

Abstract
Although DQA1*0301/DQB1*0302 is the human histocompatibility leukocyte antigen (HLA) class II gene most commonly associated with human type 1 diabetes, direct in vivo experimental evidence for its diabetogenic role is lacking. Therefore, we generated C57BL/6 transgenic mice that bear this molecule and do not express mouse major histocompatibility complex (MHC) class II molecules (DQ8+/mII−). They did not develop insulitis or spontaneous diabetes. However, when DQ8+/mII− mice were bred with C57BL/6 mice expressing costimulatory molecule B7-1 on β cells (which normally do not develop diabetes), 81% of the DQ8+/mII−/B7-1+ mice developed spontaneous diabetes. The diabetes was accompanied by severe insulitis composed of both T cells (CD4+ and CD8+) and B cells. T cells from the diabetic mice secreted large amounts of interferon γ, but not interleukin 4, in response to DQ8+ islets and the putative islet autoantigens, insulin and glutamic acid decarboxylase (GAD). Diabetes could also be adoptively transferred to irradiated nondiabetic DQ8+/mII−/B7-1+ mice. In striking contrast, none of the transgenic mice in which the diabetes protective allele (DQA1*0103/DQB1*0601, DQ6 for short) was substituted for mouse MHC class II molecules but remained for the expression of B7-1 on pancreatic β cells (DQ6+/mII−/B7-1+) developed diabetes. Only 7% of DQ−/mII−/B7-1+ mice developed diabetes at an older age, and none of the DQ−/mII+/B7-1+ mice or DQ8+/mII+/B7-1+ mice developed diabetes. In conclusion, substitution of HLA-DQA1*0301/DQB1*0302, but not HLA-DQA1*0103/DQB1*0601, for murine MHC class II provokes autoimmune diabetes in non–diabetes-prone rat insulin promoter (RIP).B7-1 C57BL/6 mice. Our data provide direct in vivo evidence for the diabetogenic effect of this human MHC class II molecule and a unique “humanized” animal model of spontaneous diabetes.
Keywords: type 1 diabetes, animal model, human MHC molecules, transgenic mice
Introduction
Like many autoimmune diseases, type 1 diabetes is associated with genes encoding the MHC 1, particularly the class II molecule DQA1*0301/DQB1*0302 (DQ8 for short) in humans and its mouse homologue I-Ag7 in the nonobese diabetic (NOD) mouse 2 3. A common feature of both DQA1*0301/DQB1*0302 and I-Ag7 molecules is the expression of a non-aspartic acid residue at position 57 of the β chain. However, if the amino acid at this residue is a charged residue, aspartic acid, then the allele has a protective effect 4 5 6. Studies also suggest that the simultaneous presence of an arginine residue at position 52 on DQA1 chain and the absence of aspartate at position 57 on the DQB1 chain further increase susceptibility to the disease 7. Although the importance of I-Ag7 in the development of murine diabetes has been unequivocally established in vivo, the role of DQA1*0301/DQB1*0302 in human disease development is mostly, if not totally, derived indirectly from genetic association analysis. Transgenic mice that carry only human MHC class II DQA1*0301/DQB1*0302 molecules and are deficient in murine MHC class II molecules (DQ8+/mII−) have been generated recently 8 9 10 11. However, in studies of diabetes they have been mainly used to identify T cell determinants of pancreatic β cell autoantigens 8 10 11. In no case did these transgenic mice (HLA-DQ8 and/or HLA-DR4) develop diabetes spontaneously, even if they were expressed on the NOD genetic background 11.
Our DQ8+/mII− mice were bred onto the C57BL/6 genetic background to maintain the specific deficiency of murine MHC class II (as C57BL/6 mice do not express MHC class II I-E molecules, the homologue of HLA-DR), thereby allowing us to more specifically assess the influence of this DQ molecule without the potential influence of the many other diabetes susceptibility genes present in NOD mice 12. However, DQ8+/mII− mice do not develop spontaneous insulitis and/or diabetes 9. To investigate if we could break the tolerance exhibited by these mice by locally providing the costimulatory signal B7-1, we generated DQ8+/mII−/rat insulin promoter (RIP).B7-1 mice by breeding DQ8+/mII− mice with RIP.B7-1 mice (also on C57BL/6 genetic background) 13. These RIP.B7-1 mice that carry murine MHC class II I-Ab molecules do not normally develop diabetes 13. Our study shows that when murine MHC class II I-Ab molecules are replaced by human DQA1*0301/DQB1*0302 molecules, this is sufficient to provoke autoimmune diabetes in the majority of non–diabetes-prone RIP.B7-1 C57BL/6 mice. Moreover, when the disease susceptibility allele DQ8 was replaced by the disease protective allele DQ6, i.e., DQA1*0103/DQB1*0601 (which does not have an arginine residue at position 52 on the DQA1 chain and does have an aspartate at position 57 on the DQB1 chain), the mice developed neither insulitis nor diabetes. These data provide direct in vivo evidence for the diabetogenic effect of HLA-DQ8 molecules and offer, for the first time, a “humanized” animal model of spontaneous diabetes.
Materials and Methods
Generation of DQ+/mII−/RIP.B7-1 Transgenic Mice.
HLA-DQA1*0301/DQB1*0302 transgenic, murine MHC class II molecule–deficient C57BL/6 mice 9 were bred with RIP.B7-1 transgenic C57BL/6 mice 13 to generate DQ8+/mII−/RIP.B7-1 transgenic mice. F1 offspring of this mating were screened for DQA1*0301/DQB1*0302 expression by flow cytometric analysis of PBLs. RIP.B7-1 transgene was screened for by PCR using genomic DNA isolated from mouse tail biopsy as described previously 14. DQ8+/RIP.B7-1+ mice were selected and intercrossed to generate DQ8+/mII−/RIP.B7-1 mice. The expression of the DQ8 transgene and murine class II I-Ab molecules was screened for by flow cytometric analysis of PBLs, and the RIP.B7-1 transgene was screened for by PCR. DQ8 and RIP.B7-1 double transgene positive and I-Ab negative (DQ8+/mII−/RIP.B7-1) mice were used for further breeding to obtain DQ8 transgene homozygosity 9. HLA-DQA1*0103/DQB1*0601 transgenic mice (also deficient for murine MHC class II molecules) were back-crossed from C57BL/10 8 to C57BL/6 background and then bred with RIP.B7-1 transgenic C57BL/6 mice. The identical breeding and screening strategy as that for the DQ8 mice was then used for generating DQ6+/mII−/RIP.B7-1 transgenic mice. DQ−/mII−/RIP.B7-1−, DQ8+/mII−/RIP.B7-1−, DQ6+/mII−/RIP.B7-1− transgenic mice, and DQ−/mII+/RIP.B7-1+ mice were used as control groups derived from the above breedings. All of the mice used in this study were littermates derived from the same breeders.
Proliferation and Cytokine Assays.
Splenocytes (2 × 105/well) from diabetic or nondiabetic mice were assayed for antigenic response against putative β cell autoantigens as described previously 9. Secreted cytokine proteins from those responses were measured by ELISA using mAbs and recommended protocols ( 9; PharMingen).
Adoptive Transfer Experiments.
Mice used for these experiments were irradiated (600 rads) 1 d before the adoptive transfer. Diabetic DQ8+/mII−/RIP.B7-1 splenocytes were injected intravenously into the recipients (107/recipient). One group of mice was injected (7 × 106/recipient) with column-purified CD8+ diabetic DQ8+/mII−/RIP.B7-1 splenocytes (>93% purity; Pierce Chemical Co.). All of the mice were monitored for glycosuria, and the experiments were terminated 8 wk after adoptive transfer unless the mice developed diabetes, which was confirmed by blood glucose, and they were killed immediately.
Immunohistology.
Pancreas, kidney, liver, and salivary gland from all of the mice used in this study were examined by immunohistochemistry as described previously 14.
Results
High Incidence of Spontaneous Diabetes Development in DQ8+/mII−/RIP.B7-1 Mice.
We recently showed that the DQ8+/mII− C57BL/6 mice could generate specific immune responses towards putative β cell autoantigens upon immunization 9. However, they did not develop spontaneous insulitis and/or diabetes 9. To investigate if the tolerance exhibited by these mice could be altered by providing the costimulatory signal B7-1 locally, we created DQ8+/mII−/RIP.B7-1 mice by breeding DQ8+/mII− mice with RIP.B7-1 mice (also on C57BL/6 genetic background) that only very rarely develop diabetes 13. In striking contrast to RIP.B7-1 mice carrying murine MHC class II I-Ab molecules, substitution of human DQA1*0301/DQB1*0302 molecules caused RIP.B7-1 mice to develop diabetes, which appeared equally in both sexes ( Fig. 1 A). Over 80% (17/21) of DQ8+/mII−/RIP.B7-1 mice developed spontaneous diabetes beginning at ∼4 mo of age. On the other hand, only 1 of 15 (6.7%) DQ8−/mII−/RIP.B7-1 mice (between 9 and 10 mo of age) and none of the DQ8+/mII+/RIP.B7-1 mice (0/17) developed spontaneous diabetes ( Fig. 1 A). Moreover, none of the DQ8−/mII+/RIP.B7-1 mice (0/18) became diabetic after 10 mo of observation, supporting previous observations in C57BL/6.RIP.B7-1 mice 13.
Figure 1.

Development of diabetes spontaneously in HLA-DQ transgenic mice. (A) Spontaneous diabetes in DQ8 experiments. Six groups of mice were used in the study as indicated. Number of mice per group was as follows: n = 21 (10 female, 11 male) DQ8+/mII−/B7+; n = 15 (8 female, 7 male) DQ8−/mII−/B7+; n = 16 (11 female, 5 male) DQ8+/mII−/B7−; n = 14 (8 female, 6 male) DQ8−/mII−/B7−; n = 17 (6 female, 11 male) DQ8+/mII+/B7+; and n = 12 (9 female, 3 male) DQ8+/mII−/B7−. (B) Spontaneous diabetes in DQ6 experiments. Four groups of mice were used in the study as indicated. Number of mice per group was as follows: n = 16 (9 female, 7 male) DQ6+/mII−/B7+; n = 12 (5 female, 7 male) DQ6−/mII−/B7+; n = 9 (4 female, 5 male) DQ6+/mII+/B7+; and n = 15 (6 female, 9 male) DQ6+/mII−/B7−. Mice were housed under specific pathogen-free conditions. Diabetes was determined by monitoring of urinary glucose and confirmed by blood glucose (>250 mg/dl).
Protection of Diabetes Development in DQ6+/mII−/RIP.B7-1 Mice.
To confirm that the diabetogenic effect seen in DQ8+/mII−/RIP.B7-1 mice was specific for HLA-DQ8 molecules, we generated HLA-DQA1*0103/DQB1*0601 transgenic mice that expressed B7-1 molecules on pancreatic β cells and were murine I-Ab deficient (DQ6+/mII−/RIP.B7-1). It is interesting that none of the DQ6+/mII−/RIP.B7-1 mice (n = 16, 9 female and 7 male) developed diabetes by 9 mo of age when the experiment was terminated ( Fig. 1 B). As was observed in the DQ8 experiments, 1 out of 12 DQ6−/mII−/RIP.B7-1 mice (8.3%) became diabetic at nearly 9 mo of age ( Fig. 1 B), whereas none of the DQ6+/mII+/RIP.B7-1 mice (0/9) developed diabetes.
Diabetes Could Be Transferred by Diabetic DQ8+/mII−/RIP.B7-1 Splenocytes.
Adoptive transfer experiments showed that diabetes in DQ8+/mII−/RIP.B7-1 mice was autoimmune mediated. Splenocytes (depleted of erythrocytes) from diabetic DQ8+/mII−/RIP.B7-1 mice were adoptively transferred into either DQ8+ or DQ8− irradiated recipients (8–10 wk of age; Fig. 2). Diabetes appeared in 78% of the DQ8+/mII−/RIP.B7-1 recipients (7/9) beginning at 3 wk after cell transfer, but in only 2 of 8 DQ8−/mII−/RIP.B7-1 recipients between 6 and 8 wk after adoptive transfer. In contrast, none of the RIP.B7-1 transgene-negative recipients, either DQ8+/mII− or DQ8−/mII−, developed diabetes 8 wk after the cell transfer ( Fig. 2). To ascertain that the diabetes, developed either spontaneously or after adoptive transfer, was not primarily CD8 mediated, since two “odd” mice became diabetic in both experiments, we performed another group of adoptive transfers using purified CD8 T cells derived from diabetic DQ8+/mII−/RIP.B7-1 splenocytes. None of the recipients (DQ8+/mII−/RIP.B7-1; n = 3) developed diabetes 8 wk after the transfer when the experiment was terminated ( Fig. 2), supporting a key role for the HLA-DQA1*0301/DQB1*0302 molecules and CD4 T cells selected by the molecules in this diabetic model.
Figure 2.

Adoptively transferred diabetes. 10 × 107 diabetic DQ8+/mII−/B7+ splenocytes (depleted red blood cells) were transferred intravenously into irradiated recipients (as indicated). Diabetes was determined as above. Number of mice per group was as follows: n = 9 (4 female, 5 male) DQ8+/mII−/B7+; n = 8 (4 female, 4 male) DQ−/mII−/B7+; n = 3 (male) DQ8+/mII−/B7−; n = 3 (male) DQ8−/mII−/B7−; and n = 3 (male) DQ8+/mII−/B7+* (these recipients were adoptively transferred with purified CD8+ splenocytes only from diabetic DQ8+/mII−/B7+ mice).
Heavy Lymphocyte Infiltration and Upregulation of Adhesion Molecule in Diabetic DQ8+/mII−/RIP.B7-1 Mice.
Pancreata from mice that developed diabetes either spontaneously or via adoptive transfer showed a profound effect of DQA1*0301/DQB1*0302 on lymphocytic infiltration. Unlike NOD mice, in which the islet infiltrates comprise CD4+ T cells and B220+ B cells, with fewer CD8+ T cells, the islet infiltrates from diabetic DQ8+/mII−/RIP.B7-1 mice contained CD8+ T cells and B220+ B cells, with fewer CD4+ T cells ( Fig. 3, top), a picture similar to that reported in recent-onset human type 1 diabetes 15. This is not entirely unexpected, as the proportion of CD8+ and CD4+ (selected by the expression of HLA-DQ molecules) T cells in DQ8+/mII−/RIP.B7-1 mice is ∼70 and 30%, respectively, of total CD3+ T cells 9. However, the TCR repertoire, as analyzed by a panel of mAbs against 15 different TCR Vβ gene products, was normal in these mice 9. In contrast to the islet infiltrates of diabetic DQ8+/mII−/RIP.B7-1 mice, the two diabetic DQ−/mII−/RIP.B7-1 mice showed heavy islet infiltration by CD8+ T cells and fewer B220+ B cells (data not shown). As expected, CD4+ T cells were absent. Thus, the larger number of B220+ B cells infiltrating the islets of DQ8+/mII−/RIP.B7-1 mice was probably recruited by DQA1*0301/DQB1*0302-selected CD4+ T cells.
Figure 3.
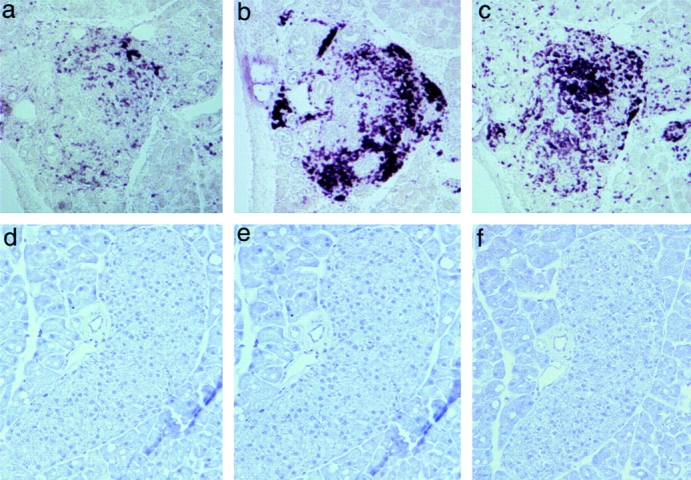
Immunohistochemis-try staining of pancreatic sections of diabetic DQ8+/mII−/B7+ mice (top panels, a–c) and diabetes-free DQ6+/mII−/B7+ mice (bottom panels, d–f). Islet infiltrates were stained positive for CD4+ T cells (a), B220+ B cells (b), and CD8+ T cells (c).
In striking contrast, pancreata from DQ6+/mII−/RIP.B7-1 mice, which were diabetes free, exhibited completely normal islets ( Fig. 3, bottom).
Optimal T cell activation requires additional ligand interactions 16 17. B7-1– or intracellular adhesion molecule (ICAM)-1–transfected fibroblasts or insect cells, acting as APCs, induce naive T cells to proliferate and secrete small amounts of IL-2 18 19. However, when both B7-1 and ICAM-1 are expressed together, IL-2 production and proliferation are greatly increased, independent of APC number or antigen concentration 18 19. Wulfing et al. 20 recently visualized the kinetic interaction of ICAM-1 with T cells after activation and found that ICAM-1 was rapidly concentrated on B cells, immediately after TCR–peptide–MHC engagement, at the interface between the two cells. Therefore, we examined the expression of ICAM-1 on lymphocytes from DQ8+/mII−/RIP.B7-1 mice. As expected, upregulation of ICAM-1 was greater on both T and B cells derived from pancreatic lymph nodes than on those from splenocytes in diabetic mice (data not shown). However, the highest expression of ICAM-1 in diabetic mice was found in the islet infiltrates ( Fig. 4). In a kinetic study, we noticed that the upregulation of ICAM-1 occurs before lymphocyte infiltration (our unpublished data). ICAM-1 expression in mice from nondiabetic control groups, including DQ6+/mII−/RIP.B7-1 mice, was much lower on lymphocytes obtained from either spleen or pancreatic lymph nodes (data not shown) and was undetectable in the islets ( Fig. 4).
Figure 4.

ICAM-1 staining of pancreatic sections of nondiabetic (a) and diabetic (b) DQ8+/mII−/B7+ mice.
Autoreactivity and Th1 Cytokine Production in Diabetic DQ8+/mII−/RIP.B7-1 Mice.
We next examined whether splenocytes from diabetic DQ8+/mII−/RIP.B7-1 mice recognize islets or the putative autoantigens, insulin and/or GAD 21 22 23 24 25. Like diabetic NOD mice, splenocytes from diabetic DQ8+/mII−/RIP.B7-1 mice showed marked autoreactivity. Specific reactivity, albeit moderate (two- to threefold), was detected toward insulin and DQ8+ islets ( Fig. 5 A). However, insulin, DQ+ islets, and GAD stimulated the production of IFN-γ (IL-4 secretion was undetectable) ( Fig. 5 B). Splenocytes from DQ6+/mII−/RIP.B7-1 mice or nondiabetic DQ8+/mII−/RIP.B7-1 mice showed much less autoreactivity, and no response to putative autoantigens (insulin and GAD) was observed in these mice (data not shown). Moreover, no autoreactivity was detected in DQ−/mII−/RIP.B7-1, DQ8+/mII+/RIP.B7-1, and DQ−/mII+/RIP.B7-1 mice. We also analyzed the TCR Vβ usage from cells derived from diabetic and nondiabetic DQ8+/mII−/RIP.B7-1 mice using a panel of mAbs specific for 15 different TCR Vβ molecules; no preferential expansion of any particular TCR Vβ subset in either CD4+ or CD8+ T cells was detected. However, higher levels of the early T cell activation marker CD69 were expressed on CD4+ than on CD8+ T cells obtained from diabetic mice (data not shown). T cells from pancreatic lymph nodes of diabetic mice secreted IFN-γ in response to anti-CD3, insulin, or GAD65, but a negligible amount of IL-4 (data not shown). Cytoplasmic cytokine staining in combination with CD4 or CD8 coreceptor staining showed that both CD4+ and CD8+ T cells were positive for cytoplasmic IFN-γ (data not shown).
Figure 5.

In vitro proliferation (A) and IFN-γ production (B) of diabetic splenocytes to islets and islet β cell autoantigens. 2 × 105/well of splenocytes (depleted red blood cells) were cultured in medium (Click's containing 5% heat-inactivated FCS) alone or with antigens (as indicated). Cytokine production was measured in the supernatants of the cultures (3 d) by ELISA.
Diabetic DQ8+/mII−/RIP.B7-1 Mice Also Suffer from Sialadenitis.
The autoimmune response in NOD mice is not restricted to islets. NOD mice, for unknown reasons, also develop autoimmune sialadenitis, which is associated with a decline in salivary gland function 26 27 28. This phenomenon has also been reported in type 1 diabetic patients. Moreover, a specific autoantibody against α-fodrin, an important autoantigen in human Sjogren's syndrome, was found recently in NOD mice 29. Moreover, the splenocytes of NOD mice produced Th1-type cytokines in response to α-fodrin 29. Interestingly, salivary glands from diabetic DQ8+/mII−/B7+ mice also exhibited significant infiltration, consisting of CD4 and CD8 T lymphocytes and, to a lesser extent, B lymphocytes ( Fig. 6, top). The development of diabetes appeared to precede the development of sialadenitis, as was found in NOD mice (our unpublished data). In addition, the sialadenitis was even more severe in DQ8+/mII−/B7+ mice that developed diabetes after adoptive transfer (data not shown). Since B7-1 is expressed in islets and not salivary glands, this may be explained by the fact that more autoreactive T cells (specific to the autoantigens shared between pancreas and salivary gland) were transferred to the recipients. Sialadenitis was much milder in nondiabetic DQ8+/mII−/RIP.B7-1 mice, and no sialadenitis was found in mice carrying other genotypes: DQ8+/mII−/B7−, DQ−/mII−/B7−, DQ8+/mII+/B7+, DQ−/mII+/B7+, and DQ6+/mII−/B7+ ( Fig. 6, bottom).
Figure 6.
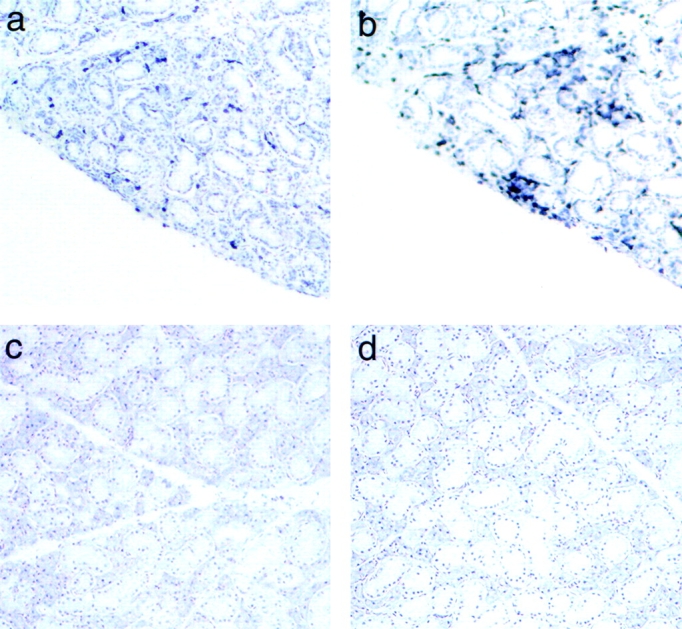
Staining of frozen sections of salivary gland from diabetic DQ8+/mII−/B7+ mice (top panels, a and b) and normal DQ6+/mII−/B7+ mice (bottom panels, c and d). Lymphocyte infiltrates were stained positive for CD4 (a and c) and CD8 (b and d) T cells.
Discussion
Tolerance to self-antigens is the principal means used by the immune system to protect against “self-destruction.” Self-tolerance involves two components: central tolerance and peripheral tolerance 30 31 32. High-affinity autoreactive T cells are deleted in the thymus by negative selection, a mechanism that depends on the affinity/avidity levels of each molecule in the TCR–MHC–peptide trimolecular complex 33. However, negative selection is not always complete: some autoreactive T cells can escape and enter the peripheral lymphoid organs 34 35 36 37 38 39. A striking feature of the MHC class II molecules in NOD mice (I-Ag7) and the human HLA-DQA1*0301/DQB1*0302 is their low-affinity binding to antigenic peptides 40 41 42. Thus, T cells selected on these MHC molecules could potentially escape from central tolerance and survive in the periphery. This characteristic of HLA-DQA1*0301/DQB1*0302 (DQ8) or I-Ag7 molecules, compared with HLA-DQA1*0103/DQB1*0601 (DQ6) or I-Ab, may allow more of these cells to escape. However, current data indicate that the presence of autoreactive CD4 T cells alone in the periphery is not sufficient to cause diabetes. Our previous study 9 as well as many studies from NOD mice 43 44 45 46 47 suggest that both activated CD4 and CD8 T cells are required for disease to occur. Autoreactive T cells in the periphery are usually “anergic,” “ignorant,” or “suppressed,” thereby preventing self-destruction. This may be because most autoantigen-expressing tissue cells express low levels of MHC class I and do not express MHC class II molecules. In addition, they do not express costimulatory signals that are needed to fully activate T cells in the periphery. From this perspective, the initiation of autoimmunity may be largely dependent on the MHC class I–expressing tissue cells and, in the case of this study, the expression of appropriate costimulatory molecules on the cells that express autoantigen(s). Using RIP transgenic mice that express a viral protein on pancreatic β cells, Ohashi et al. 48 and Oldstone et al. 49 demonstrated that these transgenic mice did not develop spontaneous diabetes even though they had a high frequency of viral (“self ”) reactive T cells in the periphery. However, when these anti-self (viral) T cells were activated by the local expression of B7-1 molecule (RIP.B7-1), spontaneous diabetes was produced 50. Unlike their transgene-encoded self (viral) model of diabetes, which is triggered by antiviral CD8-mediated autoimmunity, the β cell expression of B7-1 in our model is not sufficient for spontaneous disease development.
Our most striking observation is that the substitution of human HLA-DQ8 for murine MHC class II molecules caused RIP.B7-1 mice (on a C57BL/6 genetic background), which are normally resistant to diabetes, to spontaneously become diabetic. Moreover, replacement of human HLA-DQ6 molecules for human HLA-DQ8 molecules in these mice completely prevented the disease. Similar studies showing that induction of disease depends on the presence of particular HLA-DQ alleles have been recently reported in an arthritis model 51 52. However, in that case, the disease had to be induced by immunization with collagen (as an autoantigen) in the presence of CFA.
It should be noted that the B7-1 molecules in our mice were expressed on pancreatic β cells, which do not normally express MHC class II molecules 53. Thus, it is likely that the predominant effect of the RIP.B7-1 transgene was on CD8 T cells 54. Nonetheless, this CD8 T cell effect is not sufficient to cause diabetes in mice expressing murine I-Ab molecules. Their CD4 T cells do not support autoimmunity, and perhaps even have an inhibitory effect. By replacing I-Ab with the human DQ8 molecules, autoreactive CD4 T cells are selected that can now, in conjunction with the activated CD8 T cells, cause insulitis and diabetes. The strongest evidence for the pivotal diabetogenic role of DQ8 molecules in disease development is that the “exchange” of human DQ8 for human DQ6 molecules abolished spontaneous disease development. The result is even more impressive given the fact that the DQ transgene rescues CD4 T cells in these mice to only ∼30% of the normal level of CD4 T cells but their effect on the disease susceptibility or protection is very dominant. In terms of the DQ8 effect, more complete rescue of CD4 T cells may cause the disease to develop even earlier. This view is consistent with our data showing that T cells from the diabetic DQ8+/mII−/B7+ mice can recognize DQ8+ but not DQ8− islets as well as naturally processed and expressed autoantigens derived from pancreatic β cells, such as insulin and GAD.
The mechanism by which CD4 T cells selected on HLA-DQ8 molecules facilitate the development of diabetes is not clear. It has been reported that apoptosis participates in the remodeling of the endocrine pancreas in both rodents (shortly after birth) and humans 55 56 57. It is possible that HLA-DQ8–expressing APCs such as dendritic cells (DCs) take up autoantigens from apoptotic β cells and present them to T cells. Autoreactive CD4 T cells may recognize the same autoantigen presented by DCs and become activated. These CD4 T cells could sequentially further activate APCs via CD40L–CD40 interactions and cytokine release in the draining lymph nodes. These activated DCs could then cross-present antigens to CD8 T cells 58 as well as to other CD4 T cells, thus amplifying the self-destructive process. In addition, the CD4 T cells may facilitate the traffic of CD8 T cells to the pancreatic islets by upregulating ICAM-1/vascular cell adhesion molecule (VCAM). The upregulation of ICAM-1 on both lymphocytes infiltrating islets and pancreatic β cells provides maximal costimulation to the autoreactive T cells, which in turn further aggravate the autoimmune destruction. It is also possible that autoreactive CD8 T cells, recognizing islet-derived peptides directly, and stimulated by the presence of the RIP.B7-1 molecule, initiate the islet-directed autoimmune response. However, this autoimmune response could not be amplified to become destructive without HLA-DQ8–restricted CD4 T cell help. Regardless of the precise mechanisms involved, our model suggests that HLA-DQ8 molecules play an essential role in the disease development and provides direct in vivo evidence of the importance of human HLA-DQ8 molecules in autoimmune diabetes. Conversely, our model also implies the disease-protective role of HLA-DQ6. This is the first model demonstrating that HLA-DQ8 could promote spontaneous diabetes in a diabetes-resistant mouse strain. Thus, this partially humanized transgenic model may offer a more relevant system for (a) examining the immunopathogenesis of, and (b) testing specific therapeutic intervention for human type 1 diabetes.
Acknowledgments
We thank C.A. Janeway for his generous help, Irene Visintin for her technical advice, and Jennifer Granata and Joanne Appicelli for taking care of the animals used in this study.
This work was supported by a National Institutes of Health grant (RO1 AI44427), Diabetes Program Project Grants (National Institutes of Health/Juvenile Diabetes Foundation International POI-DK53015), and the Islet and Cell Biology Cores of Yale Diabetes Endocrinology Research Center (DERC P30-DK45735). L. Wen and F.S. Wong are recipients of Career Development Awards from Juvenile Diabetes Foundation International. R.A. Flavell is a Howard Hughes Medical Institute investigator.
Footnotes
Abbreviations used in this paper: DC, dendritic cell; GAD, glutamic acid decarboxylase; ICAM, intracellular adhesion molecule; mII, murine MHC class II; NOD, nonobese diabetic; RIP, rat insulin promoter.
References
- Todd J.A., Acha-Orbea H., Bell J.I., Chao N., Fronek Z., Jacob C.O., McDermott M., Sinha A.A., Timmerman L., Steinman L. A molecular basis for MHC class II–associated autoimmunity Science. 240 1988. 1003 1009[published erratum at 241:888] [DOI] [PubMed] [Google Scholar]
- Todd J.A. Genetic control of autoimmunity in type 1 diabetes. Immunol. Today. 1990;11:122–129 . doi: 10.1016/0167-5699(90)90049-f. [DOI] [PubMed] [Google Scholar]
- Acha-Orbea H., McDevitt H.O. The first external domain of the nonobese diabetic mouse class II I-A beta chain is unique. Proc. Natl. Acad. Sci. USA. 1987;84:2435–2439 . doi: 10.1073/pnas.84.8.2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd J.A., Bell J.I., McDevitt H.O. HLA-DQ beta gene contributes to susceptibility and resistance to insulin-dependent diabetes mellitus. Nature. 1987;329:599–604 . doi: 10.1038/329599a0. [DOI] [PubMed] [Google Scholar]
- Dorman J.S., LaPorte R.E., Stone R.A., Trucco M. Worldwide differences in the incidence of type I diabetes are associated with amino acid variation at position 57 of the HLA-DQ beta chain. Proc. Natl. Acad. Sci. USA. 1990;87:7370–7374 . doi: 10.1073/pnas.87.19.7370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorsby E., Ronningen K.S. Particular HLA-DQ molecules play a dominant role in determining susceptibility or resistance to type 1 (insulin-dependent) diabetes mellitus. Diabetologia. 1993;36:371–377 . doi: 10.1007/BF00402270. [DOI] [PubMed] [Google Scholar]
- Khalil I., d'Auriol L., Gobet M., Morin L., Lepage V., Deschamps I., Park M.S., Degos L., Galibert F., Hors J. A combination of HLA-DQ beta Asp57-negative and HLA-DQ alpha Arg52 confers susceptibility to insulin-dependent diabetes mellitus. J. Clin. Invest. 1990;85:1315–1319 . doi: 10.1172/JCI114569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raju R., Munn S.R., David C.S. T cell recognition of human pre-proinsulin peptides depends on the polymorphism at HLA DQ locusa study using HLA DQ8 and DQ6 transgenic mice. Hum. Immunol. 1997;58:21–29 . doi: 10.1016/s0198-8859(97)00212-7. [DOI] [PubMed] [Google Scholar]
- Wen L., Wong F.S., Burkly L., Altieri M., Mamalaki C., Kioussis D., Flavell R.A., Sherwin R.S. Induction of insulitis by glutamic acid decarboxylase peptide–specific and HLA-DQ8–restricted CD4+ T cells from human DQ transgenic mice J. Clin. Invest. 102 1998. 947 957[published erratum at 102:following 1463] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonderstrup G., McDevitt H. Identification of autoantigen epitopes in MHC class II transgenic mice. Immunol. Rev. 1998;164:129–138 . doi: 10.1111/j.1600-065x.1998.tb01215.x. [DOI] [PubMed] [Google Scholar]
- Liu J., Purdy L.E., Rabinovitch S., Jevnikar A.M., Elliott J.F. Major DQ8-restricted T-cell epitopes for human GAD65 mapped using human CD4, DQA1*0301, DQB1*0302 transgenic IAnull NOD mice. Diabetes. 1999;48:469–477 . doi: 10.2337/diabetes.48.3.469. [DOI] [PubMed] [Google Scholar]
- Wicker L.S., Todd J.A., Peterson L.B. Genetic control of autoimmune diabetes in the NOD mouse. Annu. Rev. Immunol. 1995;13:179–200 . doi: 10.1146/annurev.iy.13.040195.001143. [DOI] [PubMed] [Google Scholar]
- Guerder S., Picarella D.E., Linsley P.S., Flavell R.A. Costimulator B7-1 confers antigen-presenting-cell function to parenchymal tissue and in conjunction with tumor necrosis factor alpha leads to autoimmunity in transgenic mice. Proc. Natl. Acad. Sci. USA. 1994;91:5138–5142 . doi: 10.1073/pnas.91.11.5138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong S., Guerder S., Visintin I., Reich E.P., Swenson K.E., Flavell R.A., Janeway C.A., Jr. Expression of the co-stimulator molecule B7-1 in pancreatic beta-cells accelerates diabetes in the NOD mouse. Diabetes. 1995;44:326–329 . doi: 10.2337/diab.44.3.326. [DOI] [PubMed] [Google Scholar]
- Itoh N., Hanafusa T., Miyazaki A., Miyagawa J., Yamagata K., Yamamoto K., Waguri M., Imagawa A., Tamura S., Inada M. Mononuclear cell infiltration and its relation to the expression of major histocompatibility complex antigens and adhesion molecules in pancreas biopsy specimens from newly diagnosed insulin-dependent diabetes mellitus patients. J. Clin. Invest. 1993;92:2313–2322 . doi: 10.1172/JCI116835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubey C., Croft M. Accessory molecule regulation of naive CD4 T cell activation. Immunol. Res. 1996;15:114–125 . doi: 10.1007/BF02918501. [DOI] [PubMed] [Google Scholar]
- Bluestone J.A. New perspectives of CD28-B7-mediated T cell costimulation. Immunity. 1995;2:555–559 . doi: 10.1016/1074-7613(95)90000-4. [DOI] [PubMed] [Google Scholar]
- Cai Z., Brunmark A., Jackson M.R., Loh D., Peterson P.A., Sprent J. Transfected Drosophila cells as a probe for defining the minimal requirements for stimulating unprimed CD8+ T cells. Proc. Natl. Acad. Sci. USA. 1996;93:14736–14741 . doi: 10.1073/pnas.93.25.14736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubey C., Croft M., Swain S.L. Costimulatory requirements of naive CD4+ T cells. ICAM-1 or B7-1 can costimulate naive CD4 T cell activation but both are required for optimum response. J. Immunol. 1995;155:45–57 . [PubMed] [Google Scholar]
- Wulfing C., Sjaastad M.D., Davis M.M. Visualizing the dynamics of T cell activationintracellular adhesion molecule 1 migrates rapidly to the T cell/B cell interface and acts to sustain calcium levels. Proc. Natl. Acad. Sci. USA. 1998;95:6302–6307 . doi: 10.1073/pnas.95.11.6302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tisch R., Yang X.D., Singer S.M., Liblau R.S., Fugger L., McDevitt H.O. Immune response to glutamic acid decarboxylase correlates with insulitis in non-obese diabetic. Nature. 1993;366:72–75 . doi: 10.1038/366072a0. [DOI] [PubMed] [Google Scholar]
- Kaufman D.L., Clare-Salzler M., Tian J., Forsthuber T., Ting G.S., Robinson P., Atkinson M.A., Sercarz E.E., Tobin A.J., Lehmann P.V. Spontaneous loss of T-cell tolerance to glutamic acid decarboxylase in murine insulin-dependent diabetes. Nature. 1993;366:69–72 . doi: 10.1038/366069a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegmann D.R., Norbury-Glaser M., Daniel D. Insulin-specific T cells are a predominant component of islet infiltrates in pre-diabetic NOD mice. Eur. J. Immunol. 1994;24:1853–1857 . doi: 10.1002/eji.1830240820. [DOI] [PubMed] [Google Scholar]
- Eisenbarth G.S., Jackson R.A., Pugliese A. Insulin autoimmunitythe rate limiting factor in pre-type I diabetes J. Autoimmun. 5Suppl. A1992. 241 246 [DOI] [PubMed] [Google Scholar]
- Verge C.F., Stenger D., Bonifacio E., Colman P.G., Pilcher C., Bingley P.J., Eisenbarth G.S. Combined use of autoantibodies (IA-2 autoantibody, GAD autoantibody, insulin autoantibody, cytoplasmic islet cell antibodies) in type 1 diabetesCombinatorial Islet Autoantibody Workshop. Diabetes. 1998;47:1857–1866 . doi: 10.2337/diabetes.47.12.1857. [DOI] [PubMed] [Google Scholar]
- Goillot E., Mutin M., Touraine J.L. Sialadenitis in nonobese diabetic micetransfer into syngeneic healthy neonates by splenic T lymphocytes. Clin. Immunol. Immunopathol. 1991;59:462–473 . doi: 10.1016/0090-1229(91)90041-8. [DOI] [PubMed] [Google Scholar]
- Robinson C.P., Yamamoto H., Peck A.B., Humphreys-Beher M.G. Genetically programmed development of salivary gland abnormalities in the NOD (nonobese diabetic)-scid mouse in the absence of detectable lymphocytic infiltrationa potential trigger for sialoadenitis of NOD mice. Clin. Immunol. Immunopathol. 1996;79:50–59 . doi: 10.1006/clin.1996.0050. [DOI] [PubMed] [Google Scholar]
- Yamamoto H., Ishibashi K., Nakagawa Y., Maeda N., Zeng T., Robinson C.P., Oxford G.E., Chegini N., Humphreys-Beher M.G. Detection of alterations in the levels of neuropeptides and salivary gland responses in the non-obese diabetic mouse model for autoimmune sialoadenitis. Scand. J. Immunol. 1997;45:55–61 . doi: 10.1046/j.1365-3083.1997.d01-375.x. [DOI] [PubMed] [Google Scholar]
- Yanagi K., Ishimaru N., Haneji N., Saegusa K., Saito I., Hayashi Y. Anti-120-kDa alpha-fodrin immune response with Th1-cytokine profile in the NOD mouse model of Sjogren's syndrome. Eur. J. Immunol. 1998;28:3336–3345 . doi: 10.1002/(SICI)1521-4141(199810)28:10<3336::AID-IMMU3336>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- Marrack P., Kappler J. T cell tolerance. Semin. Immunol. 1990;2:45–49 . [PubMed] [Google Scholar]
- Miller J.F., Morahan G. Peripheral T cell tolerance. Annu. Rev. Immunol. 1992;10:51–69 . doi: 10.1146/annurev.iy.10.040192.000411. [DOI] [PubMed] [Google Scholar]
- Miller J.F. Self-nonself discrimination and tolerance in T and B lymphocytes. Immunol. Res. 1993;12:115–130 . doi: 10.1007/BF02918299. [DOI] [PubMed] [Google Scholar]
- Robey E., Fowlkes B.J. Selective events in T cell development. Annu. Rev. Immunol. 1994;12:675–705 . doi: 10.1146/annurev.iy.12.040194.003331. [DOI] [PubMed] [Google Scholar]
- Bonomo A., Kehn P.J., Shevach E.M. Premature escape of double-positive thymocytes to the periphery of young mice. Possible role in autoimmunity. J. Immunol. 1994;152:1509–1514 . [PubMed] [Google Scholar]
- Miller J.F., Flavell R.A. T-cell tolerance and autoimmunity in transgenic models of central and peripheral tolerance. Curr. Opin. Immunol. 1994;6:892–899 . doi: 10.1016/0952-7915(94)90010-8. [DOI] [PubMed] [Google Scholar]
- Liu G.Y., Fairchild P.J., Smith R.M., Prowle J.R., Kioussis D., Wraith D.C. Low avidity recognition of self-antigen by T cells permits escape from central tolerance. Immunity. 1995;3:407–415 . doi: 10.1016/1074-7613(95)90170-1. [DOI] [PubMed] [Google Scholar]
- Laufer T.M., DeKoning J., Markowitz J.S., Lo D., Glimcher L.H. Unopposed positive selection and autoreactivity in mice expressing class II MHC only on thymic cortex. Nature. 1996;383:81–85 . doi: 10.1038/383081a0. [DOI] [PubMed] [Google Scholar]
- Akkaraju S., Ho W.Y., Leong D., Canaan K., Davis M.M., Goodnow C.C. A range of CD4 T cell tolerancepartial inactivation to organ-specific antigen allows nondestructive thyroiditis or insulitis. Immunity. 1997;7:255–271 . doi: 10.1016/s1074-7613(00)80528-2. [DOI] [PubMed] [Google Scholar]
- Heath V., Mason D., Ramirez F., Seddon B. Homeostatic mechanisms in the control of autoimmunity. Semin. Immunol. 1997;9:375–380 . doi: 10.1006/smim.1997.0095. [DOI] [PubMed] [Google Scholar]
- Nepom B.S., Nepom G.T., Coleman M., Kwok W.W. Critical contribution of beta chain residue 57 in peptide binding ability of both HLA-DR and -DQ molecules. Proc. Natl. Acad. Sci. USA. 1996;93:7202–7206 . doi: 10.1073/pnas.93.14.7202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrasco-Marin E., Shimizu J., Kanagawa O., Unanue E.R. The class II MHC I-Ag7 molecules from non-obese diabetic mice are poor peptide binders. J. Immunol. 1996;156:450–458 . [PubMed] [Google Scholar]
- Kanagawa O., Shimizu J., Unanue E.R. The role of I-Ag7 beta chain in peptide binding and antigen recognition by T cells. Int. Immunol. 1997;9:1523–1526 . doi: 10.1093/intimm/9.10.1523. [DOI] [PubMed] [Google Scholar]
- Bendelac A., Carnaud C., Boitard C., Bach J.F. Syngeneic transfer of autoimmune diabetes from diabetic NOD mice to healthy neonates. Requirement for both L3T4+ and Lyt-2+ T cells. J. Exp. Med. 1987;166:823–832 . doi: 10.1084/jem.166.4.823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller B.J., Appel M.C., O'Neil J.J., Wicker L.S. Both the Lyt-2+ and L3T4+ T cell subsets are required for the transfer of diabetes in nonobese diabetic mice. J. Immunol. 1988;140:52–58 . [PubMed] [Google Scholar]
- Serreze D.V., Leiter E.H., Christianson G.J., Greiner D., Roopenian D.C. Major histocompatibility complex class I-deficient NOD-B2mnull mice are diabetes and insulitis resistant. Diabetes. 1994;43:505–509 . doi: 10.2337/diab.43.3.505. [DOI] [PubMed] [Google Scholar]
- Wicker L.S., Leiter E.H., Todd J.A., Renjilian R.J., Peterson E., Fischer P.A., Podolin P.L., Zijlstra M., Jaenisch R., Peterson L.B. Beta 2-microglobulin-deficient NOD mice do not develop insulitis or diabetes. Diabetes. 1994;43:500–504 . doi: 10.2337/diab.43.3.500. [DOI] [PubMed] [Google Scholar]
- Mora C., Wong F.S., Chang C.H., Flavell R.A. Pancreatic infiltration but not diabetes occurs in the relative absence of MHC class II-restricted CD4 T cellsstudies using NOD/CIITA-deficient mice. J. Immunol. 1999;162:4576–4588 . [PubMed] [Google Scholar]
- Ohashi P.S., Oehen S., Buerki K., Pircher H., Ohashi C.T., Odermatt B., Malissen B., Zinkernagel R.M., Hengartner H. Ablation of “tolerance” and induction of diabetes by virus infection in viral antigen transgenic mice. Cell. 1991;65:305–317 . doi: 10.1016/0092-8674(91)90164-t. [DOI] [PubMed] [Google Scholar]
- Oldstone M.B., Nerenberg M., Southern P., Price J., Lewicki H. Virus infection triggers insulin-dependent diabetes mellitus in a transgenic modelrole of anti-self (virus) immune response. Cell. 1991;65:319–331 . doi: 10.1016/0092-8674(91)90165-u. [DOI] [PubMed] [Google Scholar]
- von Herrath M.G., Guerder S., Lewicki H., Flavell R.A., Oldstone M.B. Coexpression of B7-1 and viral (“self ”) transgenes in pancreatic beta cells can break peripheral ignorance and lead to spontaneous autoimmune diabetes. Immunity. 1995;3:727–738 . doi: 10.1016/1074-7613(95)90062-4. [DOI] [PubMed] [Google Scholar]
- Bradley D.S., Nabozny G.H., Cheng S., Zhou P., Griffiths M.M., Luthra H.S., David C.S. HLA-DQB1 polymorphism determines incidence, onset, and severity of collagen-induced arthritis in transgenic mice. Implications in human rheumatoid arthritis. J. Clin. Invest. 1997;100:2227–2234 . doi: 10.1172/JCI119760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley D.S., Das P., Griffiths M.M., Luthra H.S., David C.S. HLA-DQ6/8 double transgenic mice develop auricular chondritis following type II collagen immunizationa model for human relapsing polychondritis. J. Immunol. 1998;161:5046–5053 . [PubMed] [Google Scholar]
- McInerney M.F., Rath S., Janeway C.A., Jr. Exclusive expression of MHC class II proteins on CD45+ cells in pancreatic islets of NOD mice. Diabetes. 1991;40:648–651 . doi: 10.2337/diab.40.5.648. [DOI] [PubMed] [Google Scholar]
- Wong F.S., Visintin I., Wen L., Granata J., Flavell R., Janeway C.A., Jr. The role of lymphocyte subsets in accelerated diabetes in nonobese diabetic–rat insulin promoter–B7-1 (NOD–RIP–B7-1) mice. J. Exp. Med. 1998;187:1985–1993 . doi: 10.1084/jem.187.12.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaglia L., Cahill C.J., Finegood D.T., Bonner-Weir S. Apoptosis participates in the remodeling of the endocrine pancreas in the neonatal rat. Endocrinology. 1997;138:1736–1741 . doi: 10.1210/endo.138.4.5069. [DOI] [PubMed] [Google Scholar]
- Hill D.J., Petrik J., Arany E. Growth factors and the regulation of fetal growth Diabetes Care. 21Suppl. 21998. B60 B69 [PubMed] [Google Scholar]
- Bouwens L., Pipeleers D.G. Extra-insular beta cells associated with ductules are frequent in adult human pancreas. Diabetologia. 1998;41:629–633 . doi: 10.1007/s001250050960. [DOI] [PubMed] [Google Scholar]
- Schoenberger S.P., Toes R.E., van der Voort E.I., Offringa R., Melief C.J. T-cell help for cytotoxic T lymphocytes is mediated by CD40-CD40L interactions. Nature. 1998;393:480–483. doi: 10.1038/31002. [DOI] [PubMed] [Google Scholar]