

Abstract
Cell death by necrosis is typically associated with inflammation, in contrast to apoptosis. We have identified additional distinctions between the two types of death that occur at the level of dendritic cells (DCs) and which influence the induction of immunity. DCs must undergo changes termed maturation to act as potent antigen-presenting cells. Here, we investigated whether exposure to apoptotic or necrotic cells affected DC maturation. We found that immature DCs efficiently phagocytose a variety of apoptotic and necrotic tumor cells. However, only exposure to the latter induces maturation. The mature DCs express high levels of the DC-restricted markers CD83 and lysosome-associated membrane glycoprotein (DC-LAMP) and the costimulatory molecules CD40 and CD86. Furthermore, they develop into powerful stimulators of both CD4+ and CD8+ T cells. Cross-presentation of antigens to CD8+ T cells occurs after uptake of apoptotic cells. We demonstrate here that optimal cross-presentation of antigens from tumor cells requires two steps: phagocytosis of apoptotic cells by immature DCs, which provides antigenic peptides for major histocompatibility complex class I and class II presentation, and a maturation signal that is delivered by exposure to necrotic tumor cells, their supernatants, or standard maturation stimuli, e.g., monocyte-conditioned medium. Thus, DCs are able to distinguish two types of tumor cell death, with necrosis providing a control that is critical for the initiation of immunity.
Keywords: dendritic cells, apoptosis, necrosis, cross-presentation, ELISPOT assay
Introduction
The immune system has to contend with two types of cell death and their consequences: necrosis and apoptosis 1 2 3. Necrotic and apoptotic cells can be phagocytosed by dendritic cells (DCs), potent initiators of immunity 4 5 6. However, uptake is restricted to the immature stage of development when DCs are well equipped to acquire antigen but express low levels of the requisite MHC and costimulatory molecules needed for T cell stimulation 4. Upon receipt of a maturation signal, DCs downregulate antigen acquisition, express higher levels of costimulatory and MHC molecules, and become stably differentiated to activate resting T cells. Maturation can be triggered by multiple stimuli, including LPS, contact allergens, bacteria and viruses, cell products (monocyte-conditioned medium [MCM], TNF-α, IL-1β, PGE2, IFN-α), immunostimulatory unmethylated CpG oligonucleotides, and poly(I:C) (7 8 9 10 11 12 13 14 15 16, and signaling molecules (CD40L [17, 18]).
We have recently shown that DCs phagocytose apoptotic cells and cross-present antigen from these sources to CD8+ T cells. These studies were performed with DCs that had undergone maturation after exposure to MCM, and which have proceeded to downregulate their phagocytic capacity 5. We subsequently found that immature DCs were over fivefold more efficient than mature DCs in phagocytosing apoptotic cells and that they could serve as targets for antigen-specific CD8+ cells 19. However, it remained unknown whether immature DCs could stimulate a resting CD8+ T cell population after acquisition of apoptotic cells. Since it is the immature DC located in the periphery that captures antigen most efficiently and which most likely needs a signal to leave the tissue and migrate to the regional lymph nodes, we investigated whether the uptake of dead or dying cells could initiate immunity by inducing DC maturation. Here, we found that exposure to necrotic but not apoptotic tumor cells provided the requisite maturation signal to DCs that resulted in the upregulation of maturation-specific markers, costimulatory molecules, and the capacity to induce antigen-specific CD4+ and CD8+ T cells. Thus, DCs are able to distinguish two types of tumor cell death, with necrosis providing a critical signal that will promote the initiation of immunity. Our data suggest new approaches to manipulate the immune system so that desired immune responses, e.g., to tumors, can be induced.
Materials and Methods
DC Culture.
DCs were prepared as described previously 20 21 22. In brief, DCs were generated from T cell–depleted PBMCs by culturing cells for 5–6 d in the presence of 1,000 U/ml GM-CSF (Immunex) and 1,000 U/ml IL-4 (R&D Systems). RPMI 1640 supplemented with 20 μg/ml gentamicin, 10 mM Hepes (GIBCO BRL), and 1% autologous plasma (heparinized) was used for cell culture. Cultures were supplemented with cytokines on days 2 and 4. On day 5 or 6, nonadherent immature DCs were collected and transferred to new 6-well plates. In some cases, DCs were left in the 6-well plates. Mature DCs were generated by the addition of 50% vol/vol MCM 20 on the day of transfer and harvested on days 7–8.
Induction of Apoptosis and Necrosis.
UV-triggered apoptosis was induced using a 60-mJ UVB lamp (Derma Control, Inc.), calibrated to provide 2 mJ/cm2/s. After 8 h, >50% of the cells were apoptotic. For the induction of secondary necrosis, the cells were cultured for 24 h after which >80% of the cells incorporated trypan blue. Primary necrosis was achieved via repeated freezing (at −80°C) and thawing (at 37°C) or by hypotonic lysis. One cycle of freezing and thawing resulted in mostly intact cells, which incorporated trypan blue. In contrast, four to five freeze–thaw cycles resulted in complete disruption of the cells into fragments. Supernatants were prepared from apoptotic or necrotic cells (0.5–5 × 105/ml), by spinning the cells or cell lysates at 1,500 rpm for 10 min. The supernatants were filtered through 0.22-μm Millipore filters and either used immediately or frozen at −80°C until further use.
Primary Cells, Cell Lines, and Media.
Human cell lines consisting of EBV-transformed B lymphocyte cell lines (B-LCL), melanoma cells (SK29), and kidney adenocarcinoma cells (293) were cultured in RPMI supplemented with 10% FCS. All cell lines were tested for Mycoplasma (Mycoplasma detection kit; American Type Culture Collection). Mouse cell lines consisted of melanoma cells (B16), fibroblasts (L cells), macrophage cell line (RAW), fibroblasts (NIH-3T3), and a thymoma line (EL-4) grown in DMEM supplemented with 10% FCS. Human primary cells consisted of keratinocytes, monocytes, γ-globulin–activated monocytes, influenza-infected monocytes, T cells, mitogen-activated T blasts, and B cells. Mouse primary cells consisted of liver cells and kidney cells.
Phagocytosis Assay.
293 cells were dyed red with PKH26 according to the manufacturer's protocol (Sigma Chemical Co.). Immature DCs were dyed green with PKH67 and then cocultured with the apoptotic and necrotic cells at cell equivalent ratios of 1:1 for 3 h at 4°C or 37°C. Phagocytosis of apoptotic and necrotic cells by immature DCs was defined by the percentage of double positive cells by FACS® analysis as described previously 23.
Phagocytosis of Latex Beads.
Immature DCs were cocultured with 5 × 106 green fluorescent microspheres (diameter 2.16 μm, 2.5% solids, carboxylate-modified latex; Sigma Chemical Co.) with or without addition of MCM for 48 h 24. They were then collected, separated from unengulfed beads by a buoyant density separation with Ficoll-Paque (Amersham Pharmacia Biotech), and stained for CD83 expression.
mAbs.
mAbs to the following antigens were used. CD83 (unconjugated CD83 or CD83-FITC; Immunotech) and DC–lysosomal-associated membrane glycoprotein (DC-LAMP; provided by Dr. S. Lebecque, Schering Plough, Dardilly, France) are markers expressed primarily by mature DCs. CD86, HLA-DR, CD40, CD25, CD8, and CD14 were obtained from Becton Dickinson. Isotype control mAbs included IgG1, IgG2a, and IgG2b (Immunotech). The secondary antibody was PE-conjugated F(ab′)2 goat anti–mouse IgG heavy and light chain (Jackson ImmunoResearch Labs). The DC populations were phenotyped with the panel of mAbs listed above and analyzed on a FACScan™ (Becton Dickinson). For intracellular staining, cells were first fixed in 1% paraformaldehyde and then permeabilized with 0.5% saponin before incubation with the primary antibody.
T Cell Proliferation Assays.
Apoptotic or necrotic cell lines or their supernatants were added at various ratios to day 5 or 6 immature DCs. After 48 h of coculture, the DCs were assayed for their T cell stimulatory capacity. For the superantigen-dependent assay, DCs were irradiated with 3,000 rad before addition to syngeneic T cells in the presence of 0.1 ng/ml staphylococcal enterotoxin A (SEA). After 3 d, 4 μCi/ml [3H]thymidine was added for 16 h. We observed background proliferation in 5-d cocultures of immature DCs and allogeneic T cells in the MLR, probably because of some DC maturation as a result of DC–T cell interactions. To avoid this, we fixed immature DCs after exposure to dying cells with 1% paraformaldehyde for 30 min on ice, washed extensively, and added them in graded doses to allogeneic T cells. After 4 d, 4 μCi/ml [3H]thymidine was added for 16 h. Immature and MCM-matured DCs not exposed to dying cells served as controls.
ELISA.
The measurement of cytokines in the supernatants of cocultures of DCs and apoptotic cells via ELISA (Endogen) was performed according to the manufacturer's protocol. 5 × 105 DCs were cocultured with 2.5 × 105 B-LCL cells for 18 h with or without addition of 1 ng/ml LPS, after which the supernatants were assayed for the release of TNF-α.
ELISPOT Assay for IFN-γ Release from Single Antigen-specific CD8+ T Cells.
Enzyme-linked immunospot (ELISPOT) assays for IFN-γ release from single antigen-specific CD8+ T cells were performed as described elsewhere 25. 96-well plates (Multiscreen; Millipore) were coated overnight at 4°C with 10 μg/ml of the primary anti–IFN-γ mAb (Mabtech), washed, and blocked with RPMI containing 5% PHS for 1 h at 37°C. Cells were added to wells for 18 h at 37°C, after which the wells were washed before a 2-h incubation with 50 μl of the secondary antibody (1 μg/ml, biotin-conjugated anti–IFN-γ mAb; Mabtech). Avidin-bound biotinylated horseradish peroxidase H was added (Vectastain Elite kit; Vector Laboratories) for 1 h at room temperature. The plates were then washed and incubated for 5 min in stable diaminobenzene (Research Genetics) for visualization of the complex. The spots were counted with a stereomicroscope (Stemi 2000; Carl Zeiss, Inc.). Responses were considered positive if >10 spot-forming cells (SFCs) were analyzed per 2 × 105 cells plated and the response was at least twofold greater than the respective controls.
Assay for Antigen-specific CTLs.
Immature DCs were exposed to supernatants from apoptotic or necrotic EL-4 cell lines or MCM (33% vol/vol) for 48 h, after which they were pulsed with 1 μg/ml of the HLA-A*0201–restricted influenza matrix peptide (GILGFVFTL) for 1 h. DCs were cultured with autologous HLA-A*0201+ T cells at varying ratios for 7 d. CTL activity was assayed using a conventional Cr-release assay. The targets were T2 cells pulsed or unpulsed with 1 μg/ml of the matrix peptide for 1 h. The E/T ratio was 20:1. Specific lysis was determined by subtracting the percent killing of unpulsed T2 cells.
Results
Both Apoptotic and Necrotic Cells Are Phagocytosed by the DCs.
To generate immature DCs, we cultured freshly isolated blood monocytes in GM-CSF and IL-4 for 6 d. These cells are characterized by low levels of HLA and costimulatory molecules (CD40, CD86) 4 and the DC-restricted, maturation-associated markers CD83 20 21 22 and DC-LAMP 26. We first verified that immature DCs phagocytosed necrotic cells comparably to apoptotic cells 19. A FACS® assay was used where green PKH67GL-labeled DCs are visually assessed for their ability to take up red PKH26GL-labeled apoptotic or necrotic tumor cells (kidney adenocarcinoma, 293 cells) at a ratio of 1:1 23. Over 40% of immature DCs phagocytosed either apoptotic or necrotic cells within 3 h (Fig. 1A–C). Phagocytosis of necrotic and apoptotic cells by DCs was also confirmed visually via immunofluorescence (data not shown). Uptake was profoundly reduced when dead cells were cocultured with DCs at 4°C, indicating that cells or cell fragments were not bound nonspecifically. As expected, mature DCs displayed poor phagocytic activity for either apoptotic or necrotic cells (Fig. 1 C). As a control for the effects of phagocytosis, we added FITC-labeled latex beads to immature DCs, which were then cultured for 48 h in the presence or absence of MCM, the latter to induce maturation. Uptake of latex beads by itself did not induce maturation, as monitored by surface CD83 expression, and furthermore did not inhibit maturation via MCM (Fig. 1D and Fig. E).
Figure 1.


DCs phagocytose apoptotic cells and necrotic cells. 293 cells were labeled red with the PKH26GL fluorescent cell linker and induced to undergo apoptosis via UVB irradiation, or necrosis via repeated freezing and thawing. Immature DCs were dyed green with PKH67GL and then cocultured with the apoptotic and necrotic cells for 3 h at 4°C or 37°C at a ratio of 1:1. Cells were analyzed by FACScan™ where double positive cells indicate uptake of the apoptotic and necrotic cells by the DCs. Dot plots are gated on the FL1 high positive cells (DCs), thus excluding the dead 293 cells from the analysis. (A) Green-dyed DCs and red-dyed apoptotic (293 UV) and necrotic (293 FT) cells alone. (B) Uptake of apoptotic and necrotic cells by immature DCs. (C) Uptake of apoptotic and necrotic cells by mature DCs. The results shown are representative of 10 different experiments. (D and E) On day 6 of culture, 7 × 105 immature DCs were cultured with 5 × 106 green fluorescent microspheres in the absence (D) or presence (E) of MCM. On day 8, the DCs were separated from unengulfed beads by density gradient centrifugation and analyzed by FACScan™ for uptake of latex beads and CD83 expression.
DCs Exposed to Necrotic Cells Acquire a Mature Phenotype.
We next monitored the effects of dying cells on the maturation of DCs. A panel of human cell lines was tested, including a melanoma cell line (SK29), kidney adenocarcinoma cells (293), and EBV-transformed B lymphocyte cell lines (B-LCL) (Fig. 2 A). Again, a 48-h coculture was carried out at different ratios of dying cells to DCs (1:5–1:2 for the cell lines, 1:5–10:1 for primary cells), after which the DCs were collected, stained for markers indicative of maturation, and analyzed on a FACScan™. Exposure to necrotic cells, but not apoptotic cells, induced the expression of the maturation-associated markers CD83 and DC-LAMP in a large percentage of DCs (Fig. 2a–C) and upregulated both costimulatory (CD86) and HLA molecules (Fig. 2D and Fig. E). Furthermore, CD40 levels almost doubled after exposure to necrotic cells (Fig. 2D and Fig. E). The induction of maturation was equivalent to that seen with MCM or poly(I:C) (data not shown) and was irreversible, as DCs did not revert to an immature phenotype in culture. Necrotic but not apoptotic mouse cell lines, including L cells, EL-4 cells, NIH-3T3 cells, and B16 melanoma cells, also induced the maturation of DCs. However, primary cells, e.g., T cells, B cells, monocytes, and keratinocytes, were ineffective in this regard (data not shown). The same was true for influenza-infected monocytes induced to undergo apoptosis or necrosis after infection, as even at a 10:1 ratio of dead cells to DCs they failed to induce maturation (Fig. 2 F). T cells activated by mitogen stimulation or monocytes stimulated by γ-globulin also did not induce maturation (data not shown).
Figure 2.

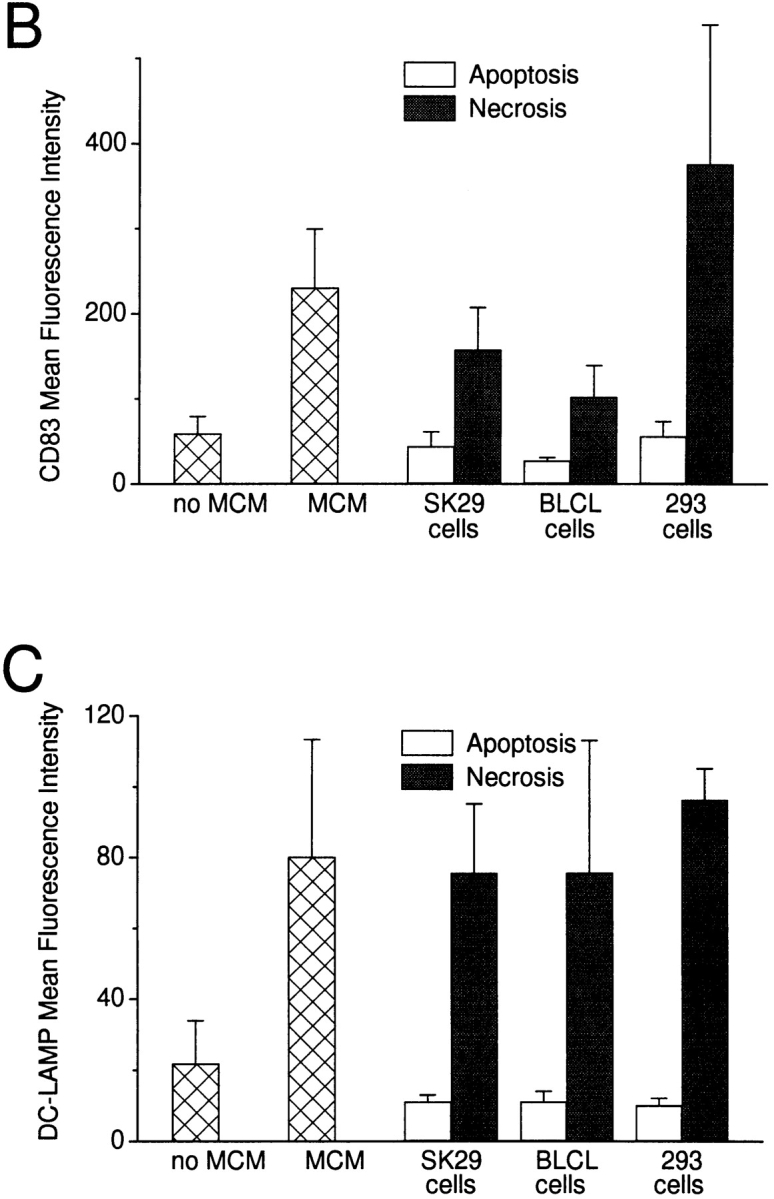

DCs exhibit a mature phenotype after exposure to necrotic but not apoptotic cell lines. (A) Apoptotic or necrotic B-LCL cells were cocultured with DCs at ratios of 1:2 or 1:5. After 48 h, the DCs were stained extracellularly for CD83 and intracellularly for CD83 and DC-LAMP. (B and C) DCs were cocultured with apoptotic or necrotic cell lines at a ratio of 1:2 for 48 h and then stained extracellularly for CD83 and intracellularly for DC-LAMP. (D) Immature and mature DCs were stained with mAbs to CD86, CD40, HLA-DR, and/or CD83. Apoptotic (Apop) or necrotic (F/T) cell lines (E), influenza-infected or uninfected macrophages (MO) (F), or supernatants from dead cells (G) were added to immature DCs. After 48 h, the DC were stained with mAbs to CD83, CD86, CD40, and HLA-DR. (H) Apoptotic and necrotic primary cells were cocultured for 48 h with DCs, after which the CD83 expression of the DCs was compared with DCs matured with MCM, LPS, or necrotic (F/T, or by hypotonic lysis) 293 cells. (I) Supernatants of apoptotic cells cultured for 8 and for 24 h or supernatants of partly ablated (1× F/T) or completely ablated (4× F/T) cells were added to immature DCs, and after 48 h the DCs were stained with an mAb to CD83. In D, E, and G, values represent the averages of the geometric mean indices from three or more experiments. Error bars mark the SD. In I, a directly labeled anti-CD83 (CD83-PE) antibody was used, accounting for the lower mean fluorescence intensities. Isotype-matched antibodies served as controls in all experiments (data not shown). In F, H, and I, a representative experiment of two to four is shown.
Supernatants from Necrotic Cells Induce the Maturation of DCs.
To define the nature of factors in necrotic cells that were responsible for inducing maturation, we collected supernatants from necrotic and apoptotic cell lines, filtered them through a 0.22-μm filter, and added these to immature DC cultures. Supernatants from necrotic but not apoptotic cells induced maturation (Fig. 2 G). In contrast, supernatants from necrotic primary cells again had no effect on DC maturation (Fig. 2 H). It was shown recently that an excess of apoptotic cells can induce maturation of a murine DC line 27. In that study, apoptotic cells were cultured for up to16 h, before their addition to immature DC cell lines. Interestingly, when we added apoptotic cells to DCs at similar ratios of 5:1 or 10:1, there was extensive DC death (data not shown). It is possible that the phagocytic capacity of our human DCs was overwhelmed and that the DCs died as a consequence. Because apoptotic cells can undergo secondary necrosis in long-term cultures, the studies of Rovere et al. 27 could be explained by the release of soluble factors that induce maturation. To test this, we compared the supernatants derived from UV-irradiated tumor cells (e.g., EL-4 cells) cultured for 8 h (when >50% of the cells are Annexin V positive) and 24 h (when >90% of cells undergo secondary necrosis as visualized by trypan blue inclusion). Supernatants from apoptotic cells cultured for 24 h failed to mature DCs (Fig. 2 I). The same was true for apoptotic cells that were lysed after 8 h (to fragment apoptotic cell bodies) or supernatant of live cells cultured for 48 h (data not shown). In addition, we were interested whether there were differences in supernatants of tumor cells undergoing limited or extensive necrosis. After one round of freeze–thaw, the cells are grossly intact but incorporate trypan blue, whereas four freeze–thaw cycles result in complete cellular fragmentation. Interestingly, only supernatants of cells that underwent four cycles of freeze–thaw were effective in maturing DCs, as determined by CD83 expression (Fig. 2 I). To define the active factor contained within the supernatants of necrotic cells, we examined the supernatants for inflammatory cytokines. Obvious candidates for maturation, such as TNF-α and IL-1β, were not detectable by ELISA (data not shown).
DCs Exposed to Necrotic Cells or Their Supernatants Acquire Potent T Cell Stimulatory Capacity.
Mature DCs are potent stimulators of T cells, the most straightforward assays being the induction of allogeneic T cell proliferation in the MLR and superantigen-dependent T cell proliferation 28 29. After a 48-h exposure to dying cells or their respective supernatants, DCs were washed and added in graded doses to 2 × 105 allogeneic or syngeneic T cells, in the latter case together with 0.1 ng/ml SEA. Strong stimulation of allogeneic and superantigen-stimulated syngeneic T cells was observed with DCs that had been matured with necrotic cells, their respective supernatants, or with MCM (Fig. 3A and Fig. B). In contrast, exposure to apoptotic cells did not induce MLR stimulatory activity or superantigen-dependent syngeneic proliferation above the low background in the absence of MCM (Fig. 3A and Fig. B). The [3H]thymidine uptake data were confirmed by analysis of T cell size on the FACScan™. In response to mature DCs (matured with MCM or with exposure to necrotic cells), >30% of the CD3+ T cells in the MLR were blasts (high forward light scattering), whereas immature DCs (no MCM, or exposure to apoptotic cells) induced <5% blast transformation (not shown).
Figure 3.


DCs gain heightened T cell stimulatory capacity after exposure to necrotic but not apoptotic cell lines. Apoptotic or necrotic cell lines (B-LCL) were added at a ratio of 1:5 to day 5 or 6 immature DCs. After 48 h of coculture, the DCs were assayed for their T cell stimulatory capacity. (A) DCs were irradiated with 3,000 rad using a cesium irradiator before addition to 2 × 105 syngeneic T cells and 0.1 ng/ml SEA. After 3 d, 4 μCi/ml [3H]thymidine was added for 16 h. (B) After coculture with apoptotic or necrotic cell lines (B-LCL) or their respective supernatants (sup), DCs were fixed with 1% paraformaldehyde for 30 min at 4°C, washed extensively, and added in graded doses to 2 × 105 allogeneic T cells. After 4 d, 4 μCi/ml [3H]thymidine was added for 16 h. Immature and MCM-matured DCs served as controls. T cells cultured alone gave counts <600 cpm. Results are representative of three or more experiments, and the values shown represent the mean of triplicate wells. (C) 5 × 105 DCs were cocultured with apoptotic B-LCL cells (ApopLCL) at a 2:1 ratio with or without the addition of 1 ng/ml LPS. After 16 h, the supernatants were tested for TNF-α release by ELISA.
Importantly, when DCs were exposed to a mixture of apoptotic and necrotic cells, they induced similar increases in T cell stimulation as DCs cultured with necrotic cells or their supernatants (data not shown). Furthermore, DCs exposed in parallel to apoptotic cells and MCM heightened T cell responses to the same extent as DCs matured with MCM. These experiments indicated that ingestion of apoptotic cells did not inhibit DC maturation or function. Experiments where DCs were stimulated with LPS after uptake of apoptotic cells revealed an uninhibited secretion of inflammatory cytokines, i.e., TNF-α (Fig. 3 C) or IL-1β, compared with control DCs that were not cocultured with apoptotic cells. This contrasts with recently published data showing that phagocytosis of apoptotic cells induces immunosuppressive and antiinflammatory effects in macrophages (e.g., release of IL-10 and PGE2 after LPS stimulation) and that apoptotic cells themselves can be sources of factors (e.g., IL-10) that skew the immune response 30 31 32.
Exposure to Necrotic Cell Lines or Their Supernatants Allows for the Induction of Antigen-specific CD8+ Effector Cells.
We next tested the effect of exposing immature DCs to necrotic or apoptotic cell lines and their respective supernatants on the induction of antigen-specific CD8+ effector cells. DCs from HLA-A*0201+ individuals were exposed to dying tumor cells, their supernatants, or MCM for 2 d. The cells were then collected, pulsed with the HLA-A*0201–restricted influenza matrix peptide (GILGFVFTL), and cocultured with syngeneic T cells for 7 d. The induction of influenza-specific IFN-γ–producing CD8+ cells was tested after restimulating the T cells with matrix peptide–pulsed autologous DCs in an ELISPOT assay, whereas the cytolytic activity of responding T cells was tested directly on matrix peptide–pulsed T2 target cells in a conventional chromium-release assay. Only DCs exposed to necrotic cell lines, their supernatants, or MCM permitted the induction of matrix peptide–specific CD8+ effector cells (Fig. 4a and Fig. b), whereas exposure to apoptotic cell lines or their supernatants was not effective.
Figure 4.


Immature DCs matured via necrotic supernatants are able to induce antigen-specific CD8+ effector cells. Cocultures of immature DCs were supplemented with nothing, MCM, or supernatants (sup) of apoptotic (UVB-irradiated B-LCL) or necrotic (frozen–thawed B-LCL) cells and incubated for 48 h. They were collected, unpulsed or pulsed with 1 μg/ml influenza matrix peptide for 1 h, and cocultured with 2 × 105 syngeneic T cells at a ratio of 1:30 for 7 d to allow for the activation of CD8+ effector cells. Responding T cells were collected and restimulated with autologous DCs that were untreated or pulsed with matrix peptide (MP) for 18 h. The induction of influenza matrix peptide–specific CD8+ T cells was measured in an IFN-γ ELISPOT assay (A). Alternatively, 7-d cocultures were directly tested for matrix peptide–specific CTL activity in a 4-h chromium-release assay using matrix peptide–pulsed T2 cells as targets (B). Results are representative of three experiments, and the values shown represent the mean of triplicate wells.
A Maturation Signal Provided by Necrotic Cell Lines, Their Respective Supernatants, or MCM Is Required for Optimal Cross-Presentation of Antigens Derived from Apoptotic Cells.
In previous studies, we found that mature DCs can present antigens from apoptotic cells or tumor lines to elicit CD8+ CTL responses 5. However, phagocytosis of apoptotic cells is maximal when DCs are immature 19. As exposure of immature DCs to apoptotic cell lines fails to mature them, we tested whether the addition of necrotic cell lines, their supernatants, or MCM would enhance antigen cross-presentation. Immature DC cultures (derived from HLA-A*0201+ monocytes) were fed with influenza-infected or uninfected apoptotic EL-4 cells and supplemented with either supernatants of apoptotic or necrotic cell lines or MCM. After 48 h, the DCs were collected and cocultured with 2 × 105 syngeneic T cells at a ratio of 1:15. 18 h later, antigen-specific (IFN-γ) production by CD8+ T cells was measured in an ELISPOT assay. We have previously shown that the primary cells making IFN-γ in this assay are CD8+ T cells 25. Only DCs matured via exposure to necrotic cell supernatants or MCM were able to induce influenza-specific CD8+ effector cells after the phagocytosis of influenza-infected EL-4 cells (Fig. 5). It was necessary to use supernatant from cell lines that had undergone four freeze–thaw cycles, consistent with our observation that extensive necrosis is required to release maturation factors (Fig. 2 I). Neither early nor late apoptotic cell supernatants allowed for the induction of significant levels of IFN-γ–producing SFCs. Fig. 5 (right) shows that once mature, DCs efficiently present the HLA-A*0201+ restricted influenza matrix peptide to syngeneic T cells. The number of SFCs obtained with matrix peptide–pulsed, MCM-matured DCs was similar to that measured when DCs were fed apoptotic influenza-infected EL-4 cells and matured with 4× necrotic cell supernatant. We conclude that immature DCs phagocytose apoptotic cells 19 but do not efficiently cross-present antigens from these cells unless they receive a maturation stimulus. Fig. 5 is notable in providing the first demonstration of cross-presentation by DCs to CD8+ T cells where xenogeneic cells are the source of antigen.
Figure 5.

Exposure of immature DCs to maturation stimuli is necessary for optimal cross-presentation of antigen derived from apoptotic cells. EL-4 cells were infected or uninfected with influenza virus and induced to undergo apoptosis by UVB irradiation. Apoptotic cells were fed to immature DCs at a ratio of 1:20 and simultaneously exposed to MCM or supernatants (sup) from EL-4 cell lines. Supernatants were derived from UVB-irradiated EL-4 lines that were cultured for 8 vs. 24 h, or lines that had undergone one (1×) vs. four (4×) freeze–thaw cycles. After 48 h, the DCs were collected and cocultured with syngeneic T cells. Cross-presentation of influenza antigen was detected by monitoring IFN-γ–producing SFCs in an ELISPOT assay. The data shown were calculated after subtracting the background values, i.e., the number of SFCs obtained when apoptotic uninfected ELA cells were fed to immature DCs. These values ranged from 0 to 30/106 T cells. Additional controls consisted of immature and MCM-matured DCs, which were pulsed with 1 μg/ml of the influenza matrix peptide (MP) before coculture with syngeneic T cells.
Discussion
While a role for macrophages in the phagocytic clearance of both apoptotic and necrotic cells is well established, less is known about how dying cells are dealt with by DCs. Macrophages phagocytose apoptotic cells via several receptors, thereby allowing for their efficient clearance 1. Concomitantly, macrophages are primed to produce immunoregulatory factors such as IL-10, TGF-β, and PGE2 while the production of proinflammatory cytokines (e.g., IL-12, TNF-α) is suppressed 30 31 32. Moreover, macrophages degrade rather than process antigens contained within apoptotic cells, as they fail to induce antigen-specific CTLs when injected in vivo 33 and are not recognized as targets of CTLs in vitro 19. Altogether, these data suggest that macrophages modulate the immune response after phagocytosis of apoptotic cells by the release of immunosuppressive factors and failure to present antigen. Exposure of macrophages to necrotic cells leads to macrophage activation, indicating the importance of distinguishing the effects of cell death upon APCs 34.
In contrast to macrophages, we find striking differences in the handling of dying tumor lines or transformed cell lines by DCs depending on the mechanism of cell death. Although DCs efficiently phagocytose both necrotic and apoptotic cells, only exposure to necrotic tumor cells or transformed cell lines, or their soluble derivatives, selectively induces the maturation of DCs. As this paper was under review, a similar phenomenon was described with murine DCs 34a. DCs also seem to distinguish between these two forms of death at the level of antigen presentation. We have previously shown that uptake of apoptotic cells by DCs is required for cross-presentation of antigens to CD8+ T cells 5, whereas both apoptotic and necrotic cells can serve as sources of antigen for the activation of CD4+ T cells 6. Our studies now add another refinement to these pathways. Optimal cross-presentation of antigens acquired from dying cells by DCs requires two steps: first, the efficient acquisition of dying cells via phagocytosis when DCs are in their immature state where uptake is severalfold more efficient than in the mature state 19; and second, the receipt of a maturation signal that can be provided by necrotic cell fragments, their soluble supernatants, or standard maturation stimuli, e.g., MCM. As a result, the antigen presentation can be improved almost 5–10-fold. Immature DCs can process antigens derived from apoptotic cells as they are lysed by CTL lines in short-term assays 19. However, to activate CD8+ T cells, either IFN-γ effector cells or CTLs, the DCs must undergo maturation, a state characterized by the upregulation of MHC and costimulatory molecules and the production of cytokines such as IL-12. In previous studies, we used mature DC populations to phagocytose apoptotic cells and cross-present viral and tumor antigens to both CD4+ and CD8+ T cells 5 6. These mature populations are not likely to be synchronous, explaining why a small percentage can still phagocytose apoptotic material 5 and permit cross-presentation of antigenic material onto MHC class I and II molecules. Another distinction between DCs and macrophages in their handling of apoptotic cells is that DCs are not inhibited in their ability to produce proinflammatory cytokines, whereas macrophages become primed to produce immunosuppressive cytokines and factors 30 31. This has been demonstrated in vivo, where DCs but not macrophages pulsed with apoptotic tumor cells induced an antitumor response 33.
Our studies uncovered an important distinction between the sources of necrotic cells required to induce DC maturation. We found that only certain types of cells, namely tumor lines, were capable of inducing DC maturation after necrosis. These included transformed cells of both human (SK29 melanoma cells, 293 cells, B-LCL cells, and HeLa cells) and murine origin (EL-4 cells, L cells, RAW cells, and NIH-3T3 cells). When we tested primary cells (keratinocytes, T cells, monocytes, and B cells) and cells derived from whole tissues (mouse liver and kidney), we were unable to detect maturation-inducing factor, even if they were added at an excess. Activation of T cells with mitogen or of monocytes with γ-globulin also failed to elicit this activity. This apparent distinction between primary cells and lines remains unexplained but may be due to the paucity of such factors in primary cells, and/or their induction in tumor lines possibly related or secondary to a transformation or cell cycle event. It also became clear from our studies that a certain extent of necrosis was required to release the maturation factors from tumor lines, i.e., four cycles of freeze–thaw versus one cycle. The former led to complete fragmentation of cells and presumably organelles and nuclei, whereas in the latter, dead but grossly intact cells were still evident. The distinction between primary and secondary necrosis also became apparent when we compared supernatants derived from necrotic versus 24-h-old apoptotic cells. The former but not the latter contained maturation factors. These data are consistent with the concept that apoptotic cells are noninflammatory, even when they are relatively “old,” at least as it pertains to DCs. Therefore, only in the setting of some inflammatory (e.g., cell lysis secondary to viral infection or release of cytotoxic cytokines) or stressful event (e.g., trauma or toxicity), where complete cell fragmentation is induced, may maturation factors be released.
The nature of the activating molecules provided by necrotic cells to DCs is the subject of ongoing investigation. The “danger model” proposed by Matzinger suggests that dendritic cells are activated by endogenous alarm signals released by stressed, damaged, or necrotic peripheral tissue cells 35. Two likely candidates known to be involved in DC maturation, namely TNF-α and IL-1β, were not detectable in our supernatants. As murine lines also served as sources of maturation factors, cytokines seem less likely to be involved here. Janeway 36 has proposed that DCs express nonclonal pattern recognition receptors (PRRs) that recognize invariant molecular structures in pathogens (pathogen-associated molecular patterns [PAMPs]). PAMPs expressed or induced by LPS, viruses, and bacteria may induce DC maturation 36. As some of our tumor lines were virally transformed, we cannot exclude the possibility that factors expressing PAMPs are involved. Double-stranded RNA (produced by RNA viruses like influenza), recently demonstrated to be a potent inducer of DC maturation 15 16, may be another such candidate. Other studies have shown that tumor cell death by nonapoptotic mechanisms, e.g., necrosis, is characterized by the induction of high levels of mRNA for heat shock proteins that may target antigens to immature DCs 37 38. Interestingly, heat shock proteins have been implicated in cross-priming pathways 39. It remains to be seen whether these molecules are selectively released in different types of tumor cell death and whether they can affect DC maturation.
Curiously, when we cocultured an excess of influenza-infected monocytes with immature DCs, this did not lead to their maturation. Macrophages infected with influenza produce GM-CSF, IL-1, and TNF-α, but only low levels of virus and undergo rapid spontaneous apoptosis 40. It is possible that insufficient levels of cytokines, virus, or double-stranded RNA are produced in vitro to induce DC maturation. The failure of infected monocytes, whether apoptotic or necrotic, to mature DCs might be circumvented in vivo by inflammatory cytokines from surrounding infected tissues. In addition, opsonizing the apoptotic cell might provide an alternative signal to target the apoptotic cells to DCs and induce their maturation 41. Since some viruses may not access professional APCs, mechanisms must exist in vivo whereby virus-specific responses are induced. A recent study indicated that bone marrow–derived cells are needed in vivo to cross-present antigens from virus-infected peripheral tissues 42; however, the mechanism remains undetermined.
We envision different scenarios depending on which type of dying cell DCs may encounter in vivo. We would predict that uptake of necrotic tumor cells would lead to the initiation of T cell responses to antigens processed by the DCs. These responses are likely to be CD4+ rather than CD8+, as antigens derived from necrotic cells do not seem to induce CTLs, at least in vitro 5. Phagocytosis of apoptotic cells by DCs failed to induce maturation, the consequences of which may be the induction of tolerance to self or tumor antigens 43 44 45. However, phagocytosis of apoptotic cells may lead to T cell immunity if followed by a maturation signal. Signals provided by necrotic cells in the environment, cytokines (e.g., TNF-α, IL-1, or IFN-α released by virus-infected cells), inflammatory products (e.g., LPS or bacterial cell walls), and CD4+ T cells (such as CD40L–CD40 interactions) 17 18 46 47 48 would be required to condition the DC, thus allowing for the full activation of T cells. In fact, in in vivo animal models of cross-priming and cross-tolerance, induction and maintenance of CTLs require CD4+ help 49 50.
In conclusion, our study reveals novel biological differences between apoptotic and necrotic tumor cells. For the most part, effective immunity is not induced to antigens in tumors, which might be expected to undergo some level of cell turnover through apoptosis, and secondary necrosis. Since successful immunotherapy in many cases seems to be reduced to the problem of creating the right inflammatory reaction (e.g., GM-CSF–transduced tumor lines or BCG injections [ 51, 52]), necrosis might be one of the elements critical for the induction of tumor immunity. In fact, murine studies have suggested that the immunogenicity of tumors is influenced by the mechanism of cell death in vivo. In situ killing of tumor cells by nonapoptotic mechanisms is associated with high immunogenicity, whereas apoptotic cell death is considerably less immunogenic 37. Our data suggest that by inducing necrosis where only apoptosis is prevalent, the immune system could be redirected to generate a favorable response.
Acknowledgments
The authors thank Ralph M. Steinman for helpful discussions, Judy Adams for graphics, and Frank Isdell for help with the FACScan™.
Birthe Sauter is supported by a fellowship from the Deutsche Forschungsgemeinschaft (grant SA 741/1-1). Matthew Albert is supported by the National Institutes of Health Medical Scientist training program (grant GM07793). Marie Larsson is supported by grants from the Swedish Medical Research Council (K98-99PK-12334-02). This study was supported by grants from the National Institutes of Health (AI39516 and AI44628) and the Systemic Lupus Erythematosus Foundation to Nina Bhardwaj.
Footnotes
Abbreviations used in this paper: DC, dendritic cell; ELISPOT, enzyme-linked immunospot; LAMP, lysosome-associated membrane glycoprotein; MCM, monocyte-conditioned medium; PAMP, pathogen-associated molecular pattern; SEA, staphylococcal enterotoxin A; SFC, spot-forming cell.
References
- Savill J. Recognition and phagocytosis of cells undergoing apoptosis. Br. Med. Bull. 1997;53:491–508 . doi: 10.1093/oxfordjournals.bmb.a011626. [DOI] [PubMed] [Google Scholar]
- Cohen J.J. Overviewmechanisms of apoptosis. Immunol. Today. 1993;14:126–130. doi: 10.1016/0167-5699(93)90214-6. [DOI] [PubMed] [Google Scholar]
- Duvall E., Wyllie A.H. Death and the cell. Immunol. Today. 1986;7:115–119. doi: 10.1016/0167-5699(86)90152-0. [DOI] [PubMed] [Google Scholar]
- Banchereau J., Steinman R.M. Dendritic cells and the control of immunity. Nature. 1998;392:245–252. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- Albert M.L., Sauter B., Bhardwaj N. Dendritic cells acquire antigen from apoptotic cells and induce class I-restricted CTLs. Nature. 1998;392:86–89. doi: 10.1038/32183. [DOI] [PubMed] [Google Scholar]
- Inaba K., Turley S., Yamaide F., Iyoda T., Mahnke K., Inaba M., Pack M., Subklewe M., Sauter B., Sheff D. Efficient presentation of phagocytosed cellular fragments on the MHC class II products of dendritic cells. J. Exp. Med. 1998;188:2163–2173. doi: 10.1084/jem.188.11.2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macatonia S.E., Knight S.C., Edwards A.J., Griffiths S., Fryer P. Localization of antigen on lymph node dendritic cells after exposure to the contact sensitizer fluorescein isothiocyanate. J. Exp. Med. 1987;166:1654–1667. doi: 10.1084/jem.166.6.1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacPherson G.G., Jenkins C.D., Stein M.J., Edwards C. Endotoxin-mediated dendritic cell release from the intestine. Characterization of released dendritic cells and TNF dependence. J. Immunol. 1995;154:1317–1322. [PubMed] [Google Scholar]
- De Smedt T., Pajak B., Muraille E., Lespagnard L., Heinen E., De Baetselier P., Urbain J., Leo O., Moser M. Regulation of dendritic cell numbers and maturation by lipopolysaccharide in vivo. J. Exp. Med. 1996;184:1413–1424. doi: 10.1084/jem.184.4.1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roake J.A., Rao A.S., Morris P.J., Larsen C.P., Hankins D.F., Austyn J.M. Dendritic cell loss from non-lymphoid tissues following systemic administration of lipopolysaccharide, tumor necrosis factor, and interleukin 1. J. Exp. Med. 1995;181:2237–2248. doi: 10.1084/jem.181.6.2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rescigno M., Citterio S., Thery C., Rittig M., Medaglini D., Pozzi G., Amigorena S., Ricciardi-Castagnoli P. Bacteria-induced neo-biosynthesis, stabilization, and surface expression of functional class I molecules in mouse dendritic cells. Proc. Natl. Acad. Sci. USA. 1998;95:5229–5234. doi: 10.1073/pnas.95.9.5229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhardwaj N., Bender A., Gonzalez N., Bui L.K., Garrett M.C., Steinman R.M. Influenza virus–infected dendritic cells stimulate strong proliferative and cytolytic responses from human CD8+ T cells. J. Clin. Invest. 1994;94:797–807. doi: 10.1172/JCI117399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender A., Albert M., Reddy A., Feldman M., Sauter B., Kaplan G., Hellmann W., Bhardwaj N. The distinctive features of influenza virus infection of dendritic cells. Immunobiology. 1998;198:64–69. doi: 10.1016/S0171-2985(98)80078-8. [DOI] [PubMed] [Google Scholar]
- Sparwasser T., Koch E.S., Vabulas R.M., Heeg K., Lipford G.B., Ellwart J.W., Wagner H. Bacterial DNA and immunostimulatory CpG oligonucleotides trigger maturation and activation of murine dendritic cells. Eur. J. Immunol. 1998;28:2045–2054. doi: 10.1002/(SICI)1521-4141(199806)28:06<2045::AID-IMMU2045>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Cella M., Salio M., Sakakibara Y., Langen H., Julkunen I., Lanzavecchia A. Maturation, activation, and protection of dendritic cells induced by double-stranded RNA. J. Exp. Med. 1999;189:821–829. doi: 10.1084/jem.189.5.821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdijk R.M., Mutis T., Esendam B., Kamp J., Melief C.J.M., Brand A., Goulmy E. Polyriboinosinic polyribocytidylic acid (poly(I:C)) induces stable maturation of functionally active human dendritic cells. J. Immunol. 1999;163:57–61. [PubMed] [Google Scholar]
- Caux C., Massacrier C., Vanbervliet B., Dubois B., Van Kooten C., Durand I., Banchereau J. Activation of human dendritic cells through CD40 cross-linking. J. Exp. Med. 1994;180:1263–1272. doi: 10.1084/jem.180.4.1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cella M., Scheidegger D., Palmer-Lehmann K., Lane P., Lanzavecchia A., Alber G. Ligation of CD40 on dendritic cells triggers production of high levels of interleukin-12 and enhances T cell stimulatory capacityT–T help via APC activation. J. Exp. Med. 1996;184:747–752. doi: 10.1084/jem.184.2.747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albert M.L., Pearce S.F.A., Francisco L.M., Sauter B., Roy P., Silverstein R.L., Bhardwaj N. Immature dendritic cells phagocytose apoptotic cells via αvβ5 and CD36, and cross-present antigens to cytotoxic T lymphocytes. J. Exp. Med. 1998;188:1359–1368. doi: 10.1084/jem.188.7.1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender A., Sapp M., Schuler G., Steinman R.M., Bhardwaj N. Improved methods for the generation of dendritic cells from nonproliferating progenitors in human blood. J. Immunol. Methods. 1996;196:121–135. doi: 10.1016/0022-1759(96)00079-8. [DOI] [PubMed] [Google Scholar]
- Cella M., Sallusto F., Lanzavecchia A. Origin, maturation and antigen presenting function of dendritic cells. Curr. Opin. Immunol. 1997;9:10–16. doi: 10.1016/s0952-7915(97)80153-7. [DOI] [PubMed] [Google Scholar]
- Romani N., Reider D., Heuer M., Ebner S., Eibl B., Niederwieser D., Schuler G. Generation of mature dendritic cells from human bloodan improved method with special regard to clinical applicability. J. Immunol. Methods. 1996;196:137–151. doi: 10.1016/0022-1759(96)00078-6. [DOI] [PubMed] [Google Scholar]
- Hess K.L., Babcock G.F., Askew D.S., Cook-Mills J.M. A novel flow cytometric method for quantifying phagocytosis of apoptotic cells. Cytometry. 1997;27:145–152. [PMC free article] [PubMed] [Google Scholar]
- Steinkamp J.A., Wilson J.S., Saunders G.C., Stewart C.C. Phagocytosisflow cytometric quantitation with fluorescent microspheres. Science. 1981;215:64–66. doi: 10.1126/science.7053559. [DOI] [PubMed] [Google Scholar]
- Larsson M., Jin X., Ramratnam B., Ogg G.S., Engelmayer J., Demoitie M.A., McMichael A.J., Cox W.I., Steinman R.M., Nixon D., Bhardwaj N. A recombinant vaccinia virus based ELISPOT assay detects high frequencies of Pol-specific CD8 T cells in HIV-1-positive individuals. AIDS. 1999;13:767–777. doi: 10.1097/00002030-199905070-00005. [DOI] [PubMed] [Google Scholar]
- de Saint-Vis B., Vincent J., Vandenabeele S., Vanbervliet B., Pin J.J., Ait-Yahia S., Patel S., Mattei M.G., Banchereau J., Zurawski S. A novel lysosome-associated membrane glycoprotein, DC-LAMP, induced upon DC maturation, is transiently expressed in MHC class II compartment. Immunity. 1998;9:325–336. doi: 10.1016/s1074-7613(00)80615-9. [DOI] [PubMed] [Google Scholar]
- Rovere P., Vallinoto C., Bondanza A., Crosti M.C., Rescigno M., Ricciardi-Castagnoli P., Rugarli C., Manfredi A.A. Bystander apoptosis triggers dendritic cell maturation and antigen-presenting function. J. Immunol. 1998;161:4467–4471. [PubMed] [Google Scholar]
- Freudenthal P.S., Steinman R.M. The distinct surface of human blood dendritic cells, as observed after an improved isolation method. Proc. Natl. Acad. Sci. USA. 1990;87:7698–7702. doi: 10.1073/pnas.87.19.7698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhardwaj N., Friedman S.M., Cole B.C., Nisanian A.J. Dendritic cells are potent antigen-presenting cells for microbial superantigens. J. Exp. Med. 1992;175:267–273. doi: 10.1084/jem.175.1.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voll R.E., Herrmann M., Roth E.A., Stach C., Kalden J.R. Immunosuppressive effects of apoptotic cells. Nature. 1997;390:350–351. doi: 10.1038/37022. [DOI] [PubMed] [Google Scholar]
- Fadok V.A., Bratton D.L., Konowal A., Freed P.W., Westcott J.Y., Henson P.M. Macrophages that have ingested apoptosis cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-β, PGE2, and PAF. J. Clin. Invest. 1998;101:890–898. doi: 10.1172/JCI1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y., Herndon J.M., Zhang H., Griffith T.S., Ferguson T.A. Anti-inflammatory effects of CD95 ligand (FasL)-induced apoptosis. J. Exp. Med. 1998;188:887–896. doi: 10.1084/jem.188.5.887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronchetti A.R., Rovere P., Iezzi G., Galati G., Heltai S., Protti M.P., Garancini M.P., Manfredi A.A., Rugarli C., Bellone M. Immunogenicity of apoptotic cells in vivorole of antigen load, antigen-presenting cells, and cytokines. J. Immunol. 1999;163:130–136. [PubMed] [Google Scholar]
- Ren Y., Savill J. Apoptosisthe importance of being eaten. Cell Death Differ. 1998;5:563–568. doi: 10.1038/sj.cdd.4400407. [DOI] [PubMed] [Google Scholar]
- Gallucci S., Lolkema M., Matzinger P. Natural adjuvantsendogenous activators of dendritic cells. Nat. Med. 1999;11:1249–1255. doi: 10.1038/15200. [DOI] [PubMed] [Google Scholar]
- Matzinger P. Tolerance, danger, and the extended family. Annu. Rev. Immunol. 1994;12:991–1045. doi: 10.1146/annurev.iy.12.040194.005015. [DOI] [PubMed] [Google Scholar]
- Janeway C.A., Jr. The immune system evolved to discriminate infectious nonself from noninfectious self. Immunol. Today. 1992;13:11–16. doi: 10.1016/0167-5699(92)90198-G. [DOI] [PubMed] [Google Scholar]
- Melcher A., Todryk S., Hardwick N., Ford M., Jacobson M., Vile R.G. Tumor immunogenicity is determined by the mechanism of cell death via induction of heat shock protein expression. Nat. Med. 1998;4:581–587. doi: 10.1038/nm0598-581. [DOI] [PubMed] [Google Scholar]
- Todryk S., Melcher A.A., Hardwick N., Linardakis E., Bateman A., Colombo M.P., Stoppacciaro A., Vile R.G. Heat shock protein 70 induced during tumor cell killing induces TH1 cytokines and targets immature dendritic cell precursors to enhance antigen uptake. J. Immunol. 1998;163:1398–1408. [PubMed] [Google Scholar]
- Suto R., Srivastava P.K. A mechanism for the specific immunogenicity of heat shock protein-chaperoned peptides. Science. 1995;269:1585–1588. doi: 10.1126/science.7545313. [DOI] [PubMed] [Google Scholar]
- Hofmann P., Sprenger H., Kaufmann A., Bender A., Hasse C., Nain M., Gemsa D. Susceptibility of mononuclear phagocytes to influenza virus infection and possible role in the antiviral response. J. Leukoc. Biol. 1997;612:408–414. doi: 10.1002/jlb.61.4.408. [DOI] [PubMed] [Google Scholar]
- Rovere P., Sabadini M.G., Vallinoto C., Fascio U., Rescigno M., Crosti M., Ricciardi-Castagnoli P., Balestrieri G., Tincani A., Manfredi A.A. Dendritic cell presentation of antigens from apoptotic cells in a proinflammatory contextrole of opsonizing anti-β2-glycoprotein I antibodies. Arthritis Rheum. 1999;42:1412–1420. doi: 10.1002/1529-0131(199907)42:7<1412::AID-ANR15>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- Sigal L.J., Crotty S., Andino R., Rock K.L. Cytotoxic T-cell immunity to virus-infected non-haematopoietic cells requires presentation of exogenous antigen. Nature. 1999;398:77–80. doi: 10.1038/18038. [DOI] [PubMed] [Google Scholar]
- Kurts C., Miller J.F.A.P., Carbone F. Cross-tolerancea pathway for inducing tolerance to peripheral tissue antigens. J. Exp. Med. 1998;187:1549–1553. doi: 10.1084/jem.187.10.1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurts C., Miller J.F.A.P., Subramaniam R.M., Carbone F.R., Heath W.R. Major histocompatibility complex class I–restricted cross-presentation is biased towards high dose antigens and those released during cellular destruction. J. Exp. Med. 1998;188:409–414. doi: 10.1084/jem.188.2.409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurts C., Heath W.R., Kosaka H., Miller J.F.A.P., Carbone F.R. The peripheral deletion of autoreactive CD8+ T cells induced by cross-presentation of self-antigens involves signaling through CD95 (Fas, Apo-1) J. Exp. Med. 1998;188:415–420. doi: 10.1084/jem.188.2.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridge J.P., Di Rosa F., Matzinger P. A conditioned dendritic cell can be a temporal bridge between a CD4+ T helper and a T-killer cell. Nature. 1998;393:474–478. doi: 10.1038/30989. [DOI] [PubMed] [Google Scholar]
- Schoenberger S.P., Toes R.E.M., van der Voort E.I.H., Offringa R., Melief C.J.M. T-cell help for cytotoxic T lymphocytes is mediated by CD40-CD40L interactions. Nature. 1998;393:480–483. doi: 10.1038/31002. [DOI] [PubMed] [Google Scholar]
- Bennett S.R.M., Carbone F.R., Karamalis F., Flavell R.A., Miller J.F.A.P., Heath W.R. Help for cytotoxic-T-cell responses is mediated by CD40 signalling. Nature. 1998;393:478–480. doi: 10.1038/30996. [DOI] [PubMed] [Google Scholar]
- Bennett S.R.M., Carbone F.R., Karamalis F., Flavell R.A., Miller J.F.A.P., Heath W.R. Induction of a CD8+ cytotoxic T lymphocyte response by cross-priming requires cognate CD4+ T cell help. J. Exp. Med. 1997;186:65–70. doi: 10.1084/jem.186.1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurts C., Carbone F.R., Barnden M., Blanas E., Allison J., Heath W.R., Miller J.F.A.P. CD4+ T cell help impairs CD8+ T cell deletion induced by cross-presentation of self-antigens and favors autoimmunity. J. Exp. Med. 1997;186:2057–2062. doi: 10.1084/jem.186.12.2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombo M.P. Immunotherapy. Icytokine gene transfer strategies. Cancer Metastasis Rev. 1997;16:421–432. doi: 10.1023/a:1005980418533. [DOI] [PubMed] [Google Scholar]
- Ratliff T.L. Role of the immune response in BCG for bladder cancer. Eur. Urol. 1992;21:17–21. doi: 10.1159/000474916. [DOI] [PubMed] [Google Scholar]