Sephedex G-50 Nick Column (Amersham Pharmacia Biotech). Samples were recovered from supernatants after sedimentation for 5 min in a Beckman GP-4 centrifuge to pellet BND-cellulose. To test for removal of ssDNA from the duplex substrates, small aliquots were treated again with [γ-32P]ATP and polynucleotide kinase, to preferentially end-label (unannealed) ssDNA, and duplex and ssDNA separated in non-denaturing 12% polyacrylamide gels. If significant (32P-labeled) ssDNA was detected by subsequent autoradiography, the BND-cellulose and spin-column steps were repeated. DNA concentrations in purified preparations were determined by binding PicoGreen dye (Molecular Probes, Eugene, OR) fluorescence.
Table 1. Oligomers used to construct mismatched-DNA duplexes.

NC, not constructed.
aItalicized letters correspond to mispairs and contexts shown in right hand columns.
bMismatched bases in boldface.
cBase pairs that flank mismatch sites.
Electrophoretic mobility shift assays of protein–DNA complexes
For direct binding assays, each 25-µl reaction mixture, made up in 1× Binding Buffer (50 mM potassium phosphate, pH 7.5, 50 mM NaCl, 0.1 mM EDTA,1 mM DTT, 5% glycerol), contained 10–15 µl AtMSH protein lysate, 5 mM AMP and 0.1 pmol [32P]DNA 51 bp linear duplexes. Mixtures were incubated at room temperature for 40 min, then loaded onto 5% polyacrylamide gels, made up in TBE buffer (50 mM Tris, 50 mM boric acid, 1 mM Na2EDTA, pH 8) containing 2.5% glycerol and electrophoresed at 10 V/cm for 180 min. After electrophoresis, gels were dried on Whatman 3MM paper and visualized by autoradiography at room temperature using Kodak Biomax X-ray film. Phosphorimaging and ImageQuant software were used to analyze the intensity of specific bands. Intensities of specific (‘A’) bands corresponding to binding of various mismatched DNA duplexes were normalized relative to respective intensities for G/T duplexes, which were bound strongly by both AtMSH2·MSH6 and AtMSH2·MSH7 heterodimers and were included in every experiment. Since the same radiolabeled top strand (oligo 1 or oligo 2, Table 1) was used for all base-mispairs in a particular experiment [except for (+T) substrates], relative radioactivities directly reflect relative DNA amounts in bands. Each determination was performed three times.
RESULTS
To prepare AtMSH2·MSH6 (AtMutSα) and AtMSH2·MSH7 (AtMutSγ) heterodimers for semi-quantitative testing of binding to mismatched DNA, we employed in vitro protein synthesis. We previously obtained ∼80% yields of discrete heterodimers from co-synthesis of AtMSH2 with AtMSH6, and AtMSH7, and lesser yields for AtMSH2 plus AtMSH3, but no dimers were formed between Arabidopsis and human polypeptides (14). Heterodimer yields were much lower when polypeptides synthesized together were mixed and incubated, so it was necessary to empirically adjust cDNA inputs and other conditions to obtain optimum but equal co-synthesis yields, such as those shown in Figure 1. After removal of low molecular weight compounds and some small proteins, we used the crude protein mixtures for electrophoretic mobility shift assays of binding to 51 bp synthetic DNA duplexes. Similar approaches have been widely employed to assess binding of different DNA mismatches by human MutS homologs synthesized in vitro (22,23) or present in nuclear extracts (24), or to compare binding by human MutSα, and Arabidopsis MutSα, MutSβ and MutSγ synthesized in vitro (14). In all these mismatch-binding studies, the electropherograms typically showed certain characteristics also seen here (Fig. 2): (i) shifting of a very small fraction (∼1–5%) of radiolabeled mismatched-DNA probes to slower mobilities, even though the molarities of the respective MutS-homolog proteins were expected to be roughly one-fifth or more of DNA molarities; (ii) the appearance in virtually every case, even with mock-synthesis lysates (not programmed with any cDNA), of a shifted but slower-moving ‘B’ band (Fig. 3 and data not shown), that may reflect binding by Ku70/Ku80 (25) to DNA ends; (iii) slower-moving bands (designated ‘A’ here) when MutS homologs were present, that were much stronger for mismatched than for heteroduplex DNA and, in gels generally showing good resolution, migrated as doublets—often of equal intensity, but sometimes unequal (upper > lower roughly as frequently as lower > upper), although doublets were not always observed [for example, shifted bands were doublet in HeLa extracts but singlet in 3T3 cell extracts (24)]. All studies yielded hierarchies of affinity of MutS homologs for different mismatches, with shifting homoduplex-DNA lowest or negligible, that have generally turned out to agree well with specificities subsequently determined by other techniques, including binding assays with purified proteins.
Figure 2.

Electrophoretic mobility shift assays of mismatched-DNA binding by AtMutSα and AtMutSγ. Incubation of equal amounts of Microcon- filtered co-synthesis mixtures containing AtMSH6 + AtMSH2 (AtMutSα, lanes 1–6) or AtMSH7 + AtMSH2 (AtMutSγ, lanes 7–12) polypeptides, with 0.1 pmol 32P-end-labeled 51 bp DNA duplexes, and electrophoresis in (non-denaturing) 3% polyacrylamide gels, autoradiography and analysis by densitometry were as described in Materials and Methods. Oligomers [(top strand)/(bottom strand)] comprising indicated substrates in representative electropherogram shown were as follows: G/T, 1/11; G/C, 1/12; +T, 3/18; C/A, 1/15; G/A, 1/14; (mC/G) G/T, 2/10. Mismatch-specific electropherogram bands used to score binding are indicated by letter A, and the major non-specific band by letter B. mC, 5-methyl-cytosine.
Figure 3.

Mismatch-recognition spectra for AtMutSα and AtMutSγ. Electrophoretic mobility shift analyses of mixtures of 0.1 pmol of indicated 51 bp DNA duplexes with equal aliquots of co-synthesized AtMSH7 + AtMSH2 (AtMutSγ) or AtMSH6 + AtMSH2 (AtMutSα) polypeptide mixtures were described in the Figure 2 legend. Summed intensities of mismatch-specific DNA–protein bands (designated by letter A in Fig. 2) for each mismatch were divided by intensities for G/T DNA. Data correspond to means for three independent determinations and standard deviations for binding to MutSγ (open bars), or MutSα (filled bars). Oligomer (upper/lower) pairs used were as indicated in the Figure 2 legend, plus those for G/G (1/16) and A/A (1/17).
Thus, despite apparent yields of MutS–DNA complexes much lower than would be expected for equilibrium mixtures, band-shift assays with protein mixtures can be highly informative when certain conditions are met, which is the case here. First, radiolabeled DNA substrates were scrupulously purified, because contaminants, such as ssDNA, might be bound by MutS homologs or other proteins in the mixtures. Second, homoduplex controls showed low or negligible bands at the mismatch-dependent ‘specific-band’ positions (Fig. 2, lanes 2 and 8). Third, affinities of a particular protein for a series of mismatches were determined relative to its affinity for one particular mismatch (G/T), so relative recognition spectra for different proteins could be compared (Figs 3 and 4). Fourth, (relative) band shifts were quantitatively reproducible (Figs 3 and 4). Fifth, no band shifts were seen when only one MSH polypeptide was synthesized (data not shown). Furthermore, when the amount of MutS-homolog protein was increased, either by using more protein-synthesis lysate or by using lysates in which synthesis was more efficient, the amounts of apparent binding increased in approximate direct proportion here (data not shown) and in our previous study (14).
Figure 4.

Effects of methylcytosine on binding of mismatched DNA. Electrophoretic mobility shift assays of binding of AtMutSγ (open bars) or AtMutSα (filled bars) performed and analyzed in legend to Figure 3. Oligomers used to construct 51 bp substrates with mispairs (boldface) and 5-methylcytosine (mC) in positions indicated were as follows: CG/GT, 1/11; mCG/GT, 2/11; CG/GA, 1/14; mCG/GA, 2/14; CG/AC, 1/15; mCG/AC, 2/15; TC/GG, 1/13; TmC/GG, 2/13.
We consider both ‘A’ bands seen in gel autoradiograms (Fig. 2) to reflect mismatch-specific binding, as did the authors of papers cited above. Both ‘A’ bands were typically reduced by the same proportions by increasing excesses of unlabeled mismatched DNA (data not shown). Neither band was observed if no proteins or only one MSH polypeptide were synthesized [(14) and data not shown]. Mismatch-bound AtMutSα or AtMutSγ might sometimes interfere with additional binding by lysate proteins, perhaps those responsible for band ‘B’, such that in a doublet one band reflected non-specific binding to one end, and the other non-specific binding to neither or both. Or specific binding of AtMutSα or AtMutSγ might enhance further non-specific binding of AtMutSα or AtMutSβ, respectively, or of another lysate protein to some but not all substrates. Previously, non-heterodimerized AtMSH6 or AtMSH7 polypeptides migrated during gel filtration at molecular weights higher than those of AtMSH2·MSH6 or AtMSH2·MSH7, respectively (14), suggesting binding to themselves or other proteins. Among all gels, there was no reproducible pattern of relative intensities of upper versus lower ‘A’ bands, with respect to particular mismatches or to AtMutSα versus AtMutSγ (data not shown). Thus, the apparently higher ratios of upper to lower bands for AtMutSγ compared to AtMutSα [except for (+T) seen in Fig. 2] are fortuitous.
Apparent efficiencies of binding of AtMutSα and AtMutSγ to a series of heteroduplex DNA substrates, relative to binding to G/T DNA in each case, are shown in Figure 3. As previously observed (14), AtMutSγ bound relatively poorly to the extra nucleotide (+T) substrate, which is bound well by bacterial MutS and by eukaryotic MutSα heterodimers, including AtMutSα here. Contradicting an hypothesis that it might be specialized for G/T (see Introduction), MutSγ (Fig. 3, open bars) showed a trend of better binding to G/G and C/A than to G/T, typically the base-mispair recognized best by other eukaryotic MutSα proteins, and also good binding to A/A and G/A. In contrast, MutSα (Fig. 3, filled bars) showed worse binding to all purine–purine mispairs, and to C/A, than to G/T. We reproducibly found much less binding of AtMutSγ than AtMutSα to homoduplex (G/C) DNA. Subtraction of the (G/C) values would make the relative advantages of AtMutSγ over AtMutSα for G/A, C/A, G/G and A/T binding even greater. We previously observed very little binding of a C/C mismatch by either AtMutSα or AtMutSγ (14). In two trials, excess unlabeled DNA reduced binding to radiolabeled (G/T) by AtMutSα in the qualitative order (+T) > G/T > G/G ≥ C/A ≈ G/A and by AtMutSγ in the order G/G > G/A ≥ G/T > (+T), generally in agreement with direct-binding data shown in Figure 3 (data not shown). For AtMutSγ, the competition order was C/A > G/T in one trial, and G/T > C/A in the other.
Because 5-methylcytosine levels in plant DNA are typically greater than those in DNA of other eukaryotes, amounting to up to 20–30% of nuclear DNA cytosines (16), plant MSH7 proteins might have evolved specialized recognition of mismatches in contexts involving 5-methylcytosine, such as mCpG. In fact, the trend of binding, for both AtMutSα and AtMutSγ, was less to mCG/GT and mCG/GA than to CG/GT and CG/GA (Fig. 4, first versus second and third versus fourth pairs of bars, respectively) (bold indicates mispaired bases). These dyads would mimic hydrolytic deamination of methylcytosine—more rapidly than deamination of cytosine (26)—in an mCpG couplet. Remarkably, binding to mC/A appeared substantially reduced relative to binding to C/A, by ∼4-fold for MutSγ and 2-fold for MutSα (Fig. 4, fifth versus sixth pairs). Finally, we tested for methylcytosine effects in a nonpalindromic dyad: AtMutSγ and AtMutSα both showed a trend of less binding to TmC/GC than to TC/GG (Fig. 4, seventh versus eighth pairs). Therefore, MSH7 seems not to have evolved specifically to promote fidelity of heavily methylated DNA. The significance of the apparently reduced MMR recognition with respect to stability of cytosine-methylated genomes remains to be determined.
DISCUSSION
MSH7 appears to have arisen early in plant evolution, most likely via duplication and divergence of an MSH6-like gene in a primitive plant (10,14), before the monocot–dicot split: MSH7 genes have been identified in Arabidopsis, maize (27), wheat (17), rice (27) and sugar cane (C.Menck, personal communication), but not in any other eukaryotes. Plant MSH6 genes branch phylogenetically with those in other eukaryotes. Here we show that relative to G/T, AtMSH2·MSH7 heterodimers may recognize several base mismatches better than AtMSH2·MSH6 heterodimers, at least in the DNA-sequence context tested. Comparisons of mismatch binding using single concentrations of crude protein mixtures can only be semi-quantitative, in contrast to titration of mismatched DNA with increasing amounts of MutS homologs. However, the results obtained here with protein-synthesis lysates have sufficed to test the hypotheses advanced. In some respects, competition with a mixture of many other proteins for binding to DNA with a mismatch may approximate the situation in vivo better than binding of purified proteins.
MutS, MSH6 and MSH7 protein sequences align very well (Fig. 5), and the mismatch-detection motif Phe-X-Glu (typically Phe-Tyr-Glu; E.coli MutS residues 36–38) is present in all MSH7 polypeptides (Fig. 5A). Crystal structures of E.coli and Thermus aquaticus MutS bound to mismatched DNA show that in one subunit of each asymmetric homodimer the Phe-Tyr-Glu motif directly contacts bases at the mismatch (18,19). Genetic evidence (28,29) supports the inference that it does so as well in the MSH6 subunits of eukaryotic MutS heterodimers. (In contrast, the MSH3 Phe-X-Glu sequence that aligns with E.coli MutS Phe40 is unlikely to directly bind DNA, because Phe40 is 7 Å distant from DNA, and Glu42 is at the other end of the domain, 20 Å from the nearest DNA atom.) The crystal structures show that in the same subunit that directly contacts the mismatch, a second domain forms a clamp that encircles the DNA while making non-specific interactions with its backbone (Fig. 6). This clamp may aid mismatch recognition by facilitating the kinking of DNA at the mismatch, in cooperation with the Phe-X-Glu domain. A 60° kink is common to structures of E.coli MutS with different mismatches (30) and thus seems integral to mismatch recognition.
Figure 5.

Conservation of DNA binding domains in MutS, MSH6 and MSH7 proteins. Sequence alignment showing mismatch recognition domain (A) and clamp domains (B) derived from an alignment performed with full sequences. Tubes and strands indicate α-helices and β-strands, respectively. Black dots indicate residues that contact the DNA through hydrogen bonds. Locations of aligned sequences are indicated in the structure of E.coli MutS in Figure 6.
Figure 6.
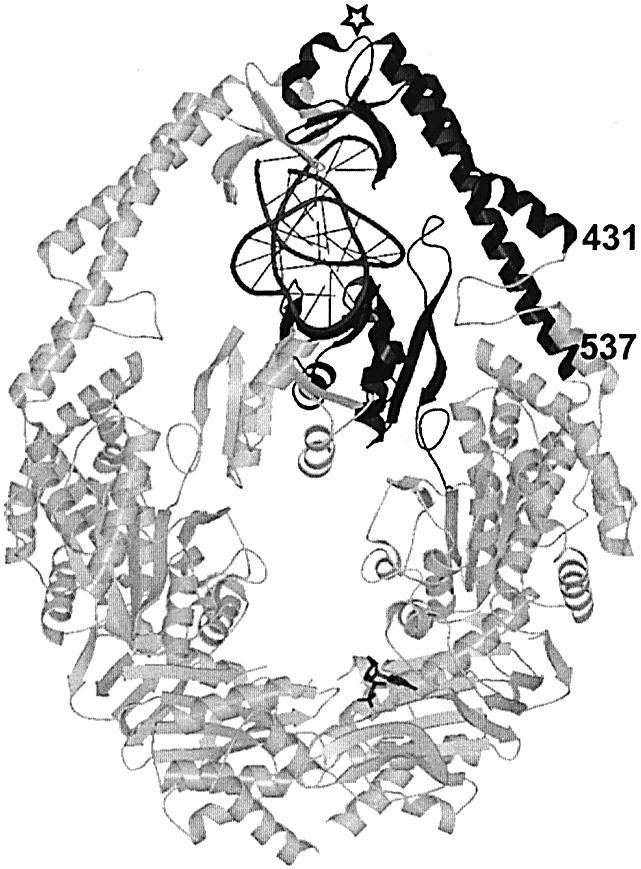
DNA binding domains in MutS. Crystal structure of E.coli MutS dimer in complex with DNA substrate. In the monomer that contacts the mismatch directly, the mismatch binding domain and clamp domain are indicated in black, DNA in dark gray, and remaining part of protein in light gray. Numbers refer to start (431) and end (537) of aligned clamp domain sequences in Figure 5B. The open star indicates the position of the insertion found in the MSH6 proteins.
The DNA clamp domain contains about 100 residues in bacterial MutS (E.coli 432 to 537), and perhaps 110 in MSH6 proteins (e.g. AtMSH6 884 to 984) (Fig. 5B). Remarkably, MSH7 lacks the amino acids in this domain. How the absence of these clamp residues in MSH7 might affect DNA binding is not immediately obvious. The deletion is certain to alter the way the vestigial domain in MSH7 contacts DNA; it might not contact DNA at all, so most non-specific contacts would be with the MSH2 subunit. If in other proteins both domains facilitate kinking of the DNA, as appears to be the case in MutS, then MutSγ might deform DNA less efficiently and therefore bind less well to homoduplex DNA. How reduced kinking efficiency might explain the paradox of reduced binding to both homoduplex DNA and to an extra looped out nucleotide, but strong binding to several base-mispairs, remains to be determined.
Previously, AtMSH7 (designated AtMSH6-2 by other workers), as well as AtMSH2 and AtMSH3, were reported to be expressed at virtually undetectable levels in Arabidopsis plant tissues, but transcripts were detected in RNA blots derived from suspension cultures (31). The whole-plant material was most likely dominated by Arabidopsis leaf tissues, in which we have previously found inactivation of AtMSH2 to have only a modest (albeit significant) effect on genomic stability (15). However, in the progeny of AtMSH2-defective plants we did find large increases in insertion–deletion mutations in nucleotide-repeat sequences (microsatellites), relative to wild-type plants, paralleling microsatellite instability observed in MMR-defective mice. Thus, the heterodimers that MSH2 forms with MSH3 and MSH6 paralogs appear to strongly antagonize spontaneous mutation in floral cells and their meristematic precursors—the plant equivalents of reserved germ lines. Since MSH2·MSH6 and MSH2·MSH3 suffice for correction of replication errors in other eukaryotes, and the data presented here argue against MSH2·MSH7 specialization for G/T or for mismatches in methylcytosine-containing contexts, how might MutSα contribute to plant genetic fidelity? The lack of a strong domain might specialize MSH7 for enhanced recognition of certain base mismatches, perhaps in certain DNA-sequence contexts. However, it is not clear that this would decisively improve plant mismatch correction relative to that in other eukaryotes. Alternatively, plant MSH7 proteins (MutSγ heterodimers) might help maintain species barriers, by antagonizing meiotic recombination between quasi-homologous (diverged but similar) DNA sequences (so-called homeologous recombination), as would be present at corresponding loci in chromosomes in hybrid progeny of interspecies crosses. The Ph2 locus in wheat (Triticum aestivum) controls chromosome pairing in its hybrids with alien species: deletion ph2a (32) and a point mutation ph2b (33), which maps under the ph2a deletion, elevate levels of homeologous recombination in hybrids with Triticum kotschyi var. variables or rye (Secale cereale), and the wheat TaMSH7 gene now appears to be closely linked to the Ph2 locus (17). Two hypotheses appear compatible with the data available at present. Both plant MutSα and MutSγ heterodimers (and MutSβ as well) might play roles in both correction of DNA replication errors and antagonism of meiotic homeologous recombination, with MSH6 and MSH7 increasing the efficiency of recognition by specializing for different subsets of mismatches and/or sequence contexts. Alternatively, the drastically reduced clamp domain of MSH7 might reflect an evolved dedication to meiotic-recombination fidelity. The very low affinity of MutSγ for linear homoduplex DNA might minimize interference with meiotic recombination between stretches of highly homologous DNA. In this case, MSH2·MSH6 and MSH2·MSH3 would mediate post-replication error correction, as in other eukaryotes, and the differences in MSH6 and MSH7 mismatch specificity might be incidental to their separate evolutionary paths. Neither effects of specific inactivation of MSH7 versus MSH6 on genomic stability or homeologous recombination, nor differential expression of MSH6 and MSH7 in mitotic versus meiotic tissues, have been examined in any plant.
Acknowledgments
ACKNOWLEDGEMENTS
This work was supported by NSF grant MCB 0078262 to J.B.H.
REFERENCES
- 1.Buermeyer A.B., Deschenes,S.M., Baker,S.M. and Liskay,R.M. (1999) Mammalian DNA mismatch repair. Annu. Rev. Genet., 33, 533–564. [DOI] [PubMed] [Google Scholar]
- 2.Harfe B.D. and Jinks-Robertson,S. (2000) DNA mismatch repair and genetic instability. Annu. Rev. Genet., 34, 359–399. [DOI] [PubMed] [Google Scholar]
- 3.Jiricny J. and Nystrom-Lahti,M. (2000) Mismatch repair defects in cancer. Curr. Opin. Genet. Dev., 10, 157–161. [DOI] [PubMed] [Google Scholar]
- 4.Wang H., Lawrence,C.W., Li,G.-M. and Hays,J.B. (1999) Specific binding of human MSH2MSH6 mismatch-repair protein heterodimers to DNA incorporating thymine- or uracil-containing UV-light photoproducts opposite mismatched bases. J. Biol. Chem., 274, 16894–16900. [DOI] [PubMed] [Google Scholar]
- 5.Mu D., Tursun,M., Puckett,D., Drummond,J.T., Modrich,P. and Sancar,A. (1997) Recognition and repair of compound DNA lesions (base damage and mismatch) by human mismatch repair and excision repair systems. Mol. Cell. Biol., 17, 760–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aquilina G. and Bignami,M. (2001) Mismatch repair in correction of replication errors and processing of DNA damage. J. Cell. Physiol., 187, 145–154. [DOI] [PubMed] [Google Scholar]
- 7.Li G.-M. (1999) The role of mismatch repair in damage-induced apoptosis. Oncol. Res., 11, 393–400. [PubMed] [Google Scholar]
- 8.Karran P. (2001) Mechanisms of tolerance to DNA damaging therapeutic drugs. Carcinogenesis, 22, 1931–1937. [DOI] [PubMed] [Google Scholar]
- 9.Young L.C., Hays,J.B., Tron,V.A. and Andrew,S.E. (2003) DNA mismatch repair proteins: guardians against UV-induced genomic instability and tumorigenesis. J. Invest. Dermatol., 121, 435–440. [DOI] [PubMed] [Google Scholar]
- 10.Culligan K.M., Meyer-Gauen,G., Lyons-Wilder,J. and Hays,J.B. (2000) Evolutionary origin, diversification and specialization of eukaryotic MutS-homolog mismatch-repair proteins. Nucleic Acids Res., 28, 463–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hays J.B. (2002) Arabidopsis thaliana, a versatile model system for study of eukaryotic genome-maintenance functions. DNA Repair, 64, 1–22. [DOI] [PubMed] [Google Scholar]
- 12.Culligan K.M. and Hays,J.B. (1997) DNA mismatch repair in plants: An Arabidopsis thaliana gene that predicts a protein belonging to the MSH2 subfamily of eukaryotic MutS homologs. Plant Physiol., 115, 833–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jean M., Pelletier,J., Hilpert,M., Belzile,F. and Kunze,R. (1999) Isolation and characterization of AtMLH1, a MutL homologue from Arabidopsis thaliana. Mol. Gen. Genet., 262, 633–642. [DOI] [PubMed] [Google Scholar]
- 14.Culligan K.M. and Hays,J.B. (2000) Arabidopsis thaliana MutS-homolog proteins—atMSH2, atMSH3, atMSH6 and a novel atMSH7 protein—form three distinct heterodimers with different specificities for mismatched DNA. Plant Cell, 12, 991–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Leonard J.M., Bollmann,S.R. and Hays,J.B. (2003) Reduction of stability of Arabidopsis genomic and transgenic DNA-repeat sequences (microsatellites) by inactivation of AtMSH2 mismatch-repair function. Plant Physiol., 133, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Adams R.I.P. and Burdon,R.H. (1985) Molecular Biology of DNA Methylation. Springer-Verlag, Berlin. [Google Scholar]
- 17.Dong C., Whitford,R. and Langridge,P. (2002) A DNA mismatch repair gene links to the Ph2 locus in wheat. Genome, 45, 116–124. [DOI] [PubMed] [Google Scholar]
- 18.Obmolova G., Ban,C., Hsieh,P. and Yang,W. (2000) Crystal structures of mismatch repair protein MutS and its complex with substrate DNA. Nature, 407, 703–708. [DOI] [PubMed] [Google Scholar]
- 19.Lamers M., Perrakis,A., Enzlin,J.H., Winterwerp,H.H.K., deWind,N. and Sixma,T. (2000) The crystal structure of DNA mismatch repair protein MutS binding to a GT mismatch. Nature, 407, 711–717. [DOI] [PubMed] [Google Scholar]
- 20.Blackwell L.J., Martik,D., Bjornson,K.P., Bjornson,E.S. and Modrich,P. (1998) Nucleotide-promoted release of hMutSα from heteroduplex DNA is consistent with an ATP-dependent translocation mechanism. J. Biol. Chem., 48, 32055–32062. [DOI] [PubMed] [Google Scholar]
- 21.Gradia S., Acharya,S. and Fishel,R. (1997) The human mismatch recognition complex hMSH2-hMSH6 functions as a novel molecular switch. Cell, 91, 995–1005. [DOI] [PubMed] [Google Scholar]
- 22.Acharya S., Wilson,T., Gradia,S., Kane,M.F., Guerrette,S., Marsischky,G.T., Kolodner,R. and Fishel,R. (1996) hMSH2 forms specific mispair-binding complexes with hMSH3 and hMSH6. Proc. Natl Acad. Sci. USA, 93, 13629–13634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Palombo F., Palombo,F., Gallinari,P., Iaccarino,I., Lettieri,T., Hughes,M., D’Arrigo,A., Truong,O., Hsuan,J.J. and Jiricny,J. (1995) GTBP, a 160-kilodalton protein essential for mismatch-binding activity in human cells. Science, 268, 1912–1914. [DOI] [PubMed] [Google Scholar]
- 24.Matton N., Simonetti,J. and Williams,K. (1999) Inefficient in vivo repair of mismatches at an oncogenic hotspot correlated with lack of binding by mismatch repair proteins and with phase of the cell cycle. Carcinogenesis, 20, 1417–1424. [DOI] [PubMed] [Google Scholar]
- 25.Mimori T. and Harding,J.A. (1986) Mechanism of interaction between Ku protein and DNA. J. Biol. Chem., 261, 10375–10379. [PubMed] [Google Scholar]
- 26.Lindahl T. and Nyberg,B. (1974) Heat-induced deamination of cytosine residues in DNA. Biochemistry, 13, 3405–3410. [DOI] [PubMed] [Google Scholar]
- 27.Horwath M., Kramer,W. and Kunze,R. (2002) Structure and expression of the Zea mayS MutS-homologs Mus1 and Mus2. Theor. Appl. Genet., 105, 423–430. [DOI] [PubMed] [Google Scholar]
- 28.Bowers J., Sokolsky,T., Quach,T. and Alani,E. (1999) A mutation in the MSH6 subunit of the Saccharomyces cerevisiae MSH2MSH6 complex disrupts mismatch recognition. J. Biol. Chem., 274, 16115–16125. [DOI] [PubMed] [Google Scholar]
- 29.Drotschmann K., Yang,W., Brownewell,F.E., Kool,L.T. and Kunkel,T.A. (2001) Asymmetric recognition of DNA local distortion. Structure-based functional studies of eukaryotic Msh2-Msh6. J. Biol. Chem., 276, 46225–46229. [DOI] [PubMed] [Google Scholar]
- 30.Natrajan G.E., Lamers,M.H., Enzlin,J.H., Winterwerp,H.H.K., Perrakis,A. and Sixma,T.K. (2003) Structures of Escherichia coli DNA mismatch repair enzyme MutS in complex with different mismatches: a common recognition mode for diverse substrates. Nucleic Acids Res., 31, 4814–4821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ade J., Belzile,F., Phillippe,H. and Doutriax,M.-P. (1999) Four mismatch repair paralogues coexist in {IArabidopsis thaliana: AtMSH2, AtMSH3, AtMSH6-1} and {IAtMSH6-2}. Mol. Gen. Genet., 262, 239–249. [DOI] [PubMed] [Google Scholar]
- 32.Sears E.R. (1976) Genetic control of chromosome pairing in wheat. Annu. Rev. Genet., 10, 31–51. [DOI] [PubMed] [Google Scholar]
- 33.Wall A.M., Riley,R. and Chapman,V. (1971) Wheat mutants permitting homeologous meiotic pairing. Genet. Res., 18, 311–328. [Google Scholar]