

Abstract
Interleukin (IL)-2 interacts with two types of functional receptors (IL-2Rαβγ and IL-2Rβγ) and acts on a broad range of target cells involved in inflammatory reactions and immune responses. For the first time, we show that a chemically synthesized fragment of the IL-2 sequence can fold into a molecule mimicking the quaternary structure of a hemopoietin. Indeed, peptide p1–30 (containing amino acids 1–30, covering the entire α helix A of IL-2) spontaneously folds into an α-helical homotetramer and stimulates the growth of T cell lines expressing human IL-2Rβ, whereas shorter versions of the peptide lack helical structure and are inactive. We also demonstrate that this neocytokine interacts with a previously undescribed dimeric form of IL-2Rβ. In agreement with its binding to IL-2Rβ, p1–30 activates Shc and p56lck but unlike IL-2, fails to activate Janus kinase (Jak)1, Jak3, and signal transducer and activator of transcription 5 (STAT5). Unexpectedly, we also show that p1–30 activates Tyk2, thus suggesting that IL-2Rβ may bind to different Jaks depending on its oligomerization. At the cellular level, p1–30 induces lymphokine-activated killer (LAK) cells and preferentially activates CD8low lymphocytes and natural killer cells, which constitutively express IL-2Rβ. A significant interferon γ production is also detected after p1–30 stimulation. A mutant form of p1–30 (Asp20→Lys), which is likely unable to induce vascular leak syndrome, remains capable of generating LAK cells, like the original p1–30 peptide. Altogether, our data suggest that p1–30 has therapeutic potential.
Keywords: interleukin 2 mimetic, synthetic hemopoietin, dimeric interleukin 2 receptor, β chain, signal transduction, natural killer cells
Introduction
Human IL-2 is a 133–amino acid (aa) polypeptide with a molecular mass of 15–18 kD depending on the degree of glycosylation 1 2. Its structure is made up of a compact core bundle of four anti-parallel α helices connected by three loops 3. IL-2 is a major cytokine regulating the immune system, with its primary biological activity consisting in promoting clonal expansion of antigen-activated T lymphocytes. Furthermore, IL-2 induces lymphokine-activated killer (LAK) and NK cell cytotoxicity against tumor cells or virus-infected cells 4.
The effects of IL-2 on its target cells are mediated through specific cell surface receptors made up of three chains (IL-2Rα, IL-2Rβ, and IL-2Rγ 5). IL-2Rα is a 55-kD protein that binds to IL-2 with a dissociation constant (K d) value of ∼10 nM 6 7. The second IL-2R component, IL-2Rβ, is a 75-kD protein with a large (286 aa) intracytoplasmic domain 8 9. The last component to be identified, IL-2Rγ, is a 64-kD protein 10 11. IL-2Rβ and IL-2Rγ belong to the hemopoietin receptor family 12, whereas IL-2Rα along with IL-15Rα belong to a distinct family of molecules 13. In the human system, two receptor complexes are functional: the association of human IL-2Rβ and IL-2Rγ forms an intermediate affinity receptor with a K d value of ∼1 nM, whereas expression of all three chains leads to the formation of a high affinity IL-2R (K d ∼ 10 pM). A structural model for the IL-2/IL-2R complex derived from the three-dimensional structure of the growth hormone–growth hormone receptor cocrystals has been proposed by Bamborough et al. 14 and is supported by experimental data 15 16 17 18.
Heterodimerization of IL-2Rβ and IL-2Rγ subunits triggers downstream signals that involve the tyrosine phosphorylation of a large array of cellular proteins. IL-2Rβ plays a critical role in this signal transduction cascade. After IL-2 stimulation, IL-2Rβ recruits protein tyrosine kinases (PTKs: p56lck, Syk, and Janus kinases [Jaks]) and an adapter protein (Shc), all of which play an essential role in lymphocyte activation. After interaction with phosphorylated Tyr338 of IL-2Rβ, Shc invokes the RAS and phosphatidylinositol 3-kinase signaling pathways that are essential in the control of cellular activation and proliferation. These pathways are dependent on two subregions of the IL-2Rβ chain, the serine-rich (S) and the acidic (A) domains. p56lck protein is associated with the A domain of IL-2Rβ and may be involved in the phosphorylation of Shc and consequently in the regulation of RAS 19. The Jak–signal transducer and activator of transcription (STAT) pathway is also implicated in the course of IL-2 activation and involves both the IL-2Rβ and IL-2Rγ subunits, which are constitutively associated with Jak1 and Jak3, respectively. As a consequence of Jak1/Jak3 phosphorylation, STAT5 is activated after binding to phosphorylated Tyr510 of IL-2Rβ. IL-2 induces the phosphorylation, dimerization, and nuclear translocation of STAT5 20.
In previous studies, we have demonstrated that the α helix A of IL-2 (aa 6–30) is essential for binding to the IL-2Rβ chain 21. Here, we report on the characterization of peptide p1–30 (comprising aa 1–30 of human IL-2), which encompasses the entire α helix A of IL-2 (see Table , top). The biological properties of this peptide, its preliminary structural features, and the signaling events it induces indicate that we have isolated a partial IL-2 mimetic, which acts specifically on IL-2Rβ dimers. At the immunological level, p1–30 is able to generate LAK cells and specifically activates NK and CD8 T cells, which express large amounts of IL-2Rβ and participate in cytotoxic responses. Moreover, p1–30 induces the production of IFN-γ, also known for its direct and indirect activity against tumor cells. For the first time, we demonstrate that a chemically synthesized cytokine fragment is able to fold into a neocytokine structure. This new molecule exhibits many of the properties of members of the hemopoietin family and has therapeutic potential.
Table 1.

| Peptides | Activity | Helix | State | K d |
|---|---|---|---|---|
| % | μM | |||
| p1–10 | <5 | 0 | M/D | 50 |
| APTSSSTKKT | ||||
| p5–15 | <5 | 2 | M/D | 113 |
| SSTKKTQLQLQ | ||||
| p10–20 | NT | 0 | M | – |
| TQLQLEHLLLD | ||||
| p1–22 | <5 | 5 | M/D | 65 |
| APTSSSTKKTQLQLEHLLLDLQ | ||||
| p10–30 | 50 | 35 | M/D | 0.2 |
| TQLQLEHLLLDLQMILNGINN | D/T | 88 | ||
| p1–30 | 100 | 50 | M/T | 5 |
| APTSSSTKKTQLQLEHLLLD LQMILNGINN | T/O | 60 | ||
| p1–30Cys | 100 | 50 | M/T | 5 |
| APTSSSTKKTQLQLEHLLLD LQMILNGINNC | T/O | 60 | ||
| p1–30Cys-FLC | NT | 55 | M/T | 2 |
| T/O | 55 |
Amino acid sequence (top) of IL-2 and p1–30. Location of α helices (A–D) is indicated by a box, and the critical region for binding to IL-2Rβ is shaded.
NT, not tested (p10–20 induced strong cellular aggregation).
Materials and Methods
Cell Lines and Proliferation Assays.
TS1 is a murine T cell line expressing mouse IL-2Rγ chain only and grows in IL-4 or IL-9. TS1β cells are TS1 cells transfected with human IL-2Rβ and are in addition able to grow in IL-2. The murine cell line 8.2 expresses mouse IL-2Rβ and mouse IL-2Rγ and grows in IL-4 only 22. Kit 225 is an IL-2–dependent human CD4 T cell line, originally derived from a human adult T cell lymphoma 23.
For proliferation assays, cells were stimulated in 96-well flat-bottomed microtiter plates at various concentrations of cytokines, IL-2 peptides, or both, and after 36 h of stimulation, [3H]TdR incorporation was measured. For testing p1–30 activity, cells were stimulated with either peptide alone or in combination with IL-2 or IL-4. Certain p1–30 effects were more readily detectable when peptide was used in combination with cytokines. The inhibitory effect of neutralizing anti–human IL-2Rβ mAb A41 and neutralizing anti–mouse IL-2Rγ mAb 3E12 plus 4G3 was tested on proliferation induced by cytokines or peptide p1–30 alone.
Peptides, Cytokines, and Soluble IL-2Rβ 31–230.
Peptides were synthesized by the step-wise solid-phase reaction using the boc/trifluoroacetic acid method 24, on a p-methylbenzhydrylamine resin with an Applied Biosystems 430A peptide synthesizer, as described previously 21. After purification, peptides were verified by mass spectrometry and amino acid analysis after total hydrolysis. The following IL-2 peptides were used in this study: p1–30, p1–22, p10–30, p1–10, p5–15, p10–20, p15–25, p20–30, p1–31, p1–31(Lys20), and p1–30Cys.
p1–30Cys was labeled on the terminal Cys31 with a fluorescein group (FLC) and purified by two successive rounds of exclusion chromatography on a G25 resin column (Amersham Pharmacia Biotech). Fractions with greater absorbance ratios at 242 and 490 nm were selected. The purified labeled peptide p1–30Cys-FLC (mol. wt. 3805, ε242nm 2,104 M−1cm−1, ε490nm 83,000 M−1cm−1, ν = 0.732 ml/mg) consisted of >92% of the total peptide amount.
The cytokines used in this work were human rIL-2 (Chiron), mouse rIL-4 (obtained from Dr. T. Honjo, University of Kyoto, Kyoto, Japan) or purified murine IL-9 (provided by Dr. J. Van Snick, Institut Ludwig pour la Recherche sur le Cancer, Brussels, Belgium).
The 31–230 aa residues of the IL-2Rβ chain (IL-2 Rβ 31–230; mol. wt. 23,352) were produced from a transfected Chinese hamster ovary (CHO) cell line and immunopurified according to established techniques 25. The molar extinction coefficient was calculated from the UV absorption spectrum at 280 nm (59,450 M−1cm−1) and amino acid analysis.
After stimulation with IL-2, p1–30, or both, IFN-γ production was measured on PBMC supernatants with a commercial enzyme immunoassay (Immunotech).
Circular Dichroism.
Molar extinction coefficients were calculated from the UV absorption spectrum and amino acid analysis (p1–30: 12,133 and 2,047 M−1cm−1 at 230 and 242 nm, respectively). Circular dichroism (CD) measurements were recorded on a spectropolarimeter (model CD6; Jobin Yvon) from 5–20 scans in far UV from 180 to 260 nm at 20°C in 20 mM sodium phosphate buffer (pH 7.2). The percentage of helix content was estimated from CD spectra according to an established procedure 26.
Molecular Weight Analysis.
Size exclusion chromatography analysis was performed with a fast protein liquid chromatography (FPLC) system. p1–30 solutions (100 μl at 150 μM in sodium phosphate 20 mM, pH 7.2) were injected on a Superdex S200 HR 10–30 column (24 ml; Amersham Pharmacia Biotech) equilibrated with the same buffer at room temperature at a constant flow rate (0.5 ml/min) and calibrated with molecular weight markers. Concentration profiles were recorded at 230 nm for peptides and 254 nm for proteins.
Diffusion–sedimentation equilibrium studies were performed at 20°C with peptide solutions (10 and 150 μM) or IL-2Rβ 31–200 (1 and 14 μM) in 20 mM sodium phosphate (pH 7.2), with a Beckman XL-A analytical ultracentrifuge at 25 and 42 krpm using standard double sector cells with 1.2-mm-thick aluminum centerpieces. Absorbances were recorded at 230, 242, and 360 nm for peptides and 280 nm for IL-2Rβ 31–200 as a function of radial distance. After at least 18 h, we verified that equilibrium was reached and recorded 10–99 successive scans. Base lines were recorded at 360 nm and 42 krpm. Protein concentration profiles at equilibrium were fitted to different models with the Origin-based Optima™ XL-A data analysis software from Beckman (single ideal component, two ideal components, and association of identical ideal components). Best fits were retained on the basis of both the χ2 value and the lack of systematic deviation of the residuals. In all cases, the base line was fixed at zero absorbance and not allowed to float during the fitting procedure. The value of the partial specific volume (ν), used for molecular weight calculations, was calulated from the amino acid composition of peptides (p1–10 [in ml/mg], n = 0.710; p5–15, 0.722; p10–20, 0.759; p1–22, 0.720; p10–30, 0.752; p1–30, 0.710; p1–30Cys, 0.710; and p1–30Cys-FLC, 0.732) and IL-2Rβ 31–200 (0.719 ml/mg). The solvent density was taken as 1.001 g/ml.
Binding of p1–30, p1–30Cys, and p1–30Cys-FLC on IL-2Rβ 31–200 was tested by analytical ultracentrifugation in 20 mM sodium phosphate, pH 7.2, at 20°C. For each experiment, one cell was filled with peptide, one with IL-2Rβ 31–200, and the last with the mixture of peptide and IL-2Rβ 31–200 in the three-cell rotor. Peptides and IL-2Rβ 31–200 concentrations were 150 and 14 μM, respectively. Absorbance was recorded at a wavelength of 242, 280, and 490 nm after reaching equilibrium at 22, 25, 28, and 42 krpm.
Immunoprecipitation and Western Blot Analysis.
Kit 225 cells were starved for 48 h in IL-2–free medium and then stimulated at 37°C with the following ligands: IL-2 (10 nM), p1–30 (60 μM), or IFN-α (1,000 U/ml). 5 × 106 cells were lysed in 125 μl of lysis buffer (50 mM Tris, pH 8, 10% glycerol, 200 mM NaCl, 0.5% NP-40, and 0.1 mM EDTA), supplemented with each of the protease inhibitors leupeptin, aprotinin, and PMSF at 10 μg/ml, and with the phosphatase inhibitors sodium fluoride (50 mM) and sodium orthovanadate (1 mM). For immunoprecipitation, lysates of 10 × 106 cells were immunoprecipitated with the indicated mAbs for 1 h at 4°C. After electrophoresis on an 8% SDS-polyacrylamide gel, the proteins were transferred to Immobilon membranes (Millipore Corp.). The immunoblots were incubated with antiphosphotyrosine mouse mAb 4G10 (Upstate Biotechnology), mAb to human Shc (Transduction Laboratories), or mAb to human Jak1, Jak2 (Upstate Biotechnology), Jak3 (Santa Cruz Biotechnology), or Tyk2. After incubation with an anti–mouse Ig peroxidase-conjugated mAb (Amersham Pharmacia Biotech) or an anti–rabbit Ig peroxidase-conjugated mAb (Biosys), reactive protein bands were visualized by enhanced chemiluminescence (ECL; Amersham Pharmacia Biotech).
For the in vitro kinase assay, p56lck was immunoprecipitated from Kit 225 lysates with an anti–human p56lck mAb. Immunoprecipitates thus obtained were suspended for 5 min at 25°C in kinase buffer, pH 7 (200 mM Mops, 50 mM Na-O-Ac, 10 mM EDTA, 10 mM MgCl2), containing 38 μg of enolase (Sigma-Aldrich), [γ-32P]ATP, and unlabeled ATP (10 μM). The samples were then electrophoresed on an 8% (SDS-PAGE) gel before drying and exposure to film.
Induction of LAK Cells.
PBMCs were stimulated for 3 or 6 d in complete medium (RPMI 1640 supplemented with 10% normal human serum) in the presence of p1–30, p1–31(Lys20), or IL-2. K562 or Daudi target cells (5 × 103 in 0.1 ml) labeled with Na2 51CrO4 (Amersham Pharmacia Biotech) were mixed into round-bottomed microwells with an equal volume of effector cells at various E/T ratios. After 4 h of incubation at 37°C, the plates were centrifuged at 2,000 g for 2 min, and cell-free supernatants were collected using a LumaPlate 96 cell harvester (Lumac-LSC). Supernatant radioactivity was assayed using an automated MicroBeta 1450 TriLux γ-counter (Wallac). Spontaneous release was determined by incubating target cells in medium alone. Maximum release was determined by adding 0.1 ml of 1 M HCl to the target cell suspension. The percentage of specific lysis was calculated as follows: 100 × (experimental 51Cr release − spontaneous 51Cr release)/(maximum 51Cr release − spontaneous 51Cr release).
FACS® Analysis.
Lymphocyte subsets were detected by direct labeling using the following mAbs labeled with R-PE: anti-CD4 (MT310; Dako), anti-CD8 (DK25; Dako), anti-CD20 (B-Ly1; Dako), and anti-CD56 (Moc-1; Dako). Anti-CD69 (Klon FN50; Dako) labeled with FITC was used as an early activation marker for the different lymphocyte subsets. Tricolor mouse anti–human CD14 (MHCD1406; Caltag Laboratories) was used to exclude monocytes from the analysis.
After 0, 24, 48, or 72 h of activation in the presence of the indicated concentration of IL-2 and/or p1–30, cells (2 × 105 in 200 μl) were simultaneously labeled with anti-subset– and anti-CD69–specific mAbs. After the staining procedure, cells were washed and fixed in 1% paraformaldehyde. A total of 2 × 104 cells per sample were analyzed with a FACScan™ flow cytometer using CELLQuest™ 1.2 software (Becton Dickinson).
Results
Biological Activity of IL-2 Peptide p1–30 and the Role of Human IL-2Rβ in p1–30 Activity.
The biological activity of peptide p1–30 was assayed on the proliferation of a mouse T cell line, which was engineered to express the human IL-2Rβ (TS1β). p1–30 induced a proliferative response in TS1β (maximal proliferation at 60 μM; Fig. 1 A). When tested in the presence of 1 or 10 nM of IL-2, a synergistic effect was observed for all tested concentrations of p1–30 compared with either p1–30 or IL-2 alone (Fig. 1 B). At 60 μM p1–30 and either 1 or 10 nM IL-2, the proliferative response was twofold higher than the sum of each individual response. Remarkably, although 10 nM of IL-2 alone induced optimal TS1β cell proliferation, addition of p1–30 led to a large increase in the proliferative response. Synergy was also observed with IL-4 (Fig. 1 E). The effect of shorter peptides in the helix A region was also evaluated on proliferation and compared with that of p1–30 (Table ).
Figure 1.

p1–30 induces cell proliferation and acts in synergy with IL-2. Role of human IL-2β. TS1β cells were stimulated with p1–30 (up to 100 μM) in the absence (A) or presence (B) of 1 or 10 nM IL-2. This was followed by measuring [3H]TdR incorporation. Proliferative responses induced with 1 or 10 nM of IL-2 were 38,148 and 72,370 cpm, respectively. Optimal responses were obtained with 60 μM of p1–30. Background was subtracted. (C) Effects of neutralizing anti–human IL-2Rβ mAb (A41) were tested on TS1β cell proliferation induced by p1–30 (60 μM), IL-2 (1 nM), or IL-9 (10 nM). (D) Effects of two neutralizing anti–mouse IL-2Rγ mAbs (3E12 and 4G3 mAbs) on the IL-2– and p1–30-induced proliferation of TS1β cells. Abs isolated from mouse ascitic fluids were diluted and added to the cell culture. (E) Synergy between p1–30 and IL-4. Proliferative responses of TS1 (expressing mouse IL-2Rγ), TS1β (expressing human IL-2Rβ and mouse IL-2Rγ), and 8.2 cells (expressing mouse IL-2Rβ and mouse IL-2Rγ) were induced at various concentrations of IL-4 (up to 100 U/ml) in the presence or absence of 60 μM of p1–30. Values obtained for p1–30 responses (Bkg) were subtracted from the IL-4 plus p1–30 response.
The critical role of human IL-2Rβ in the p1–30 effect was studied using neutralizing anti–human IL-2Rβ mAb A41 (Fig. 1 C). As expected, mAb A41 inhibited the IL-2–induced TS1β proliferation. This antibody also inhibited the p1–30-induced proliferation with a comparable efficiency, but had no effect on IL-9–induced proliferation (Fig. 1 C). In contrast, as shown in Fig. 1 D, IL-2Rγ plays a minor role if any in p1–30 activity, as measured on TS1β cells.
Further analysis of the role of IL-2Rβ on the synergistic effect induced by p1–30 was undertaken using various cell lines expressing different combinations of IL-2R chains. TS1 expresses murine IL-2Rγ, TS1β expresses human IL-2Rβ and murine IL-2Rγ, and 8.2 expresses murine IL-2Rβ and murine IL-2Rγ (Fig. 1 E). Synergy between IL-4 and p1–30 was observed only in TS1β cells, thus demonstrating that the introduction of a human IL-2Rβ is sufficient to confer responsiveness to p1–30. Results obtained with 8.2 cells are in agreement with the observation that human IL-2 does not bind murine IL-2Rβ 22.
Structure–Function Analysis of p1–30.
The secondary structure of various peptide segments in the region of aa residues 1–30 was studied by CD. Fig. 2 A shows the spectra obtained in the far UV range with a peptide concentration of 150 μM. Only two peptides showed >5% helical conformation: p10–30 (35 ± 5%) and p1–30 (50 ± 5%). The concentration dependence of the dichroic signal was evaluated between 3 and 150 μM for p1–30 (Fig. 2 A, insert) and p10–30 (data not shown). The helix content was dependent on the peptide concentration, and a maximum helix content was obtained at 30 μM of p1–30 and at 100 μM of p10–30.
Figure 2.

Structure–function analysis of p1–30. (A) The CD spectra from 180 to 260 nm are shown for p1–10, p5–15, p10–20, p1–22, p10–30, and p1–30 at 150 μM. The p1–30 concentration dependence (3–150 μM) of the CD signal at 192 nm is shown in the insert. (B) Size exclusion chromatography. Peptide p1–30 was eluted as a single main peak of molecular mass 13 kD (according to column calibration). Arrows indicate molecular weight (mw) markers, void volume (V0) and total volume (Vc). Monomeric, tetrameric, and octameric forms are shown. (C) TS1β cell proliferation was tested at various concentrations of IL-2 (from 5 × 10−3 to 10 nM) with 60 μM of either p1–30, p1–22, or p10–30. The response to IL-2 or peptide alone was subtracted from that obtained with both IL-2 and peptide. (D) Role of Asp20 in p1–31 activity. TS1β cells were stimulated with various concentrations of IL-2 (from 5 × 10−3 to 10 nM) in the presence of 60 μM of peptide p1–31 or p1–31(Lys20). Results are presented as in C. (E) The biological activity of mutant IL-2(Lys20) is shown for comparison.
The quaternary structure of p1–30 was first analyzed by size exclusion chromatography. Most of the material migrated as a single peak and eluted just before the 12.4-kD marker, indicating that the major form of the peptide (3 kD) could be a tetramer (Fig. 2 B). The chromatography pattern also showed two small “shoulders” on both sides of the main peak that could correspond to monomers (3 kD) and octamers (26 kD).
The activity of peptide fragments (p1–22 and p10–30) was studied on TS1β cells in the presence of IL-2 (Fig. 2 C). Synergy was observed with peptides p10–30 and p1–30 only, which are the ones folded in an α-helical structure (Fig. 2 A). Since p1–22 was not active, this strongly suggested a role for the helical structure in the induction of the biological effects observed.
Previous work reported that Asp20 is an essential residue for both IL-2 activity and IL-2/IL-2Rβ interaction 17 21. Peptide p1–31 (p1–30 with an additional COOH-terminal tyrosine) was mutated at position 20 (Asp→Lys) and was analyzed as above (peptides p1–31 and p1–30 had a similar activity at the proliferation level). The results showed that the Asp20 substitution had no impact on p1–31 activity (Fig. 2 D), whereas the biological activity of IL-2 (Asp20→Lys20) mutant was clearly reduced (Fig. 2 E).
The structure–function relationship was further analyzed by studying other peptides covering different portions of the p1–30 sequence. Table summarizes the most salient data concerning peptide activity, helicity, and quaternary structure. The quaternary structure of the peptides was evaluated by sedimentation–diffusion experiments. At 10 μM, p1–30 behaved essentially as a tetramer, whereas at 150 μM, p1–30 showed significant amounts of octamer in equilibrium with the tetramer (see also Fig. 3 A). At 10 and 150 μM, the molecular masses obtained for the lightest species ranged from 10,340 to 16,480 daltons, providing an estimated number of 4.0 ± 0.9 peptides per oligomer. These results are in close agreement with those obtained by chromatography and with preliminary crystallization trials (crystals of the peptide, which diffract at low resolution, were obtained reproducibly), which confirm that the p1–30 tetramer has a compact globular conformation. As described in Table , p1–10, p5–15, and p1–22 are dimers at 150 μM with K d values of 50, 113, and 60 μM, respectively. The p10–30 peptide is a tetramer at 150 μM by association of two dimers with a K d value of 88 μM. Altogether, the results shown in Table suggest that in p1–30, residues 10–30 may be responsible for the association of tetramers, and residues 1–10 may be responsible for the dimerization of tetramers to form octameric species.
Figure 3.

Sedimentation–diffusion equilibrium of p1–30 and soluble IL-2Rβ. Profiles of absorbance versus radius from the center of rotation were obtained by sedimentation–diffusion equilibrium at 25 krpm in 20 mM sodium phosphate, pH 7.2, at 20°C. Residual errors as a function of radial distance from the center of rotation show the fitting quality of the data to the selected models. (A) p1–30 initial concentration was 150 μM. Absorbance was recorded at 242 nm. A better fit was obtained with a tetramer–octamer model of equilibrium (bottom) than with a tetramer model (top). (B) IL-2Rβ 31–230 initial concentration was 14 μM. Absorbance was recorded at 295 nm. An equilibrium monomer–dimer model (bottom) seemed slightly better than a dimer model (top). (C) p1–30Cys-FLC and IL-2Rβ 31–230 initial concentrations were 150 and 14 μM, respectively. Absorbance of p1–30Cys-FLC (at 490 nm) in the presence or absence of IL-2Rβ 31–230 is shown (top). Profile at 295 nm (aromatic residue absorbance of IL-2Rβ 31–230) of the peptide–protein mixture is also shown (bottom).
Binding of p1–30Cys-FLC to IL-2Rβ 31–230.
To study directly the interaction of p1–30 with IL-2Rβ, a truncated IL-2Rβ representing most of the extracellular part (residues 31–230) of the molecule was first produced and studied by analytical ultracentrifugation. At 1 μM of protein, experiments of sedimentation–diffusion at equilibrium showed an association of two monomers with a K d value of 3 × 10−6 M. At 14 μM, the molecular mass obtained was very close to the dimer mass (Fig. 3 B). The presence of p1–30 (150 μM) along with IL-2Rβ 31–230 (1 μM) did not influence IL-2Rβ 31–230 dimerization in solution (data not shown).
To verify that p1–30 binds to IL-2Rβ 31–230 dimers, we labeled the peptide with an FLC group on a COOH-terminal cysteine (p1–30Cys-FLC) and analyzed its absorbance profile at 490 nm. p1–30Cys and p1–30Cys-FLC showed a helical conformation depending on their concentrations and formed octamers and tetramers with K d values close to those of genuine p1–30 (Table , and Fig. 3 A). The absorbance profiles of p1–30Cys-FLC (150 μM) at 490 nm and of IL-2Rβ 31–230 (14 μM) at 295 nm mixed in the same cell are displayed in Fig. 3 C (top and bottom, respectively). The data show that in the presence of IL-2Rβ 31–230, p1–30Cys-FLC had a greater molecular weight. The number of species at equilibrium in solution did not allow the accurate calculation of the K d value, but it was found to be significantly higher than the peptide concentration used in these experiments (150 μM). As expected, the molecular weight of IL-2Rβ 31–230 was not greatly influenced by the presence of p1–30Cys-FLC.
Activation of Shc and p56lck by Peptide p1–30.
Early events of activation by p1–30 were investigated on Kit 225 cells. Like TS1β cells, Kit 225 cells proliferated in response to p1–30 (maximal proliferation was obtained at 60 μM of p1–30; data not shown). We first analyzed the protein phosphorylation pattern induced by IL-2, p1–30, or a combination of both molecules by Western blot (Fig. 4 A). Unlike in unstimulated cells, the phosphorylation of four major proteins (p47, p52, p60, and p66) was clearly upregulated with p1–30 stimulation. The same proteins were also hyperphosphorylated after IL-2 stimulation.
Figure 4.

Signal transduction induced by p1–30. (A) General pattern of protein tyrosine phosphorylation. Kit 225 cells were stimulated for 10 min with IL-2 (10 nM), p1–30 (60 μM), or both. Cells were lysed, and the pattern of protein tyrosine phosphorylation was visualized by SDS-PAGE and immunoblotted with mAb 4G10, specific for phosphotyrosine (P-TYR). Arrowheads indicate the phosphorylated proteins upregulated by p1–30. (B) Phosphorylation of Shc. Kit 225 cells were stimulated for 1, 2, 5, 10, and 15 min. After lysis, phosphotyrosine-containing proteins were detected with 4G10 mAb (top). The identity and equal loading of the two Shc isoforms were confirmed by probing with the mAb to human Shc (bottom). (C) Induction of p56lck kinase activity. Kit 225 cells were stimulated for 15 min with 10 nM of IL-2, 60 μM of p1–30, or a combination of both. p56lck was immunoprecipitated and tested for its ability to phosphorylate the exogenous enolase substrate (top). The bottom panel shows that equal amounts of immunoprecipitated p56lck were used in the kinase assay.
These results led us to investigate the activation state of molecules known to participate in the IL-2–induced signal transduction cascade. Stimulation with IL-2 is known to involve the p21ras pathway via phosphorylation of the adapter protein, Shc. Therefore, Kit 225 cells were stimulated for 1, 2, 5, 10, and 15 min with p1–30 (Fig. 4 B). Blotting of the membrane with the antiphosphotyrosine mAb (top) showed an increase in the phosphorylation of two bands (p47 and p52) that reached a maximum after 10 min of stimulation. The two phosphorylated proteins corresponded to the two Shc isoforms (p47 and p52) and were loaded in equal quantity, as demonstrated by anti-Shc blotting (bottom). As control, IL-2 stimulation (5 min) also induced Shc phosphorylation.
Another important PTK implicated early during IL-2 signaling is p56lck. This protein is constitutively associated with IL-2Rβ and is presumed to phosphorylate Shc. We examined the effect of p1–30 on the tyrosine kinase activity of p56lck. After similar stimulation of Kit 225 cells and immunoprecipitation of p56lck, the immunoprecipitates were tested for their ability to phosphorylate the exogenous substrate, enolase (Fig. 4 C). Treatment with p1–30 resulted in an increased enolase phosphorylation, indicating that p1–30 activates p56lck kinase activity. As control, IL-2 also induced the kinase activity of p56lck. Fig. 4 C (bottom) also shows that an equal quantity of p56lck was immunoprecipitated and used in the in vitro kinase assay.
Implication of the Jak Proteins in p1–30 Signaling.
IL-2 is known to induce the tyrosine phosphorylation of Jak1 and Jak3. Therefore, we investigated whether Jak proteins were also phosphorylated in response to p1–30 (Fig. 5A–F). After stimulation, protein lysates of Kit 225 cells were immunoprecipitated with mAbs specific for different Jaks followed by antiphosphotyrosine (4G10) immunoblotting (top panels) or by anti-Jak immunoblotting (bottom panels). Interestingly, as shown in Fig. 5A and Fig. B, Jak1 and Jak3 were not phosphorylated after p1–30 stimulation, whereas IL-2, as expected, induced the phosphorylation of these two proteins. However, similar to IL-2, the peptide was unable to activate Jak2 (Fig. 5 C). Finally, we examined the activation status of Tyk2 (Fig. 5 D). Antiphosphotyrosine blotting showed activation of Tyk2 after p1–30 stimulation. Unexpectedly, IL-2 also induced Tyk2 phosphorylation in the Kit 225 cell line. Under the same experimental conditions, IFN-α–induced phosphorylation of Tyk2 was only twofold greater. Hence, the kinetics of Tyk2 activation was studied (Fig. 5E and Fig. F). Maximal Tyk2 phosphorylation was reached after 15 min of p1–30 stimulation and after 5 min of IL-2 stimulation.
Figure 5.
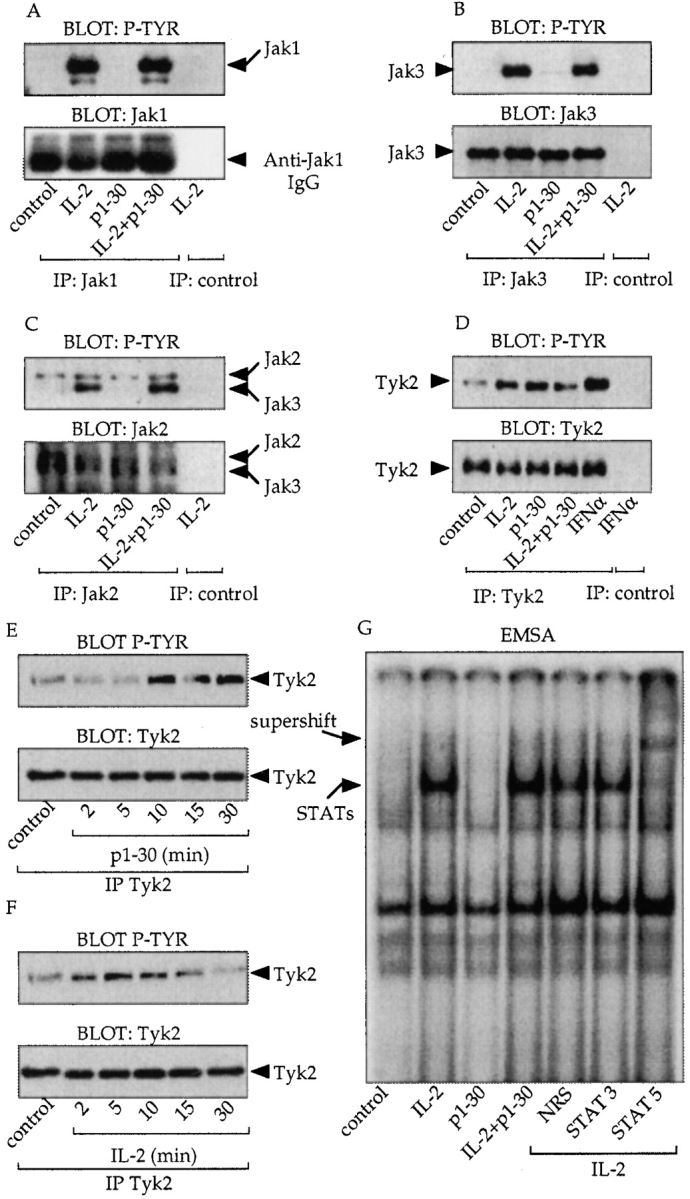
Implications of the Jak proteins in p1–30 signaling. (A–D) Analysis of Jak protein activation. Cells were stimulated for 10 min as indicated and immunoprecipitated with anti-Jak1 (A), Jak3 (B), Jak2 (C), and Tyk2 (D) mAbs. Antiphosphotyrosine blotting (P-TYR, top) and anti-Jak blotting (bottom) are shown. For C, the immunoblot displays two bands because the commercial anti-Jak2 serum cross-reacts with Jak3. However, the two kinases could be distinguished by their molecular weights. For D, cells were stimulated with IFN-α (0.3 nM) as a positive control for Tyk2 activation. (E and F) Analysis of Tyk2. Kit 225 cells were stimulated for 2, 5, 10, 15, and 30 min with p1–30 (60 μM) or IL-2 (10 nM). After lysis and anti-Tyk2 immunoprecipitation, phosphotyrosine-containing Tyk2 was detected (top). The total amount of immunoprecipitated Tyk2 corresponding to each stimulation was verified (bottom). (G) Activation of STAT proteins by IL-2 but not by p1–30. EMSA was carried out on nuclear extracts of Kit 225 stimulated for 10 min with IL-2 (10 nM), p1–30 (60 μM), or both. Nuclear extracts were incubated with the GAS probe. Supershifted complexes were analyzed with antibodies directed against STAT5, STAT3, or a nonrelevant serum (NRS) after IL-2 stimulation.
In the IL-2 system, STAT5 is activated in a manner dependent on Jak1 and Jak3. We studied the capacity of p1–30 to induce the STAT pathway. Kit 225 cells were stimulated, and nuclear extracts were subjected to electrophoretic mobility shift assay (EMSA) using an Fc-γ gene–derived IFN-γ activation sequence (GAS) probe. Strikingly, no bandshift was visualized after p1–30 stimulation, unlike IL-2, which did induce complex formation. mAb to STAT5 but not to STAT3 “supershifted” the complexes formed after IL-2 stimulation. The absence of STAT activation by p1–30 was confirmed using an additional probe (β-casein; data not shown).
p1–30 Activity on Human PBMCs.
Since IL-2Rβ is expressed on a proportion of low CD8-expressing (CD8low) lymphocytes and more extensively on NK cells 27, we investigated the effect of p1–30 on PBMCs. Human PBMCs were first tested for p1–30-induced proliferation after 3 or 6 d of stimulation. Incorporation of thymidine was dependent on the concentration of p1–30 and reached a maximum by day 3, which was maintained until day 6. In comparison, the IL-2 response was more efficient by day 3 but declined by day 6 (Fig. 6 A). To further evaluate the activity of p1–30, we tested its ability to generate LAK cells, a property attributed to IL-2 4. PBMCs were stimulated with IL-2 or p1–30 for 3 or 6 d, and the lysis of K562 and Daudi target cells was evaluated (Fig. 6 B). p1–30 was able to induce LAK cells in a concentration-dependent manner. However, the response was weaker than that of IL-2 on day 3, but values were comparable on day 6.
Figure 6.
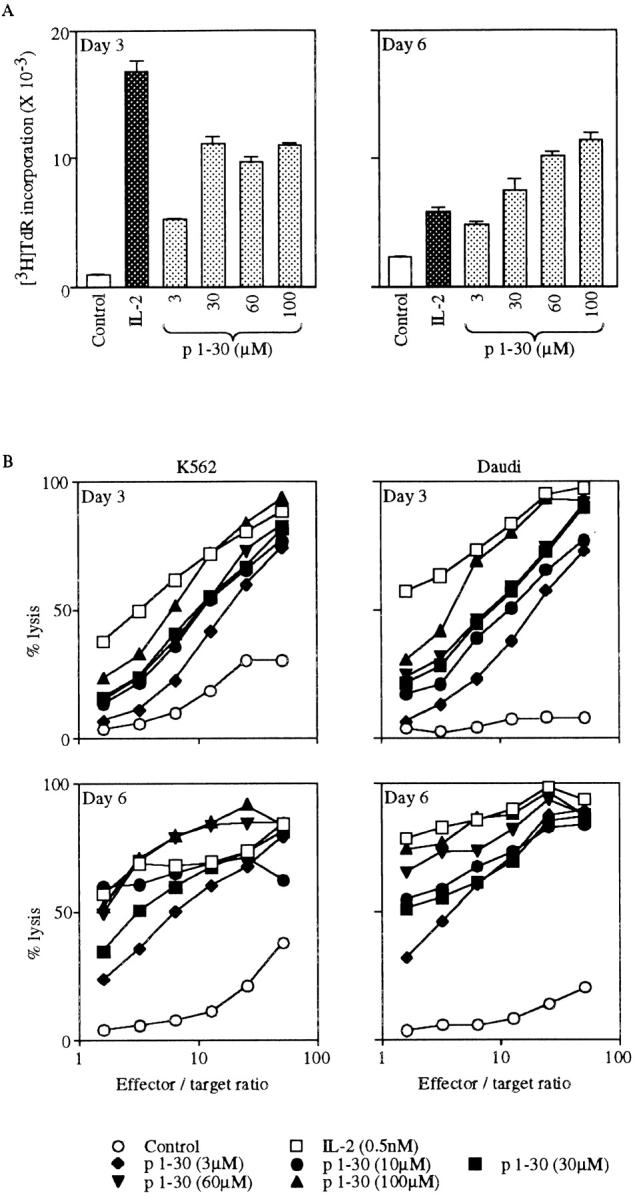

p1–30 activity on PBMCs. (A) Proliferation of PBMCs. PBMCs from healthy human donors were stimulated with IL-2 (0.5 nM) or p1–30 (3, 30, 60, or 100 μM) for 3 or 6 d followed by measurement of thymidine incorporation. The graph shows the negative control (white bars), IL-2–induced proliferation (black bars), and the dose-dependent p1–30-induced proliferation (gray bars). (B) Induction of LAK cells. PBMCs stimulated with IL-2 (0.5 nM) or p1–30 (3, 10, 30, 60, or 100 μM) for 3 or 6 d were tested for their ability to lyse K562 or Daudi target cells. Percent lysis at different effector/target ratios is shown. (C) LAK cell induction by p1–31(Lys20). The lysis of K562 or Daudi target cells by PBMCs stimulated with p1–30 or p1–31(Lys20) (30 μM) for 6 d was measured. Results obtained at different effector/target ratios are presented. Data of representative experiments are shown.
Induction of LAK cells was also studied with the peptide p1–31(Lys20). Fig. 6 C shows a comparison of p1–30 and p1–31(Lys20) activities at day 6 against K562 and Daudi cells. The two peptides displayed a similar efficiency at 30 μM when LAK cells were tested on K562 targets.
The influence of p1–30 on PBMC subset (B, T CD4, T CD8, and NK cells) activation was also studied. Expression of the early activation marker CD69 was followed after stimulation with p1–30 or IL-2 by flow cytometry (Fig. 7 A). As testified by the increased CD69 expression, p1–30 appeared to specifically activate the CD8low and the NK cell subsets as well as, to a lesser extent, the high CD8-expressing lymphocytes (CD8high). As positive control, IL-2 also induced CD69 expression on these populations. Marginal effects were observed on the other subpopulations tested. p1–30 was also shown to induce IFN-γ production in PBMCs. Interestingly, under some experimental conditions (2 d of culture), p1–30 synergizes with IL-2 in this phenomenon (Fig. 7 B).
Figure 7.

Activation of NK and CD8 T cells by p1–30 and induction of IFN-γ. (A) PBMCs were stimulated with IL-2 (0.5 nM) or p1–30 (10, 30, or 100 μM) for 1, 2, or 3 d. Subpopulations labeled with anti-CD4 (Th lymphocytes), CD8 (CTLs), CD20 (B lymphocytes), or CD56 (NK cells) were assayed for CD69 expression. Graphs represent the percentage of CD69+ cells for each population with respect to stimulant (IL-2 or p1–30) and time (day) of stimulation. Data of a representative experiment are shown. (B) IFN-γ production was measured on supernatants from PBMCs stimulated with IL-2 (0.5 nM) or p1–30 (30 μM) at day 6. In a second panel, synergy between IL-2 (0.5 nM) and p1–30 (30 μM) is shown after 2 d of stimulation.
Discussion
Reconstitution of a neocytokine from the first 30 aa of IL-2 is very novel in the field of peptide agonists. Recently characterized peptides, with agonist activities related to cytokines of the hemopoietin family, have consisted up to this point of small mimetics of erythropoietin (EPO 28) or thrombopoietin 29 selected by random phage display peptide libraries and a small nonpeptidyl mimic of G-CSF 30. These compounds act as full agonists in various biological assays, and their signaling pathways appear to be identical to those induced by the natural ligand, yet their amino acid sequences are not found in the primary sequence of the natural protein. The crystal structure of the 20-aa mimetic peptide of EPO and of the extracellular domain of the EPO receptor (EPOR) reveals a dimerization of the peptide that is bound to the homodimeric EPOR. In this study, it was shown that the peptide p1–30 contains a part of the native aa sequence of IL-2, forms a tetrameric structure that mimics a cytokine with a configuration of four α helices, and acts on cells expressing human IL-2Rβ. The peptide p1–30 also acts in synergy with IL-2 and IL-4. We also demonstrate that p1–30 induces the phosphorylation of Shc, an essential adapter in the initiation of the Ras/mitogen-activated protein kinase (MAPK) pathway, and the activation of the PTK p56lck. Unlike IL-2, however, Jak1, Jak3, and the STAT pathway were not activated after p1–30 treatment. Surprisingly, Tyk2 is phosphorylated upon both p1–30 and IL-2 stimulation of Kit 225 cells. Finally, p1–30 is able to specifically activate CD8 T lymphocytes and NK cells, and induces LAK cell–mediated cytotoxicity.
Studies of secondary and quaternary structures of p1–30 were undertaken. CD studies performed with p1–30 indicated that a high proportion of the residues was in an α-helical configuration for concentrations ranging from 3 to 150 μM. The link between the secondary structure and the biological effect was revealed by testing peptide fragments p1–22 and p10–30. Peptide p10–30 was biologically active and folded into an α-helical configuration, whereas peptide p1–22, which did not form α helices, failed to exert any biological effect. The helicity of p1–30 depends on the concentration, suggesting that oligomerization of the peptide stabilizes its folding. Quaternary structure studies of p1–30 indicated that, in a concentration range where its biological activity was observed, most of the molecules were in a compact tetrameric form. The amino acid sequence of p1–30 shows 7 leucine residues (positions 12, 14, 17, 18, 19, 21, and 25) and 2 isoleucine residues (positions 24 and 28) among the 18 COOH-terminal residues. The periodicity of these amino acids suggests that most of these side chains form a hydrophobic face on the helix containing residues 10–30. The molecular weight of p1–30 suggests that four of these faces could constitute a hydrophobic core, which stabilizes the homotetramer. Consequently, four basic p1–30 peptides could form a molecule resembling a cytokine of the hemopoietin family, composed of a compact core bundle of four α helices.
The peptide p1–30 was initially designed for binding to human IL-2Rβ 21. Studies of its biological activities revealed that p1–30 bound human IL-2Rβ specifically and that this binding was necessary to induce proliferation. The role of the IL-2Rα and γ subunits in direct binding to p1–30 was eliminated, since the effects of p1–30 existed in the absence of IL-2Rα expression (TS1β cells) and was not significantly affected by neutralizing mAbs to IL-2Rγ. Therefore, it appears that, unlike IL-2, p1–30 interacts with IL-2Rβ only. Since the biological effects of most cytokines are mediated after hetero- or homodimerization of their receptor chains, we hypothesized that the mode of action of tetrameric p1–30 was through an induction of IL-2Rβ dimerization or binding to preformed dimers. These interactions are possible, since tetrameric p1–30 has four potential binding sites for IL-2Rβ. Nevertheless, we cannot exclude that other membrane molecules can also participate in the p1–30 receptor.
To further analyze this hypothesis, we produced a soluble form of IL-2Rβ and tested its ability to dimerize and bind p1–30. Analytical ultracentrifugation showed that IL-2Rβ (31–230) alone was dimeric in solution at concentrations <1 μM. Using the FLC-labeled peptide p1–30Cys-FLC, we studied its association with IL-2Rβ (31–230) at 490 nm. The ultracentrifugation profile clearly showed the association of p1–30Cys-FLC to dimeric IL-2Rβ (31–230). In view of these results, we propose that IL-2Rβ dimers (IL-2Rβ)2 are formed before the binding of the p1–30 tetramer (p1–30)4, although induction of dimerization by p1–30 cannot be excluded. The equilibrium constant for IL-2Rβ dimerization and (p1–30)4/(IL-2Rβ)2 binding could be significantly lower at the membrane level due to the bidimensional diffusion of preoriented receptors. Under these conditions, p1–30 could induce signals by changing the conformation of (IL-2Rβ)2 complexes. Several reports concerning chimeric receptors constructed from the intracellular region of IL-2Rβ and the extracellular region of other homodimeric receptors indicate that IL-2Rβ dimers can transduce signals. A chimeric receptor EPOR/IL-2Rβ induces cell proliferation 31 and intracytoplasmic protein phosphorylation 32. Similarly, the chimeric receptor G-CSFR/IL-2Rβ induces the expression of reporter gene constructs 33, and a chimeric receptor c-kit/IL-2Rβ induces cell proliferation 34.
Characterization of the specific signals induced by p1–30 revealed that the pattern of phosphorylation induced by p1–30 was similar to that induced by IL-2. However, more detailed investigations revealed clear differences. As in stimulation with IL-2, Shc and p56lck were activated, but on the contrary and consistent with the hypothesis that p1–30 does not recruit IL-2Rγ, Jak1 and Jak3 were not phosphorylated and the STAT pathway was not involved. In the IL-2/IL-2R model, Shc and p56lck are dependent on the A and S domains of IL-2Rβ. The STAT pathway, however, is dependent on the respective association of Jak1 and Jak3 with box1 domains of IL-2Rβ and IL-2Rγ. Therefore, p1–30 signals appear to be dependent only on IL-2Rβ, and this supports the proposed (p1–30)4/(IL-2Rβ)2 model described above. Surprisingly, it was shown that p1–30, as well as IL-2, induced Tyk2 phosphorylation. Signals were weaker than the classical IFN-α–induced phosphorylation but were confirmed by studying IL-2– and p1–30–stimulated PBMCs (data not shown). Previous work has reported on the absence of IL-2–induced Tyk2 phosphorylation, but the data were obtained under different experimental conditions 35.
Even if the implications of Tyk2 are of secondary importance in IL-2 signaling, this PTK could play an essential role in p1–30 signal transduction, because no other Jaks were found to be activated.
The relationship between p1–30 and IL-2, both of which bind to the IL-2Rβ protein and are able to act in synergy, deserves further discussion. In the presence of IL-2, it has been shown that the three receptor subunits interact to form the high affinity receptor 36. The induction of the trimeric receptor (α, β, γ) may lead to the dissociation of IL-2Rβ dimers that could exist at the cell surface. When p1–30 and IL-2 were present simultaneously, a synergistic effect was observed. This may indicate that at the cell surface there is an excess of IL-2Rβ and a limited number of IL-2Rγ molecules, allowing a distribution of IL-2Rβ either in the p1–30R (IL-2Rβ)2 or in the IL-2R. On TS1β cells stimulated with p1–30 and IL-2, one may suggest that some of the IL-2Rβ molecules are included in the intermediate affinity IL-2/IL-2Rβγ complex (K d = 10−9 M), whereas free (IL-2Rβ)2 complexes, which bind IL-2 very weakly (K d = 10−7 M), remain accessible for p1–30 binding. Simultaneous utilization of IL-2Rβγ and p1–30R may produce a coupled signal and explain the synergistic effects. The signaling events leading to the synergy are not yet understood.
The bioactivity of p1–30, particularly its ability to induce LAK cells and IFN-γ production, strongly suggests that it may have therapeutic potential against tumor cells. As IL-2, p1–30 induces a potent LAK cell response, as measured on Daudi and K562 targets. Among immune cells, NK cells and some CD8 T cells express large amounts of IL-2Rβ 27. Therefore, they were thought to constitute the major targets of p1–30. Concordant with this hypothesis, analysis of the p1–30 effects on human PBMCs clearly demonstrated the peptide's capacity to upregulate the expression of CD69, especially on NK cells and CD8low lymphocytes. To further confirm our results, we have verified that p1–30 acts on purified NK cells either alone or in synergy with IL-2 (data not shown). Moreover, we demonstrated the progression of NK cells towards S and G2/M phases of the cell cycle after p1–30 stimulation alone and in synergy with IL-2 (data not shown). We have shown that p1–30 induces IFN-γ production, which also exhibits a strong activity against tumor cells 37. This cytokine is able to upregulate surface expression of MHC antigens (both class I on tumor cells and class II on antigen-presenting cells), and this results in the potentiation of antitumor immunity. In addition, IFN-γ is capable of stimulating cytokine receptors, proteasomes, and tumor-associated antigen expression. More interestingly, IFN-γ has a direct inhibitory effect on tumor cell proliferation and may play a role in the triggering of programmed cell death 38.
IL-2 is also known to induce various side effects in vivo. An important aspect of toxicity mediated by IL-2 is vascular leak syndrome (VLS). It involves damage of vascular endothelial cells leading to vascular leak, edema, and organ failure. Recent work 39 provides evidence that a structural motif in IL-2 and other VLS-inducing proteins may be responsible for binding to endothelial cells and initiating VLS. This motif is located in the α helix A of IL-2, centered on Asp20. Short peptides containing aa 15–23 of IL-2 and exhibiting this motif induce VLS, whereas mutated peptides do not. We generated a mutated peptide, p1–31(Asp20→Lys), abrogating this motif and showed that its inductive capacity on proliferation was maintained, contrary to IL-2 mutated at the same position. This suggested that the interaction of p1–30 with IL-2Rβ is somewhat different from the IL-2/IL-2Rβ interactions. p1–31(Asp20→Lys) also maintained its capacity to generate LAK cells (Fig. 6 C) and to induce IFN-γ production (data not shown). We demonstrate here the possibility of generating IL-2 mimetics, which maintain stimulatory activity on lymphocytes but lack the potential to damage vascular endothelial cells.
In this work, we have shown that a single α helix of IL-2 keeps its properties of binding to one of the IL-2R chains, (IL-2Rβ)2. The α helix (p1–30) is a neocytokine directed towards cells expressing IL-2Rβ, including those (NK and CD8low lymphocytes) involved in cytotoxic responses. The p1–30/IL-2Rβ and IL-2/IL-2Rβ interactions are different, and this may allow for the generation of different sets of signals having different biological consequences. In general terms, we have shown that a cytokine fragment can act as a selective agonist of a critical cytokine receptor chain. The present observations may be extended to other cytokine–cytokine receptor systems, and this may have fundamental and practical implications.
Acknowledgments
Dr. T. Malek (University of Miami, Miami, FL), who provided anti–murine IL-2Rγ mAbs, and Dr. T. Hori (University of Kyoto, Kyoto, Japan), who gave us permission to use the Kit 225 cell line, are kindly acknowledged. IFN-α and anti-Tyk2 were kindly provided by Dr. S. Pellegrini (Institut Pasteur, Paris). We are indebted to Dr. Marko Kryworuchko for critical reading of the manuscript. We also thank F. Gay for valuable technical advice.
This work was supported by Association de Recherche sur le Cancer (ARC), Caisse Nationale Assurance Maladie (CANAM), and by a grant from Comité Consultatif de Valorisation de l'Institut Pasteur (CCV-IP).
Footnotes
R. Eckenberg and T. Rose contributed equally to this work.
Abbreviations used in this paper: A domain, acidic domain; aa, amino acid; CD, circular dichroism; EMSA, electrophoretic mobility shift assay; EPO, erythropoietin; FLC, fluorescein; GAS, IFN-γ–activated sequence; Jak, Janus kinase; K d, dissociation constant; LAK, lymphokine-activated killer cell; PTK, protein tyrosine kinase; S domain, serine-rich domain; STAT, signal transducer and activator of transcription; VLS, vascular leak syndrome.
References
- Robb R.J., Kutny R.M., Panico M., Morris H.R., Chowdhry V. Amino acid sequence and post-translational modification of human interleukin-2. Proc. Natl. Acad. Sci. USA. 1984;81:6486–6490 . doi: 10.1073/pnas.81.20.6486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi T., Matsui H., Fujita T., Takaoka C., Kashima N., Yoshimoto R., Hamuro J. Structure and expression of a cloned cDNA for human interleukin-2. Nature. 1983;302:305–310. doi: 10.1038/302305a0. [DOI] [PubMed] [Google Scholar]
- MacKay D. Unraveling the structure of IL-2reply. Science. 1992;257:410–413. doi: 10.1126/science.1631562. [DOI] [PubMed] [Google Scholar]
- Smith K.A. Interleukin-2inception, impact and implications. Science. 1988;240:1169–1176. doi: 10.1126/science.3131876. [DOI] [PubMed] [Google Scholar]
- Thèze J., Alzari P.M., Bertoglio J. Interleukin 2 and its receptorsrecent advances and new immunological functions. Immunol. Today. 1996;10:481–486. doi: 10.1016/0167-5699(96)10057-c. [DOI] [PubMed] [Google Scholar]
- Leonard W.J., Depper J.M., Crabtree G.R., Rudikoff S., Punphery J., Robb R.J., Kronke M., Svetlik P.B., Peffer N.J., Waldmann T.A., Greene W.C. Molecular cloning and expression of cDNAs for the human interleukin-2 receptor. Nature. 1984;311:626–631. doi: 10.1038/311626a0. [DOI] [PubMed] [Google Scholar]
- Uchiyama T., Broder S., Waldmann T. A monoclonal antibody (anti-Tac) reactive with activated and functionally mature human T cells. J. Immunol. 1981;126:1293–1297. [PubMed] [Google Scholar]
- Hatakeyama M., Tsudo M., Minamoto S., Kono T., Doi T., Miyata T., Miyasaka M., Taniguchi T. Interleukin-2 receptor β chain genegeneration of three receptor forms by cloned human α and β chain cDNA's. Science. 1989;244:551–556. doi: 10.1126/science.2785715. [DOI] [PubMed] [Google Scholar]
- Teshigawara K., Wang H.M., Kato K., Smith K.A. Interleukin-2 high-affinity receptor expression requires two distinct binding proteins. J. Exp. Med. 1987;165:223–238. doi: 10.1084/jem.165.1.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeshita T., Asao H., Ohtani K., Ishii N., Kumaki S., Tanaka N., Munakata H., Nakamura M., Sugamura K. Cloning of the γ chain of the human IL-2 receptor. Science. 1992;257:379–382. doi: 10.1126/science.1631559. [DOI] [PubMed] [Google Scholar]
- Noguchi M., Nakamura Y., Russell S.M., Ziegler S.F., Tsang M., Cao X., Leonard W.J. Interleukin-2 receptor gamma chaina functional component of the interleukin-7 receptor. Science. 1993;262:1877–1880. doi: 10.1126/science.8266077. [DOI] [PubMed] [Google Scholar]
- Jacques Y., Minty A., Fradelizi D., Thèze J. Interleukins 2, 4, 7, 9, 13 and 15. In: Jacques Thèze, editor. The Cytokine Network and Immune Functions. Oxford University Press; Oxford: 1999. pp. 17–30. [Google Scholar]
- Giri J.G., Kumaki S., Ahdieh M., Friend D.J., Loomis A., Shanebeck K., DuBose R., Cosman D., Park L.S., Anderson D.M. Identification and cloning of a novel IL-15 binding protein that is strucurally related to the α chain of the IL-2 receptor. EMBO (Eur. Mol. Biol. Organ.) J. 1995;14:3654–3663. doi: 10.1002/j.1460-2075.1995.tb00035.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamborough P., Hedgecock C.J., Richards W.G. The IL-2 and IL-4 receptors studied by molecular modelling. Structure. 1994;2:839–851. doi: 10.1016/s0969-2126(94)00085-9. [DOI] [PubMed] [Google Scholar]
- Sauvé K., Nachman M., Spence C., Bailon P., Campbell E., Tsien W.-H., Kondas J.A., Hakimi J., Ju G. Localization in human interleukin 2 of the binding site to the α chain (p55) of the interleukin 2 receptor. Proc. Natl. Acad. Sci. USA. 1991;88:4636–4640. doi: 10.1073/pnas.88.11.4636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z., Zheng Z., Sun L., Liu X. Substitutions at the Glu62 residue of human interleukin-2 differentially affect its binding to the α chain and the βγ complex of the interleukin-2 receptor. Eur. J. Immunol. 1995;25:1212–1216. doi: 10.1002/eji.1830250512. [DOI] [PubMed] [Google Scholar]
- Collins L., Tsien W.-H., Seals C., Hakimi J., Weber D., Bailon P., Hoskings J., Greene W.C., Toome V., Ju G. Identification of specific residues of human interleukin-2 that affect binding to the 70-kDa subunit (p70) of the interleukin-2 receptor. Proc. Natl. Acad. Sci. USA. 1988;85:7709–7713. doi: 10.1073/pnas.85.20.7709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voss S.D., Leary T.P., Sondel P.M., Robb R.J. Identification of a direct interaction between interleukin 2 and the p64 interleukin 2 receptor γ chain. Proc. Natl. Acad. Sci. USA. 1993;90:2428–2432. doi: 10.1073/pnas.90.6.2428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gesbert F., Delespine-Carmagnat M., Bertoglio J. Recent advances in the understanding of interleukin-2 signal transduction. J. Clin. Immunol. 1998;18:307–320. doi: 10.1023/a:1023223614407. [DOI] [PubMed] [Google Scholar]
- Taniguchi T. Cytokine signaling through nonreceptor protein tyrosine kinases. Science. 1995;268:251–255. doi: 10.1126/science.7716517. [DOI] [PubMed] [Google Scholar]
- Eckenberg R., Xu D., Moreau J.L., Bossus M., Mazie J.C., Tartar A., Liu X.Y., Alzari P.M., Bertoglio J., Thèze J. Analysis of human IL-2/IL-2 receptor β chain interactionsmonoclonal antibody H2-8 and new IL-2 mutants define the critical role of alpha helix-A of IL-2. Cytokine. 1997;9:488–498. doi: 10.1006/cyto.1996.0192. [DOI] [PubMed] [Google Scholar]
- Chastagner P., Moreau J.-L., Jacques Y., Tanaka T., Miyasaka M., Kondo M., Sugamura K., Thèze J. Lack of intermediate affinity IL-2 receptor in mice leads to a dependence on IL-2 receptor α, β and γ chain expression for T cell growth. Eur. J. Immunol. 1996;26:201–206. doi: 10.1002/eji.1830260131. [DOI] [PubMed] [Google Scholar]
- Hori T., Uchiyama T., Tsudo M., Umadome H., Ohno H., Fukuhara S., Kita K., Uchino H. Establishment of an interleukin 2-dependent human T cell line from a patient with T cell chronic lymphocytic leukemia who is not infected with human T cell leukemia/lymphoma virus. Blood. 1987;70:1069–1072. [PubMed] [Google Scholar]
- Merrifield R.B. Solid phase synthesis. The synthesis of a tetrapeptide. J. Am. Chem. Soc. 1963;85:2145–2149. [Google Scholar]
- Francois C., Sorel M., Cherel M., Brailly H., Minvielle S., Blanchard D., Jacques Y. Immunological characterization of antigenic domains on human IL-2 receptor β subunitepitope-function relationships. Int. Immunol. 1995;7:1173–1181. doi: 10.1093/intimm/7.8.1173. [DOI] [PubMed] [Google Scholar]
- Manavalan P., Johnson W.C., Jr. Variable selection method improves the prediction of protein secondary structure from circular dichroism spectra. Anal. Biochem. 1987;167:76–85. doi: 10.1016/0003-2697(87)90135-7. [DOI] [PubMed] [Google Scholar]
- David D., Bani L., Moreau J.-L., Demaison C., Sun K., Salvucci O., Nakarai T., De Montalembert M., Chouaïb S., Joussemet M. Further analysis of interleukin-2 receptor subunit expression on the different human peripheral blood mononuclear cell subsets. Blood. 1998;91:165–172. [PubMed] [Google Scholar]
- Wrighton N.C., Farrell F.X., Chang R., Kashyap A.K., Barbone F.P., Mulcahy L.S., Johnson D.L., Barrett R.W., Jolliffe L.K., Dower W.J. Small peptides as potent mimetics of the protein hormone erythropoietin. Science. 1996;273:458–464. doi: 10.1126/science.273.5274.458. [DOI] [PubMed] [Google Scholar]
- Cwirla S.E., Balasubramanian P., Duffin D.J., Wagstrom C.R., Gates C.M., Singer S.C., Davis A.M., Tansik R.L., Mattheakis L.C., Boytos C.M. Peptide agonist of the thrombopoietin receptor as potent as the natural cytokine. Science. 1997;276:1696–1699. doi: 10.1126/science.276.5319.1696. [DOI] [PubMed] [Google Scholar]
- Tian S.S., Lamb P., King A.G., Miller S.G., Kessler L., Luengo J.I., Averill L., Johnson R.K., Gleason J.G., Pelus L.M. A small, nonpeptidyl mimic of granulocyte-colony-stimulating factor. Science. 1998;281:257–259. doi: 10.1126/science.281.5374.257. [DOI] [PubMed] [Google Scholar]
- Chiba T., Nagata Y., Machide M., Kishi A., Amanuma H., Sugiyama M., Todokoro K. Tyrosine kinase activation through the extracellular domains of cytokine receptors. Nature. 1993;362:646–648. doi: 10.1038/362646a0. [DOI] [PubMed] [Google Scholar]
- Jiang N., He T.C., Miyajima A., Wojchowski D.M. The box1 domain of the erythropoietin receptor specifies Janus kinase 2 activation and functions mitogenically within an interleukin 2 beta-receptor chimera. J. Biol. Chem. 1996;271:16472–16476. doi: 10.1074/jbc.271.28.16472. [DOI] [PubMed] [Google Scholar]
- Morella K.K., Lai C.F., Kumaki S., Kumaki N., Wang Y., Bluman E.M., Witthuhn B.A., Ihle J.N., Giri J., Gearing D.P. The action of interleukin-2 receptor subunits defines a new type of signaling mechanism for hematopoietin receptors in hepatic cells and fibroblasts. J. Biol. Chem. 1995;270:8298–8310. doi: 10.1074/jbc.270.14.8298. [DOI] [PubMed] [Google Scholar]
- Nelson B.H., Lord J.D., Greenberg P.D. Cytoplasmic domains of the interleukin-2 receptor beta and gamma chains mediate the signal for T-cell proliferation. Nature. 1994;369:333–336. doi: 10.1038/369333a0. [DOI] [PubMed] [Google Scholar]
- Russel S.M., Johnston J.A., Noguchi M., Kawamura M., Bacon C.M., Friedmann M., Berg M., McVicar D.W., Witthuhn B.A., Silvennoinen O. Interaction of IL-2Rβ and γc chains with Jak1 and Jak3implications for XSCID and XCID. Science. 1994;266:1042–1045. doi: 10.1126/science.7973658. [DOI] [PubMed] [Google Scholar]
- Damjanovich S., Bene L., Matko J., Alileche A., Goldman C.K., Sharrow S., Waldmann T.A. Preassembly of interleukin 2 (IL-2) receptor subunits on resting Kit 225 K6 T cells and their modulation by IL-2, IL-7, and IL-15a fluorescence resonance energy transfer study. Proc. Natl. Acad. Sci. USA. 1997;94:13134–13139. doi: 10.1073/pnas.94.24.13134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan D.H., Shankaran V., Dighe A.S., Stockert E., Aguet M., Old L.J., Schreiber R.D. Demonstration of an interferon gamma-dependent tumor surveillance system in immunocompetent mice. Proc. Natl. Acad. Sci. USA. 1998;95:7556–7561. doi: 10.1073/pnas.95.13.7556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan D.H., Schreiber R.D. The interferonsbiochemistry and biology. In: Jacques Thèze, editor. The Cytokine Network and Immune Functions. Oxford University Press; Oxford: 1999. pp. 111–124. [Google Scholar]
- Baluna R., Rizo J., Gordon B.E., Ghetie V., Vitetta E.S. Evidence for a structural motif in toxins and interleukin-2 that may be responsible for binding to endothelial cells and initiating vascular leak syndrome. Proc. Natl. Acad. Sci. USA. 1999;96:3957–3962. doi: 10.1073/pnas.96.7.3957. [DOI] [PMC free article] [PubMed] [Google Scholar]