

Abstract
We examined the effectiveness of cytotoxic T lymphocyte–associated antigen 4 (CTLA-4) blockade, alone or in combination with a granulocyte/macrophage colony-stimulating factor (GM-CSF)–expressing tumor cell vaccine, on rejection of the highly tumorigenic, poorly immunogenic murine melanoma B16-BL6. Recently established tumors could be eradicated in 80% (68/85) of the cases using combination treatment, whereas each treatment by itself showed little or no effect. Tumor rejection was dependent on CD8+ and NK1.1+ cells but occurred irrespective of the presence of CD4+ T cells. Mice surviving a primary challenge rejected a secondary challenge with B16-BL6 or the parental B16-F0 line. The same treatment regimen was found to be therapeutically effective against outgrowth of preestablished B16-F10 lung metastases, inducing long-term survival. Of all mice surviving B16-BL6 or B16-F10 tumors after combination treatment, 56% (38/68) developed depigmentation, starting at the site of vaccination or challenge and in most cases progressing to distant locations. Depigmentation was found to occur in CD4-depleted mice, strongly suggesting that the effect was mediated by CTLs. This study shows that CTLA-4 blockade provides a powerful tool to enhance T cell activation and memory against a poorly immunogenic spontaneous murine tumor and that this may involve recruitment of autoreactive T cells.
Keywords: CTLA-4, immunotherapy, autoimmunity, vitiligo, melanoma
Recent work has shown that unaltered self-antigens aberrantly expressed in tumors or expressed in a tissue-specific fashion can be recognized by T cells isolated from mice or human cancer patients (for review see references 1 and 2). This finding suggests that autoreactive T cells escape thymic deletion and reach the periphery, where they can in some instances be activated and involved in antitumor immune responses. It is generally believed that these autoreactive T cells display relatively low avidity 3 but can be effective when activated under proper circumstances 4 5 6 7. In addition to the characterization of these self-antigens targeted in antitumor responses, our understanding of the requirements for proper T cell activation have provided possible explanations for the absence of tumor-specific immunity.
Full activation of naive T cells requires stimulation of the TCRs by corresponding peptide–MHC complexes, as well as costimulation through engagement of CD28 by B7.1 or B7.2 (B7) on the APCs (for review see reference 8). Stimulation of T cells by antigen in the absence of costimulatory signals can result in unproductive T cell stimulation or T cell tolerance 9. The lack of expression of B7 by tumor cells was shown to be one factor that can contribute to their failure to elicit productive immune responses 10 11. CTLA-4 is a second counterreceptor for B7 that plays an inhibitory role in T cell activation. Accumulating data suggests that CTLA-4 engagement downregulates T cell responses by raising the threshold of signals needed for effective T cell activation, although it is possible that CTLA-4 might also play a role in terminating ongoing T cell responses 12. In vivo, monoclonal antibodies that block CTLA-4/B7 interactions have been shown to enhance CD4+ T cell expansion in response to a variety of stimuli, including peptide antigens, superantigen, and parasites, and can exacerbate and accelerate autoimmune disease in murine models of diabetes and experimental autoimmune encephalitis (for review see reference 12). It has been reported that blockade of CTLA-4/B7 interactions prevents induction of peripheral T cell tolerance upon vaccination with peptides under tolerogenic conditions, suggesting that CTLA-4 might be actively involved in the induction of anergy 13.
We have previously shown that CTLA-4–blocking antibodies accelerate rejection of B7-transfected tumor cells and can induce rejection of large, established B7-negative tumors 14. When applied to a variety of tumor models, we found that susceptibility to anti–CTLA-4–induced rejection correlated with susceptibility to B7-induced rejection (Leach, D.R., manuscript in preparation; reference 15). This suggests that susceptibility to CTLA-4–induced regression is related to the inherent immunogenicity of the tumor. Thus, immunogenic tumors such as the fibrosarcoma Sa1/N, 51BLim10, RENCA, and the prostate carcinoma TRAMP/C1 were completely rejected by injection of CTLA-4–blocking antibodies, whereas outgrowth of poorly immunogenic tumors such as the melanoma B16-BL6 or the mammary tumor SM1 was minimally affected (14 16; Leach, D.R., manuscript in preparation). Synergy with a GM-CSF tumor cell vaccine was demonstrated in the case of the SM1 tumor 17. Although these studies did not directly demonstrate enhanced tumor-specific T cell activity as a result of CTLA-4 blockade, in vivo depletion experiments demonstrated that both CD4+ and CD8+ T cells were required for rejection of the immunogenic tumors 51BLim10, Sa1/N, and SM1 17. NK1.1+ cells were found to also play an intriguing but not yet defined role in the eradication of TRAMP/C1 by CTLA-4 (Hurwitz, A.A. and J.P. Allison, unpublished observations).
In this study, we show that the combination of CTLA-4 blockade and GM-CSF–producing vaccines is therapeutically effective against the highly tumorigenic and poorly immunogenic melanoma B16-BL6 in a mechanism dependent on CD8+ and NK1.1+ cells but independent of CD4+ T cells. Mice cured from established subcutaneous B16-BL6 tumors are immune to rechallenge with B16-BL6 or the parental line B16-F0 after 4 mo. We further show that B16-F10 pulmonary metastases can be eradicated by the combination treatment and that metastatic lesions from these mice show extensive infiltration by mononuclear cells. In both the subcutaneous and metastatic melanoma models, we found that surviving mice developed depigmentation, indicating that autoimmunity directed against pigmented cells was concurrently induced. As animals depleted of CD4+ T cells also developed depigmentation, it is very likely that this autoimmune phenomenon is induced by CD8+ T cells directed against pigmentation antigens. This model is well suited to studying the significance of autoreactive CD8+ T cells in antitumor responses as well as investigating the role of CTLA-4 in peripheral tolerance in a preclinical setting relevant to the immunotherapy of cancer.
Materials and Methods
Mice.
C57BL/6 female mice (obtained from Charles River Labs/National Cancer Institute) were maintained and treated in accordance with National Institutes of Health and American Association of Laboratory Animal Care regulations and used for tumor experiments when 8–12 wk old. All subcutaneous injections were performed after mice inhaled of the anaesthetic methoxyflurane.
Antibodies.
Generation and purification of the hamster anti-murine CTLA-4 antibody 9H10 has been described in previous work 18. Similarly, GK1.5 (anti-CD4), 2.43 (CD8), PK136 (NK1.1), and 116.3 (Lyt2.1; rat IgG, obtained from B.J. Fowlkes, National Institute of Allergy and Infectious Diseases, Bethesda, MD) were prepared in our laboratory as ascites or purified from supernatant using standard procedures. Mouse IgG and hamster IgG were purchased from Jackson ImmunoResearch Labs., Inc., and rat IgG was from Sigma Chemical Co. RM4.4–PE (CD4), anti-CD8b2–PE, and DX5 (pan-NK) were obtained from PharMingen and were used to confirm depletions of the relevant population.
Cell Lines and GM-CSF Gene Transduction.
B16-BL6, B16-F10 (obtained from Dr. I. Fidler, MD Anderson Cancer Center, Houston, TX), B16-F0 (American Type Culture Collection), and DC2.4 19 were cultured in DMEM supplemented with 1 U/ml penicillin, 1 μg/ml streptomycin, 50 μg/ml gentamycin, 2 μM l-glutamine, and 8% FCS (hereafter referred to as complete DMEM). The C57Bl/6-derived tumor cell lines EL4 (thymoma) and MC38 (colorectal carcinoma; obtained from Dr. N. Restifo, National Cancer Institute, Bethesda, MD) were maintained in RPMI supplemented with antibiotics, l-glutamine, 20 μM β-ME, and 8% FCS. GM-CSF–producing B16-BL6 and B16-F10 were obtained by retroviral transduction 20. GM-CSF production by short-term lines (F10) or clones (BL6) was tested by ELISA using commercially available antibodies to murine GM-CSF (PharMingen). Clones BL6/GM-E, BL6/GM-18, BL6/GM-45, BL6/GM-52 (producing 5, 20, 40, or 50 ng GM-CSF/106 cells/24 h, respectively), and the line F10/g (producing 30–40 ng/106 cells/24 h) were cultured using complete DMEM. GM-CSF production was routinely confirmed in vitro during the course of vaccination experiments.
Subcutaneous Challenge and Treatment Experiments.
Mice were shaved on the back and challenged subcutaneously with 104 B16-BL6 cells in PBS. At the same day or later as indicated, treatment was initiated by injecting 106 irradiated (16,000 rads) GM-CSF–producing cells (in PBS) subcutaneously into the left flank and repeated 3 and 6 d later. The vaccine consisted of a 1:1 mixture of clones BL6/GM-E and BL6/GM-18. Treatment with 9H10 or control hamster IgG was started simultaneously or 3 d later with similar results. Antibodies were delivered intraperitoneally at 100 μg in PBS, usually followed by two 50-μg injections every 3 d. Tumor growth was scored by measuring perpendicular diameters. Mice were killed when the tumors displayed severe ulceration or reached a size of 300 mm2. Depletion of T or NK cells was accomplished by injection of the relevant antibodies (500 μg, i.p.) 7, 6, and 5 d before tumor challenge and maintained by injections every 10 d during the experiment. Depletions were confirmed in lymph nodes and spleens 1 d before challenge by flow cytometry using noncross-reactive antibodies. Routinely, <1% CD4+ T cells, CD8+ T cells, or NK1.1+ cells were detected in lymph nodes (after CD4 or CD8 depletion) or spleens (NK1.1 depletion), whereas mice treated with control antibodies (mouse IgG, rat IgG, or 116.3) demonstrated unchanged lymphocyte profiles as compared with untreated mice.
Treatment of Lung Metastases.
To establish lung metastases, mice were injected intravenously with 5 × 104 or 105 B16-F10 cells. Treatment using irradiated F10/g cells and antibodies was started after 24 h, following the same protocol as outlined for treatment of subcutaneous tumors. After 25 d, lungs were harvested from each treatment group and surface metastases were counted using a dissection microscope. Paraffin-embedded lung sections were stained with hematoxylin–eosin using standard procedures. For survival experiments, 5 × 104 B16-F10 cells were injected intravenously and treatment was started the next day.
Generation of CTL Cultures and IFN-γ Release Assay.
Spleens were harvested from mice rejecting B16-BL6 and restimulated in vitro with B16-BL6/B7.1 or a mixture of B16-F0 and the dendritic cell line DC2.4 after overnight coculture. 5 × 106 spleen cells were mixed with 105 irradiated (16,000 rads) stimulator cells, and recombinant human IL-2 was added to a final concentration of 30 IU/ml. After 7 d, cells were collected and purified by Histopaque (Sigma-Aldrich) gradient centrifugation. Live cells (2.5 × 105 per well) were stimulated with target cells (5 × 104 per well) in 96-well round-bottom plates for 24 h, after which supernatant was collected and tested for the presence of IFN-γ by sandwich ELISA (PharMingen).
Results
CTLA-4 Blockade Together with GM-CSF–producing Cellular Vaccines Causes Rejection of Established B16-BL6 Tumors.
B16-BL6 was originally derived from the spontaneous murine melanoma cell line B16-F0 by in vivo selection for invasiveness 21. Both the parental line and its variant express low levels of H-2Kb and Db, and MHC class II is undetectable by flow cytometry in vitro and ex vivo (data not shown). Vaccination with irradiated B16-BL6 does not protect against subsequent challenge with live B16-BL6 cells, nor does B7.1 expression result in any significant change in tumor growth in vivo (20 22; our unpublished results). By these criteria, B16-BL6 is a very poorly immunogenic tumor. In previous experiments, we had found that CTLA-4 blockade was not therapeutically effective against poorly immunogenic tumors such as B16-BL6. We also found that vaccination with irradiated B16-BL6 cells in combination with anti–CTLA-4 was ineffective (data not shown). We hypothesized that this might be due to insufficient presentation of tumor antigens by host APCs. Therefore, we chose to combine CTLA-4 blockade with GM-CSF–producing irradiated B16-BL6 whole cell vaccine, which was described by others as the most effective prophylactic vaccine against B16 20 and augmented immunity against SM1 17. Presumably, GM-CSF production at the site of vaccination might attract host APCs and enhance their function in vivo. C57BL/6 mice were challenged with 104 B16-BL6 cells subcutaneously and subsequently treated starting on the same day or 4–12 d later. A representative experiment is shown in Fig. 1. Administration of anti–CTLA-4 antibody 9H10 or control hamster IgG by themselves had no effect on growth of B16-BL6 tumors. Vaccination with irradiated GM-CSF–producing B16-BL6 cells along with control antibody delayed growth when initiated at the time of tumor implantation but had no effect when treatment was delayed. However, the combination of GM-CSF–producing vaccine and CTLA-4 blockade induced rejection of all tumors injected the same day or 4 d earlier. One of five mice carrying a day 8 B16-BL6 tumor rejected a small palpable tumor after combination treatment including CTLA-4 blockade. The growth of tumors established 12 d earlier was also delayed by the combination treatment, although rejection was not obtained. When the data from a series of 10 experiments were combined, an overall success rate of combination treatment of 80% was achieved (68/85 mice cured) when treatment was begun at day 0 or 4 d after tumor implantation (Table ). These results corroborate the finding that CTLA-4 blockade and GM-CSF–producing vaccines act synergistically to cause rejection of poorly immunogenic tumors 17.
Figure 1.

Successful treatment of preestablished B16-BL6 using anti–CTLA-4 and GM-CSF–producing BL6 vaccine. C57BL/6 female mice (five per group) were injected with 104 B16-BL6 cells subcutaneously on the back, on the same day (A) or 4, 8, or 12 d (B–D) before treatment was started. Treatment consisted of three consecutive injections (in a 6-d time frame as indicated in Materials and Methods) of anti–CTLA-4 antibody 9H10 intraperitoneally (•), control hamster IgG (100, 50, 50 μg; ○), or 106 irradiated BL6/g cells subcutaneously, in combination with 9H10 (▪) or hamster IgG (□). Tumor growth (mm2) was scored by measuring perpendicular diameters and was averaged for all mice within each group. In some treatment groups, only a fraction of the mice (indicated between brackets) developed a tumor.
Table 1.
Combination Treatment of B16-BL6 Using Anti–CTLA-4 Plus GM-CSF–producing Vaccine
| Experiment no. | B16-BL6 Day of challenge | Treatment schedule | Fraction of mice responding per treatment group | Depigmentation(fraction of responding mice) | |||
|---|---|---|---|---|---|---|---|
| Control IgG | Anti–CTLA-4 | BL6/GM-CSF plus control IgG | BL6/GM-CSF plus anti–CTLA-4 | ||||
| 1 | 0 | A | 0/10 | — | — | 7/9 | 4/7 |
| 2 | 0 | A | 0/5 | 0/5 | 2/5 | 5/5 | 3/5 |
| B | 5/5 | 3/5 | |||||
| C | 5/5 | 1/5 | |||||
| 3 | −4 | A | 0/5 | 0/5 | 0/5 | 3/5 | 1/3 |
| 4 | 0 | A | 0/5 | — | 0/5 | 6/8 | 3/6 |
| D | 2/5 | 3/5 | 2/3 | ||||
| 5 | 0 | E | 0/5 | 0/5 | 0/5 | 2/5 | 2/2 |
| F | 2/5 | 3/5 | 2/3 | ||||
| 6 | 0 | A | 0/5 | — | — | 6/10 | 4/6 |
| 7 | 0 | A | 0/5 | 0/5 | 1/5 | 4/5 | 1/4 |
| 8 | 0 | A | 0/5 | 0/5 | 0/5 | 9/9 | 6/9 |
| 9 | −4 | A | 0/5 | 0/5 | 0/5 | 5/5 | 3/5 |
| 10 | 0 | A | 0/5 | 0/5 | 1/5 | 5/5 | 3/5 |
| Total number of mice responding: | 0/55 | 0/35 | 8/50 | 68/85 | 36/68 | ||
| Percentage: | 0 | 0 | 16 | 80 | 56 | ||
A single dose of GM-CSF–producing vaccine administered on the same day as tumor challenge was sufficient to eradicate tumors in all of the mice when combined with CTLA-4 blockade (Fig. 2). Similarly, a single dose of anti–CTLA-4 after three vaccinations with GM-CSF–producing cells was sufficient to induce B16-BL6 rejection (not shown). GM-CSF production by the vaccine was found to be critical for the synergistic effect, as vaccination with irradiated untransduced B16-BL6 cells in combination with anti–CTLA-4 antibodies was not effective, as had been found previously for synergistic treatment of SM1 (data not shown; reference 17).
Figure 2.

A single dose of GM-CSF–producing vaccine cooperates with CTLA-4 blockade to induce 100% cure of B16-BL6. Mice were inoculated subcutaneously with 104 B16-BL6 cells. On the same day, combination treatment was initiated using triple BL6/g vaccine (days 0, 3, and 6) combined with either hamster IgG (100, 50, 50 μg on days 3, 6, and 9; □) or anti–CTLA-4 (▪). Control treatments consisted of antibody injections alone: hamster IgG (○) or anti–CTLA-4 (•). Also, anti–CTLA-4 treatment was combined with a single (▴) or double injection (♦) of the BL6/g vaccine. Average tumor size was calculated for all mice within a treatment group (mm2). The fraction of mice developing tumors is shown between brackets.
Combination of CTLA-4 Blockade and GM-CSF–producing Vaccines Induces Effective Immunity to Rechallenge with B16-BL6.
To determine whether mice cured from the initial challenge of B16-BL6 had developed immunity to rechallenge, surviving mice received a second challenge of 2 × 104 B16-BL6 on the left flank 128 d after the primary challenge. Also, resistance to the parental B16-F0 melanoma cell line was tested by injecting 2 × 104 cells into the right flank. Naive age-matched control mice grew both tumors and required euthanasia within 30 d. All mice cured from a primary challenge with B16-BL6 rejected B16-F0. Within the first experiment, the two mice that had rejected the primary challenge after BL6/g vaccination alone were unable to reject a secondary B16-BL6 challenge (Table ). In contrast, seven out of nine mice that received BL6/g vaccine plus anti–CTLA-4 also rejected B16-BL6 (Table ). In two rechallenge experiments combined, 20/24 mice cured from B16-BL6 by combination treatment were immune to secondary challenge with B16-BL6, and 11 mice were resistant to rechallenge with B16-F0. Only four of eight mice cured upon vaccination with GM-CSF–producing cells alone were resistant to rechallenge, but the few mice surviving a primary tumor after treatment with BL6/GM-CSF vaccine alone did not allow any conclusion to be drawn as to the possible enhancement of memory formation by anti–CTLA-4 (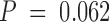 , NS; Table ). Although immunity to rechallenge with B16-BL6 was not found in 100% of mice cured by the combination treatment, the fact that B16-F0 was rejected by all suggests that mice surviving a primary challenge with B16-BL6 had mounted adequate memory to an antigen(s) shared between the parental line and its more invasive variant.
, NS; Table ). Although immunity to rechallenge with B16-BL6 was not found in 100% of mice cured by the combination treatment, the fact that B16-F0 was rejected by all suggests that mice surviving a primary challenge with B16-BL6 had mounted adequate memory to an antigen(s) shared between the parental line and its more invasive variant.
Table 2.
Treatment of B16-BL6 Using Anti–CTLA-4 Facilitates Development of Memory to Rechallenge
| Primary treatment | Tumor incidence at secondary challenge | |
|---|---|---|
| B16-BL6 | B16-F0 | |
| Experiment 1 (rechallenge on day 128) | ||
| BL6/GM plus hamster IgG | 2/2 | 0/2 |
| BL6/GM plus anti–CTLA-4 | 2/9* | 0/9 |
| None | 5/5 | 5/5 |
| Experiment 2 (rechallenge on day 100–130) | ||
| BL6/GM plus hamster IgG | 2/6 | — |
| BL6/GM plus anti–CTLA-4 | 2/15 | — |
| None | 5/5 | — |
CD8+ and NK1.1+ but not CD4+ Cells Are Required for Combination Treatment of B16-BL6.
To determine the involvement of T and NK cells in the rejection of B16-BL6, mice were depleted of CD4+, CD8+, or NK1.1+ cells before tumor challenge. Treatment was started on the same day as tumor implantation following the general schedule of three simultaneous injections of vaccine and anti–CTLA-4. Depletion of CD8+ cells abrogated the effect of treatment (Table ; 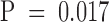 compared with control rat IgG). Mice depleted of NK1.1+ cells were also largely unable to reject their tumors (8/10). We observed that the tumor-bearing, NK-depleted mice had developed multiple tumors at the site of challenge, suggesting that NK cells could be involved in the first line of defense against the MHC class Ilo B16-BL6 challenge. Surprisingly, CD4+ T cells were not required for tumor rejection. In fact, 80% of the CD4-depleted mice rejected their tumors after treatment with anti–CTLA-4 and GM-CSF vaccine under suboptimal conditions where 50–60% of the control groups rejected B16-BL6 (Table ). Depletion of both CD4+ and CD8+ cells abolished the therapeutic effect. It is apparent that CD8+ T cells and, most likely, NK1.1+ cells are necessary for rejection of B16-BL6 using CTLA-4 blockade and GM-CSF–producing vaccines. Activation of CD8+ T cells involved in rejection of B16-BL6 does not appear to be dependent on CD4 help in this system.
compared with control rat IgG). Mice depleted of NK1.1+ cells were also largely unable to reject their tumors (8/10). We observed that the tumor-bearing, NK-depleted mice had developed multiple tumors at the site of challenge, suggesting that NK cells could be involved in the first line of defense against the MHC class Ilo B16-BL6 challenge. Surprisingly, CD4+ T cells were not required for tumor rejection. In fact, 80% of the CD4-depleted mice rejected their tumors after treatment with anti–CTLA-4 and GM-CSF vaccine under suboptimal conditions where 50–60% of the control groups rejected B16-BL6 (Table ). Depletion of both CD4+ and CD8+ cells abolished the therapeutic effect. It is apparent that CD8+ T cells and, most likely, NK1.1+ cells are necessary for rejection of B16-BL6 using CTLA-4 blockade and GM-CSF–producing vaccines. Activation of CD8+ T cells involved in rejection of B16-BL6 does not appear to be dependent on CD4 help in this system.
Table 3.
Involvement of Lymphocyte Subsets in Rejection of B16-BL6 through Cotreatment with Anti–CTLA-4 and BL6/GM Vaccine
| Depletion | B16-BL6 tumor take | Remarks |
|---|---|---|
| CD4 | 2/10 | Depigmentation (4/8 survivors) |
| CD8 | 9/10 | — |
| CD4 plus CD8 | 5/5 | — |
| NK1.1 | 8/10 | Multiple tumors developed at injection site, no depigmentation |
| Control mouse IgG | 5/10 | Depigmentation (3/5 survivors) |
| Control rat IgG | 4/10 | Depigmentation (4/6 survivors) |
| No depletion | 5/10 | Depigmentation (3/5 survivors) |
| No depletion, no treatment | 10/10 | — |
Depletion of lymphocyte subsets was achieved by injecting depleting antibodies GK1.5 (anti-CD4), 2.43 (CD8), PK136 (NK1.1), or control antibodies at days −8, −7, −6, and every 7 (GK1.5) to 10 d thereafter. Depletion was checked at day −1. Results are compiled from two experiments.
Generation of B16-specific T Cells Is Strongly Enhanced by CTLA-4 Blockade In Vivo.
To determine if tumor-reactive T cells were induced by the combination therapy, mice were immunized with BL6/g plus anti–CTLA-4 or control IgG and challenged with B16-BL6 after 4 wk. 10 d after challenge, spleens from four mice in each group were pooled and restimulated with B16-BL6/B7.1 or a mixture of B16-F10 and the dendritic cell line DC2.4 19. After one round of restimulation in vitro, specific IFN-γ release was tested using different variants of B16 and two unrelated tumor cell lines expressing the H-2b haplotype, the thymoma EL4 and the colorectal carcinoma MC38. As shown in Fig. 3T cells from mice vaccinated with BL6/g in the presence of control hamster IgG produced very low levels of IFN-γ in this assay. T cells from mice treated with anti–CTLA-4 in vivo had greatly enhanced B16-specific IFN-γ secretion. These results indicate that CTLA-4 blockade during vaccination with BL6/g specifically enhances reactivity toward an antigen (or antigens) expressed by B16 and its variants. In addition, all splenocyte cultures established from mice that were long-term (3–10 mo) survivors after combination treatment were found to specifically react with B16 and its variants, as tested by IFN-γ release after one round of restimulation in vitro (data not shown). Successful rejection of B16-BL6 coincides with the generation of tumor-specific T cell activity.
Figure 3.


Anti–CTLA-4 enhances IFN-γ production by B16-specific T cells induced in vivo. Mice (four per group) were vaccinated with irradiated BL6/g (106 per mouse) and cotreated with control hamster IgG (A) or anti–CTLA-4 (B). After 4 wk, mice were challenged with 2 × 104 B16-BL6, and 10 d later, splenocytes were pooled and restimulated in vitro using B16-BL6/B7.1 (open bars) or a mixture of B16-F10 and DC2.4 dendritic cells (filled bars). On day 8, cultures were tested for tumor-specific IFN-γ release as described in Materials and Methods. Targets included B16 sublines -F0, -F10, and -BL6, as well as unrelated H-2b tumors EL4 and MC38.
Suppression of B16-F10 Lung Metastases and Induction of Long-Term Survival by Combination Treatment.
We next sought to determine whether anti–CTLA-4 combined with vaccination would be effective against metastatic disease. 105 B16-F10 cells (selected for metastasis exclusively to the lungs) were injected intravenously, and treatment was started 1 d later. On day 25, mice were killed and surface lung metastases were counted. Treatment with anti–CTLA-4 alone did not have any appreciable effect on the lung metastasis count as compared with control IgG (Table ). Immunization with F10/g reduced the number of metastases in a few mice. Treatment of F10/g-vaccinated mice with anti–CTLA-4 further suppressed lung colonization and completely inhibited pulmonary metastases in two of five mice sampled. Histological analysis of these lung samples demonstrated that CTLA-4 blockade in combination with F10/g vaccination was associated with infiltration of mononuclear cells in all of the metastases stained and observed in three of the five tumor-bearing lungs (the two remaining sets of lungs were found to be tumor free) (Fig. 5). Neither anti–CTLA-4 nor F10/g vaccination alone resulted in lymphocytic infiltration in lung tumors or surrounding tissue. A few polymorphonuclear cells were observed in the smaller metastases from mice vaccinated with F10/g in the presence of control IgG, but there were no extensive infiltrates in larger lesions in any of the control groups. The observation that the combination therapy had at least some effect in enhancing infiltration and reducing lung metastases led us to test its effectiveness in increasing survival, as shown in Fig. 4. Mice challenged with 5 × 104 B16-F10 cells and treated with control hamster IgG all (10/10) succumbed to lung failure due to extensive metastatic disease by day 75 after injection. Anti–CTLA-4 by itself prolonged survival, as did vaccination with F10/GM. However, 13/13 mice receiving the combination treatment were still alive by day 80 (Fig. 4). Lungs taken from these surviving mice did not demonstrate metastatic lesions on their surfaces. This is the first demonstration that CTLA-4 blockade in vivo is therapeutically effective against disseminated disease.
Table 4.
Reduced Number of B16-F10 Lung Metastases after Combination Treatment with Anti–CTLA-4 and F10/GM Vaccine
| Treatment of lung metastases | Lung metastasis count |
|---|---|
| Control hamster IgG | >200, >200, >200, 25, 16 |
| Anti–CTL-4 | >200, >200, >200, >200, >200 |
| Hamster IgG plus F10/GM vaccine | >200, >200, 35, 49, 4 |
| Anti–CTLA-4 plus F10/GM vaccine | 87, 28, 6, 0, 0 |
B16-F10 (105, i.v.)-induced lung metastases were treated with hamster IgG, 9H10, and F10/GM vaccine in combination with either antibody on days 1, 4, and 7 after challenge. Surface lung metastases were counted under a dissecting microscope 25 d after inoculation. Counts are shown for each individual mouse; all counts over 200 were scored as >200.
Figure 5.
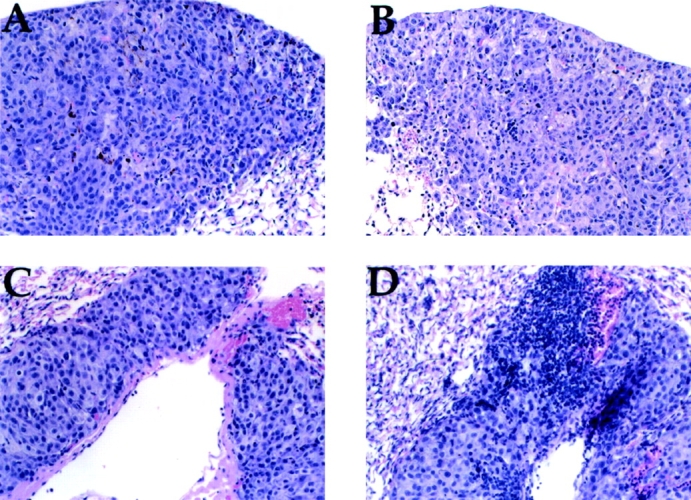
B16-F10 metastases demonstrate lymphocytic infiltration after treatment with anti–CTLA-4 and F10/g vaccine. Mice injected with 105 B16-F10 intravenously and treated with control hamster IgG (A), 9H10 (B), or F10/g vaccine in combination with either hamster IgG (C) or 9H10 (D) on days 1, 4, and 7, as outlined in the Fig. 4 legend. On day 25, lungs were harvested, fixed in 10% neutral-buffered formalin, and processed for hematoxylin–eosin staining.
Figure 4.

Mice bearing B16-F10 lung metastases show enhanced survival when treated with anti–CTLA-4 and F10/g vaccine. B16-F10 cells (5 × 104 per mouse) were injected into the tail vein and 24 h later, treatment was started using control hamster IgG (10 mice, ○), anti–CTLA-4 antibody 9H10 (9 mice; •), irradiated F10/g (106 subcutaneously) in combination with hamster IgG (10 mice; □) or 9H10 (13 mice; ▪) on days 1, 4, and 7, according to the dosing schedule used for subcutaneous tumors (see Fig. 1 legend). Mice were followed for survival, and in some subjects death due to extensive pulmonary metastasis was confirmed by harvesting lungs postmortem.
Mice Surviving Subcutaneous B16-BL6 Tumors or B16-F10 Lung Metastases Develop Skin and Hair Depigmentation.
Within 4–8 wk after challenge, 56% (38/68 cured mice) of the surviving mice developed depigmentation, starting at the sites of vaccination (left flank) and challenge (back) (Fig. 6 A). Moreover, depigmentation was observed at the site of vaccination in a similar proportion of mice surviving B16-F10 lung metastases (Fig. 6 B). Rejection of a B16-BL6 tumor established 8 d before start of treatment (Fig. 1) induced fast and progressive depigmentation appearing within 25 d after challenge and spreading to distant sites, indicating that a relatively strong antitumor response resulted in rapid manifestation of progressive depigmentation (Fig. 6 C). Depigmentation did occur in mice that received combination treatment in a prophylactic setting but at reduced frequency (not shown). Interestingly, depigmentation was not dependent on the presence of CD4+ T cells, as four of eight CD4-depleted mice rejecting their tumors also developed progressive depigmentation (Table ). In some cases, tumor-bearing mice (moribund despite treatment with anti–CTLA-4 and BL6/GM) were found to develop small areas of hair depigmentation at the site of progressive tumor growth. Depigmentation was never observed in the mice that were treated by BL6/GM-CSF vaccination without CTLA-4 blockade or in any of the other treatment groups. These findings suggest that CTLA-4 blockade allows for the activation of autoreactive lymphoid cells that are involved in rejection of a tumor derived from the melanocytic lineage and may also mediate rejection of normal pigment-containing cells in the skin and hair follicles expressing pigmentation antigens.
Figure 6.



Rejection of B16-BL6 or B16-F10 as a result of treatment with anti–CTLA-4 and GM-CSF–producing vaccines causes autoimmune skin and hair depigmentation. After successful treatment for B16-BL6 subcutaneously or B16-F10 intravenously, C57Bl/6 mice developed skin and hair depigmentation. (A) Depigmentation of both sites of vaccination and challenge, after rejection of a day 0 tumor. (B) Progressive depigmentation found in a mouse rejecting a B16-BL6 subcutaneous tumor, established 8 d before treatment started. (C) Depigmentation at the site of vaccination of a mouse cured from preestablished B16-F10 lung metastases.
Discussion
In this study, we show that administration of anti–CTLA-4 antibody, when combined with an irradiated GM-CSF–producing tumor cell vaccine, results in rejection of previously established primary tumors and resistance to secondary challenge in mice inoculated with the nonimmunogenic melanoma B16-BL6. Similarly, this combination treatment led to the eradication of B16-F10 lung metastases. The combination treatment induced massive infiltration of mononuclear cells into the remaining lung metastases. Tumor rejection by the combination treatment was reflected by an enhancement of B16-specific T cell responses in vitro. After tumor eradication, 56% of the surviving mice developed depigmentation of the hair (Table ). Both tumor rejection and subsequent depigmentation were dependent on the presence of CD8+ T cells and NK1.1+ cells but did not require CD4+ T cells.
We have found that treatment with anti–CTLA-4 is sufficient to obtain rejection of many, but not all, experimental tumors 14 16 17. The effectiveness of CTLA-4 blockade appears to correlate with that of B7-positive tumor cell vaccines, suggesting that it is most effective against tumors with a significant degree of intrinsic immunogenicity. The lack of therapeutic effectiveness of CTLA-4 blockade by itself on B16-BL6 can most likely be attributed to the poor capacity of this tumor to provide antigens to host APCs. GM-CSF–transduced tumor cells have been shown to induce potent immunity to a variety of tumors, including B16 20 22. The effectiveness of GM-CSF in these systems can probably be attributed to the capacity of this cytokine to attract host bone marrow–derived APCs and enhance their differentiation, thereby increasing their capacity to capture tumor-derived antigens in the local environment of the irradiated tumor cell vaccine 23 24 25. Although this immunization strategy has been shown to greatly enhance the immunogenicity of B16 cells and leads to resistance to subsequent challenge with viable tumor cells, the response elicited by irradiated GM-CSF tumor cells is only marginally if at all effective in the treatment of preestablished tumors. We obtained tumor rejection in 16% of mice treated with the vaccine alone (Table ), and then only when it was administered on the same day as tumor challenge. These results suggest that GM-CSF vaccine has a limited potential to elicit an effector cell response of sufficient potency to obtain rejection in tumor-bearing mice. The potency of the combination of the vaccine and anti–CTLA-4 antibody can likely be attributed to enhanced cross-priming of T cells by host APCs by the vaccine, together with a highly potentiated T cell response as a result of the removal of the inhibitory effects of CTLA-4 by antibody blockade. This results in a synergistic enhancement of the T cell response to a level capable of eliminating the preexisting tumor cell mass. This could occur as a consequence of activation of a larger number of naive T cells due to a lowering of the threshold for activation or a more sustained response due to temporary removal of signals involved in terminating the response 12. Rejection is accompanied by long-lived memory, as indicated by the fact that cured mice reject rechallenge in the absence of treatment 4 mo after the initial treatment.
Whereas the combination treatment resulted in an overall cure rate of 80% in mice treated on day 4 or before, effectiveness was much lower when initiated at day 8 and was essentially ineffective at day 12 or later. This is in contrast to our previous findings that CTLA-4 blockade by itself was quite effective in the treatment of well established tumors in other model systems. Subcutaneous tumors of the colon carcinoma 51Blim10 or the fibrosarcoma Sa1N could be eradicated when antibody was administered beginning as late as 2 wk after tumor inoculation, and complete eradication was obtained even when treatment was delayed until the tumors reached a size of 100–140 mm2 (Leach, D.R., manuscript in preparation). The difference in the responses obtained in these experiments and in this study may be related to the relative antigenicity of the systems—more immunogenic targets may also be better targets f or effector T cells than the poorly immunogenic B16-BL6, which might simply be able to outstrip the emerging T cell response. It may be that tumors grow beyond a critical size than can be effectively dealt with by the immune response. It is also possible that loss of effectiveness of the vaccine is a consequence of induction of nonresponsiveness or tolerance in tumor-reactive T cells. It has been reported that treatment with anti–CTLA-4 resulted in rejection of two fibrosarcomas when begun 1–2 wk after inoculation but that late-stage tumors (7–10 wk) were resistant to treatment 26. This loss of effectiveness was accompanied by a loss of in vitro antitumor responses, suggestive of deletion or inactivation of T cells. It has also been shown that growth of a B cell lymphoma engineered to express influenza hemagglutinin results in the progressive inactivation of adoptively transferred T cells bearing hemagglutinin-specific TCRs 27. In this system, administration of anti–CTLA-4 greatly enhanced T cell priming if begun before responses were totally lost but could not reverse tolerance once established 27a. The basis for loss of responsiveness of more established B16-BL6 tumors to the combined treatment regimen remains to be established.
Our results demonstrate that both the therapeutic effect and the subsequent depigmentation obtained with the combination treatment required CD8+ and NK1.1+ cells but was independent of CD4+ T cells. The involvement of NK1.1+ cells in prophylaxis induced by B16/GM-CSF vaccines has been previously noted, especially when MHC Class Iio or I− cells were used 28 29. It is therefore not surprising that eradication of B16-BL6 in our model might require NK1.1+ cells. An important contribution of the NK1.1+ cells may be to lyse cells in the vaccine, thereby enhancing antigen uptake by host APCs recruited to the site of vaccination by GM-CSF in the vaccine.
Our previous studies have revealed that tumor rejection after anti–CTLA-4 treatment, given alone in the case of immunogenic tumors or together with a GM-CSF–transduced vaccine for a poorly immunogenic mammary carcinoma, required both CD4+ and CD8+ cells 14 17. This result could be interpreted as indicative of a requirement for CD4+ T cell help for the effective induction of CD8+ CTLs. However, several observations have suggested a role for CD4+ T cells in antitumor responses beyond provision of help for CTLs. Depletion of CD4+ or CD8+ T cells after immunization but before tumor challenge abrogates the ability of irradiated GM-CSF–producing B16 cells to induce protective immunity 20. CD4+, but not CD8+, T cells were required for the induction of immunity with a GM-CSF–expressing, class I MHC–negative tumor cell vaccine 28. Finally, an extensive analysis using a variety of knockout mice as hosts has shown that CD4+ T cells were absolutely required for the induction of protective immunity using GM-CSF–expressing B16 cells but that absence of CD8+ T cells resulted in only a partial loss of effectiveness 22. This, together with the observation that cytokines elaborated by CD4+ T cells resulted in the recruitment and activation of eosinophiles and macrophages, suggested an additional role for CD4+ T cells in orchestrating CD8+ T cell–independent protective mechanisms when GM-CSF–expressing B16 cell vaccine is used in the setting of prophylaxis.
In the therapeutic setting, our finding that CD4+ T cells are dispensable for obtaining tumor rejection suggests that the combination treatment in this system can allow for direct induction of CD8+ T cell responses, in agreement with what has recently been reported for antiparasite responses 30. One contributing factor might be a high dose and persistence of antigen due to the use of three doses of tumor cell vaccine. It is also possible that CTLA-4 blockade lowers the threshold of stimulation or costimulation that is required for activation of naive T cells. It has recently been shown that a very important mechanism of CD4+ T cell help for the generation of CTLs is an enhancement of antigen presentation and costimulatory activity of dendritic cells as a consequence of engagement of CD40 on the dendritic cell by CD40L on activated CD4+ cells 31 32 33. It is possible that CTLA-4 blockade lowers the threshold of signals needed for CD8+ T cell activation to a level that can be provided by GM-CSF–stimulated dendritic cells in the absence of “licensing” by activated CD4+ T cells.
After eradication of B16-BL6 tumors, 56% of the surviving mice developed depigmentation starting at the sites of vaccination and challenge and spreading to distant sites. Loss of coat color indicated that systemic and progressive autoimmunity had developed toward pigment-bearing cells. For human melanoma patients, a good correlation between autoimmune depigmentation and improved clinical response has been documented 34 35. Melanoma-associated hypopigmentation closely resembles vitiligo, an autoimmune phenomenon that possibly involves antibody and T cell responses against melanocyte antigens 36 37. Genes encoding proteins associated with pigment synthesis or with melanosomes have been cloned and characterized as targets for CTLs in human melanoma patients 1 2. Reinfusion of autologous tumor-infiltrating lymphocytes specifically recognizing gp100/Pmel-17 or tyrosinase led to tumor regressions in some cases, although the value of targeting such antigens is unclear from such clinical studies because the adoptive transfers were performed in the presence of high-dose systemic IL-2 38. Apparently, T cell tolerance against these melanocyte antigens can be broken to induce antitumor reactivity. Currently, several approaches (peptide or genetic vaccination), to (re-)direct melanocyte-reactive CTLs against melanoma are clinically evaluated. The consequences of breaking tolerance to pigment antigens are largely unknown and could be studied in an appropriate murine model.
Two murine melanocyte antigens (TRP2 and Pmel-17/gp100) have been found to serve as CTL antigens in the immune response to B16 tumors 6 39. T cell tolerance toward Pmel-17/gp100 could only be broken by using xenogeneic human gp100. No autoimmune depigmentation was reported after vaccination with peptide or recombinant vaccinia virus or after adoptive transfer of specific CTL clones. In addition to CTLs, potent antibody responses were generated against gp75/TRP1 by vaccinating naive mice with recombinant human gp75, hgp75 DNA, human melanoma cells expressing gp75, or recombinant vaccinia virus expressing human gp75 but not using murine gp75 formulations 40 41 42. Apparently, B cell tolerance toward gp75 was broken by using xenogeneic antigen. Follow-up studies demonstrated that tumor protection required CD4+ and NK1.1+ cells but not CD8+ cells, whereas depigmentation developed in CD4−/− and FcRγ2/− mice in the absence of tumor protection, suggesting that the phenomena are caused by different mechanisms 43. It should be noted that gp75/TRP1 is the most abundant protein in melanocytes and some melanomas, and it can be detected on the cell surface (in contrast to the other melanocyte antigens), which could explain the finding of autoimmune depigmentation associated with anti-gp75 antibodies.
In our system, tumor rejection induced by a combination of the BL6/GM-CSF vaccine and CTLA-4 blockade was followed by depigmentation, which can occur in the absence of CD4+ T cells. Depigmentation was not observed in any of the small number of mice whose tumors were rejected after treatment with the vaccine alone, nor was depigmentation noted in previous studies of GM-CSF/B16 vaccines used for prophylaxis 20 22. It seems likely that depigmentation occurs in our system because the GM-CSF vaccine, when enhanced by CTLA-4 blockade, can elicit CTLs directed to normal melanocyte antigens expressed by the tumor cells, and the same cells responsible for tumor rejection also mediate autoimmune destruction of normal melanocytes. However, it remains possible that antibodies to gp75 or other antigens have some role in depigmentation in intact mice.
In our view, there are at least two nonexclusive explanations for our observation that anti–CTLA-4 antibodies synergize with BL6/GM-CSF vaccine to induce rejection and autoimmunity: (a) CTLA-4 blockade greatly increases the burst size of T cells responding to the GM-CSF vaccine, thus enhancing the mobilization of effector cells, and (b) CTLA-4 blockade lowers the threshold for T cell activation, thereby allowing the recruitment and activation of low-affinity autoreactive T cells that might have escaped central tolerance induction. In either case, autoreactive CTLs involved in tumor rejection could find targets in melanocytes exposed through local inflammation or skin destruction. Although it is an unwanted side effect of treatment, depigmentation or vitiligo is considered to be an acceptable risk for the treatment of melanoma in clinical situations. To our knowledge, this report is the first describing T cell–dependent depigmentation after successful treatment of murine melanoma. Rejection of B16-BL6 through CTLA-4 blockade plus GM-CSF–producing vaccines could serve as a model to study the relationship between tumor immunity and autoimmunity in a setting relevant to the treatment of human cancer.
Acknowledgments
We thank Dana Leach for helpful discussions and support, Jennifer Ziskin and Stan Grell for technical assistance, and Jerry Kapler for expert photography. We also thank Rienk Offringa for critically reading the manuscript.
Andrea van Elsas is the recipient of a postdoctoral fellowship from the Dutch Cancer Society (Nederlandse Kankerbestrijding), Andy Hurwitz is the recipient of the US Army Breast Cancer Initiative fellowship, and James Allison is an investigator of the Howard Hughes Medical Institute. This work was supported in part by National Cancer Institute grant CA57986.
Footnotes
Andrea van Elsas' present address is Department of Immunohematology and Bloodbank, Tumor Immunology lab. E3-Q, Leiden University Medical Center, P.O. Box 9600, 2300 RC Leiden, The Netherlands.
References
- Boon T., Cerottini J.C., Van den Eynde B., van der Bruggen P., Van Pel A. Tumor antigens recognized by T lymphocytes. Annu. Rev. Immunol. 1994;12:337–365. doi: 10.1146/annurev.iy.12.040194.002005. [DOI] [PubMed] [Google Scholar]
- Rosenberg S.A. Cancer vaccines based on the identification of genes encoding cancer regression antigens. Immunol. Today. 1997;18:175–182. doi: 10.1016/s0167-5699(97)84664-6. [DOI] [PubMed] [Google Scholar]
- Liu G.Y., Fairchild P.J., Smith R.M., Prowle J.R., Kioussis D., Wraith D.C. Low avidity recognition of self-antigen by T cells permits escape from central tolerance. Immunity. 1995;3:407–415. doi: 10.1016/1074-7613(95)90170-1. [DOI] [PubMed] [Google Scholar]
- Poplonski L.B., Vukusic B., Pawling J., Clapoff S., Roder J., Hozumi N., Whither J. Tolerance is overcome in beef insulin-transgenic mice by activation of low-affinity autoreactive T cells. Eur. J. Immunol. 1996;26:601–606. doi: 10.1002/eji.1830260315. [DOI] [PubMed] [Google Scholar]
- Morgan D.J., Kreuwel H.T.C., Fleck S., Levitsky H.I., Pardoll D.M., Sherman L.A. Activation of low avidity CTL for a self-epitope results in tumor rejection but not autoimmunity. J. Immunol. 1998;160:643–651. [PubMed] [Google Scholar]
- Overwijk W.W., Tsung A., Irvine K.E., Parkhurst M.R., Goletz T.J., Tsung K., Carroll M.W., Liu C., Moss B., Rosenberg S.A. gp100/pmel 17 is a murine tumor rejection antigeninduction of “self”-reactive tumoricidal T cells using high-affinity, altered peptide ligand. J. Exp. Med. 1998;188:277–286. doi: 10.1084/jem.188.2.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehl S., Hombach J., Aichele P., Rulicke T., Odermatt B., Hengartner H., Zinkernagel R., Pircher H. Viral and bacterial infections interfere with peripheral tolerance induction and activate CD8+ T cells to cause immunopathology. J. Exp. Med. 1998;187:763–774. doi: 10.1084/jem.187.5.763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allison J.P. CD28-B7 interactions in T-cell activation. Curr. Opin. Immunol. 1994;6:414–419. doi: 10.1016/0952-7915(94)90120-1. [DOI] [PubMed] [Google Scholar]
- Schwartz R.H. A cell culture model for T lymphocyte clonal anergy. Science. 1990;248:1349–1356. doi: 10.1126/science.2113314. [DOI] [PubMed] [Google Scholar]
- Townsend S., Allison J.P. Tumor rejection after direct costimulation of CD8+ T cells by B7-transfected melanoma cells. Science. 1993;259:368–370. doi: 10.1126/science.7678351. [DOI] [PubMed] [Google Scholar]
- Chen L., Ashe S., Brady W.A., Hellstrom I., Hellstrom K.E., Ledbetter J.A., McGowan P., Linsley P.S. Costimulation of antitumor immunity by the B7 counterreceptor for the T lymphocyte molecules CD28 and CTLA-4. Cell. 1992;71:1093–1102. doi: 10.1016/s0092-8674(05)80059-5. [DOI] [PubMed] [Google Scholar]
- Thompson C.B., Allison J.P. The emerging role of CTLA-4 as an immune attenuator. Immunity. 1997;7:445–450. doi: 10.1016/s1074-7613(00)80366-0. [DOI] [PubMed] [Google Scholar]
- Perez V.L., Van Parijs L., Biuckians A., Zheng X.X., Strom T.B., Abbas A.K. Induction of peripheral T cell tolerance in vivo requires CTLA-4 engagement. Immunity. 1997;6:411–417. doi: 10.1016/s1074-7613(00)80284-8. [DOI] [PubMed] [Google Scholar]
- Leach D., Krummel M., Allison J.P. Enhancement of antitumor immunity by CTLA-4 blockade. Science. 1996;271:1734–1736. doi: 10.1126/science.271.5256.1734. [DOI] [PubMed] [Google Scholar]
- Hurwitz, A.A., A. van Elsas, D.R. Leach, J. Ziskin, J. Villasenor, T. Truong, and J.P. Allison. 1999. Manipulation of T cell activation to generate anti-tumor CTL. In Cytotoxic Cells: Basic Mechanisms and Medical Applications. M.V. Sitkovsky and P.A. Henkart, editors. Lippincott, Philadelphia. In press.
- Kwon E.D., Hurwitz A.A., Foster B.A., Madias C., Feldhaus A.L., Greenberg N.M., Burg M.B., Allison J.P. Manipulation of T cell costimulatory and inhibitory signals for immunotherapy of prostate cancer. Proc. Natl. Acad. Sci. USA. 1997;94:8099–8103. doi: 10.1073/pnas.94.15.8099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurwitz A.A., Yu T.F., Leach D.R., Allison J.P. CTLA-4 blockade synergizes with tumor-derived GM-CSF for treatment of an experimental mammary carcinoma. Proc. Natl. Acad. Sci. USA. 1998;95:10067–10071. doi: 10.1073/pnas.95.17.10067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krummel M.F., Allison J.P. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J. Exp. Med. 1995;182:459–465. doi: 10.1084/jem.182.2.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Z., Reznikoff G., Dranoff G., Rock K.L. Cloned dendritic cells can present exogenous antigens on both MHC class I and class II molecules. J. Immunol. 1997;158:2723–2730. [PubMed] [Google Scholar]
- Dranoff G., Jaffee E., Lazenby A., Golumbek P., Levitsky H., Brose K., Jackson V., Hamada H., Pardoll D., Mulligan R.C. Vaccination with irradiated tumor cells engineered to secrete GM-CSF stimulates potent, specific, and long lasting anti-tumor immunity. Proc. Natl. Acad. Sci. USA. 1993;90:3539–3543. doi: 10.1073/pnas.90.8.3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart I.R. The selection of characterization of an invasive variant of the B16 melanoma. Am. J. Pathol. 1979;97:587–600. [PMC free article] [PubMed] [Google Scholar]
- Hung K., Hayashi R., Lafond-Walker A., Lowenstein C., Pardoll D., Levitsky H. The central role of CD4+ T cells in the antitumor immune response. J. Exp. Med. 1998;188:2357–2368. doi: 10.1084/jem.188.12.2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inaba K., Inaba M., Romani N., Aya H., Deguchi M., Ikehara S., Muramatsu S., Steinman R.M. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J. Exp. Med. 1992;176:1693–1702. doi: 10.1084/jem.176.6.1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang A.Y., Golumbek P., Ahmadzadeh M., Jaffee E., Pardoll D., Levitsky H. Role of bone marrow-derived cells in presenting MHC class I-restricted tumor antigens. Science. 1994;264:961–965. doi: 10.1126/science.7513904. [DOI] [PubMed] [Google Scholar]
- Banchereau J., Steinman R.M. Dendritic cells and the control of immunity. Nature. 1998;392:245–252. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- Yang Y., Zou J., Mu J., Wijesuriya R., Ono S., Walunas T., Bluestone J., Fujiwara H., Hamaoka T. Enhanced induction of antitumor T-Cell responses by cytotoxic T lymphocyte associated molecule-4 blockadethe effect is manifested only at the restricted tumor-bearing stages. Cancer Res. 1997;57:4036–4041. [PubMed] [Google Scholar]
- Staveley-O'Carroll K., Sotomayor E., Montgomery J., Borrello I., Hwang L., Fein S., Pardoll D., Levitsky H. Induction of antigen-specific T cell anergyan early event in the course of tumor progression. Proc. Natl. Acad. Sci. USA. 1998;95:1178–1183. doi: 10.1073/pnas.95.3.1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotomoyer E.M., Borrello I.M., Tubb E., Allison J.P., Levitsky H.I. In vivo blockade of CTLA-4 enhances the priming of responsive T-cells, but fails to prevent the induction of tumor antigen-specific tolerance. Proc. Natl. Acad. Sci. USA. 1999;In press doi: 10.1073/pnas.96.20.11476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levitsky H.I., Lazenby A., Hayashi R.J., Pardoll D.M. In vivo priming of two distinct antitumor effector populationsthe role of MHC class I expression. J. Exp. Med. 1994;179:1215–1224. doi: 10.1084/jem.179.4.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu T.C., Huang A.Y.C., Jaffee E.M., Pardoll D.M. A reassessment of the role of B7-1 expression in tumor rejection. J. Exp. Med. 1995;182:1–7. doi: 10.1084/jem.182.5.1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy K.D., Hermans I.F., Fraser J.H., Le Gros G., Ronchese F. Cytotoxic T lymphocyte–associated antigen 4 (CTLA-4) can regulate dendritic cell–induced activation and cytotoxicity of CD8+ T cells independently of CD4+ T cell help. J. Exp. Med. 1999;189:1157–1162. doi: 10.1084/jem.189.7.1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoenberger S.P., Toes R.E., van der Voort E.I., Offringa R., Melief C.J. T-cell help for cytotoxic T lymphocytes is mediated by CD40-CD40L interactions. Nature. 1998;393:480–483. doi: 10.1038/31002. [DOI] [PubMed] [Google Scholar]
- Bennett S.R., Carbone F.R., Karamalis F., Flavell R.A., Miller J.F., Heath W.R. Help for cytotoxic-T-cell responses is mediated by CD40 signalling. Nature. 1998;393:478–480. doi: 10.1038/30996. [DOI] [PubMed] [Google Scholar]
- Ridge J.P., Di Rosa F., Matzinger P. A conditioned dendritic cell can be a temporal bridge between a CD4+ T-helper and a T-killer cell. Nature. 1998;393:474–478. doi: 10.1038/30989. [DOI] [PubMed] [Google Scholar]
- Richards J.M., Mehta N., Ramming K., Skosey P. Sequential chemoimmunotherapy in the treatment of metastatic melanoma. J. Clin. Oncol. 1992;10:1338–1343. doi: 10.1200/JCO.1992.10.8.1338. [DOI] [PubMed] [Google Scholar]
- Rosenberg S.A., White D.E. Vitiligo in patients with melanomanormal tissue antigens can be targets for cancer immunotherapy. J. Immunother. Emphasis Tumor Immunol. 1996;19:81–84. [PubMed] [Google Scholar]
- Okamoto T., Irie R.F., Fujii S., Huang S.K., Nizze A.J., Morton D.L., Hoon D.S. Anti-tyrosinase-related protein-2 immune response in vitiligo patients and melanoma patients receiving active-specific immunotherapy. J. Invest. Dermatol. 1998;111:1034–1039. doi: 10.1046/j.1523-1747.1998.00411.x. [DOI] [PubMed] [Google Scholar]
- Ogg G.S., Rod Dunbar P., Romero P., Chen J.L., Cerundolo V. High frequency of skin-homing melanocyte-specific cytotoxic T lymphocytes in autoimmune vitiligo. J. Exp. Med. 1998;188:1203–1208. doi: 10.1084/jem.188.6.1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawakami Y., Eliyahu S., Delgado C.H., Robbins P.F., Sakaguch K., Appella E., Yannelli J.R., Adema G., Miki T., Rosenberg S.A. Identification of a human melanoma antigen recognized by tumor-infiltrating lymphocytes associated with in vivo tumor rejection. Proc. Natl. Acad. Sci. USA. 1994;91:6458–6462. doi: 10.1073/pnas.91.14.6458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloom M.B., Perry-Lalley D., Robbins P.F., Li Y., el-Gamil M., Rosenberg S.A., Yang J.C. Identification of tyrosinase-related protein 2 as a tumor rejection antigen for the B16 melanoma. J. Exp. Med. 1997;185:453–459. doi: 10.1084/jem.185.3.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naftzger C., Takechi Y., Kohda H., Hara I., Vijayasaradhi S., Houghton A.N. Immune response to a differentiation antigen induced by altered antigena study of tumor rejection and autoimmunity. Proc. Natl. Acad. Sci. USA. 1996;93:14809–14814. doi: 10.1073/pnas.93.25.14809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara I., Takechi Y., Houghton A.N. Implicating a role for immune recognition of self in tumor rejectionpassive immunization against the brown locus protein. J. Exp. Med. 1995;182:1609–1614. doi: 10.1084/jem.182.5.1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overwijk W.W., Lee D.S., Surman D.R., Irvine D.R., Touloukian C.E., Shan C.-C., Carroll M.W., Moss B., Rosenberg S.A., Restifo N.P. Vaccination with a recombinant vaccinia virus encoding a “self” antigen induces autoimmune vitiligo and tumor cell destruction in micerequirement for CD4+ T lymphocytes. Proc. Natl. Acad. Sci. USA. 1999;96:2982–2987. doi: 10.1073/pnas.96.6.2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber L.W., Bowne W.B., Wolchok J.D., Srinivasan R., Qin J., Moroi Y., Clynes R., Song P., Lewis J.J., Houghton A.N. Tumor immunity and autoimmunity induced by immunization with homologous DNA. J. Clin. Invest. 1998;102:1258–1264. doi: 10.1172/JCI4004. [DOI] [PMC free article] [PubMed] [Google Scholar]