

Abstract
IκB kinase (IKK) α and β phosphorylate IκB proteins and activate the transcription factor, nuclear factor (NF)-κB. Although both are highly homologous kinases, gene targeting experiments revealed their differential roles in vivo. IKKα is involved in skin and limb morphogenesis, whereas IKKβ is essential for cytokine signaling. To elucidate in vivo roles of IKKα in hematopoietic cells, we have generated bone marrow chimeras by transferring control and IKKα-deficient fetal liver cells. The mature B cell population was decreased in IKKα−/− chimeras. IKKα−/− chimeras also exhibited a decrease of serum immunoglobulin basal level and impaired antigen-specific immune responses. Histologically, they also manifested marked disruption of germinal center formation and splenic microarchitectures that depend on mature B cells. IKKα−/− B cells not only showed impairment of survival and mitogenic responses in vitro, accompanied by decreased, although inducible, NF-κB activity, but also increased turnover rate in vivo. In addition, transgene expression of bcl-2 could only partially rescue impaired B cell development in IKKα−/− chimeras. Taken together, these results demonstrate that IKKα is critically involved in the prevention of cell death and functional development of mature B cells.
Keywords: gene targeting, B cells, IκB kinase α, germinal center, nuclear factor κB
Introduction
The nuclear factor (NF)1-κB family of transcription factors plays critical roles in the activation of inflammatory immune responses 1 2. In resting cells, NF-κBs associate with IκBs and are retained in the cytoplasm as inactive forms. Proinflammatory stimuli, such as LPS, IL-1, or TNF-α, cause phosphorylation and degradation of IκBs, which lead to NF-κB activation. The protein kinases that phosphorylate IκB have been identified by three independent groups 3 4 5 6 7. Two IκB kinases (IKKs), IKKα and IKKβ, are main components of the IκB kinase complex and homologous with each other in the amino acid structures. Both contain a kinase domain, a leucine zipper, and a helix-loop-helix. However, gene targeting experiments revealed that they function differentially depending on the tissue. IKKα is essential for limb patterning and for epidermal keratinocyte proliferation and differentiation 8 9 10. Meanwhile, IKKβ-deficient mice showed a massive apoptosis of hepatocytes 11 12 13. Furthermore, IKKβ, but not IKKα, was found to be essential for NF-κB activation by proinflammatory cytokines.
B lineage cells also contain NF-κB activity, which can be augmented by LPS or CD40 ligation. All known mammalian members of the NF-κB/Rel transcription factor family, including p50(NF-κB1), p52(NF-κB2), p65(RelA), RelB, and c-Rel, are expressed in B cells 14 15 16 17 18. Gene targeting of these components caused various levels of impairment of B cell generation and function 19 20 21 22 23 24 25 26 27 28. However, because IKKα- and IKKβ-deficient mice died in an early neonatal period, it is unclear whether IKKα or IKKβ is involved in NF-κB activation in B lineage cells.
To investigate how IKKα is involved in lymphocytes, we have established bone marrow (BM) chimeras with a transfer of fetal liver cells from mice obtained by intercrossing IKKα+/− mice. IKKα−/− chimeras showed a reduction of mature B cell population, impairment of basal and Ag-specific Ig production, and disruption of splenic microarchitecture including germinal center (GC) formation. Our results revealed the critical roles of IKKα in peripheral B cell survival and maturation.
Materials and Methods
Generation of BM Chimeras.
Generation of IKKα−/− mice was described previously 10. H2K-bcl-2 transgenic (tg) mice overexpressing Bcl-2 29 were provided by Dr. I. Weissman (Stanford University, Stanford, CA). IKKα+/− mice were intercrossed to obtain embryos with IKKα1/+, IKKα1/−, or IKKα−/− genotype. bcl-2-tg-IKKα−/− embryos were obtained from bcl-2-tg-IKKα1/− × IKKα+/− mating. For each chimera, 5–10 × 106 fetal liver cells from the embryos at day 13.5–15.5 of gestation were transferred intravenously into a recombination activating gene (RAG)2-deficient C57BL/6 (B6) mice 30 that had received 12 Gy from an x-ray irradiation system, MBR-1520R (Hitachi Medical Corporation) before transfer, as described previously 31. The recipient mice were given 1 mg/ml neomycin sulfate and 1,000 U/ml polymyxin B in their drinking water after irradiation and analyzed 6–10 wk after reconstitution.
Flow Cytometric Analysis.
Single cell suspensions were incubated first with anti-CD16/32 to minimize nonspecific staining and stained with cocktails of mAbs conjugated to FITC, PE, or biotin for 20 min at 4°C. The biotinylated Abs were developed with streptavidin conjugated to PE or Cychrome. All mAbs, except PE-labeled anti-IgD (Southern Biotechnology Associates, Inc.), were purchased from BD PharMingen. Flow cytometric analysis was performed using a FACSCalibur™ (Becton Dickinson) with CELLQuest™ software (Becton Dickinson).
Analysis for Cell Survival.
Cells were cultured at 2 × 106 per ml in 24-well plates for the indicated periods with or without 25 μg/ml LPS (055:B5; Difco) or 0.5 μg/ml anti-CD40 (BD PharMingen). Harvested cells were stained with PE-B220 and further stained with annexin-V-FLUOS staining kit (Boehringer) by following the manufacturer's protocol. Stained cells were analyzed with a FACSCalibur™.
Lymphocyte Activation in Culture.
Splenic B cells were purified by depletion of non-B cells with a Magnetic Cell Sorter (MACS; Miltenyi Biotec). In brief, splenocytes were first stained with biotinylated anti-CD43 mAb and incubated with streptavidin microbeads. Then CD43− cells were purified on MACS AS column and used as splenic B cells 32. Splenic T cells were also purified by MACS with biotinylated anti-CD8 mAb, and CD4 and streptavidin microbeads. The resulting B and T cell preparations contained >90% B220+ cells and 95% CD3+ cells, respectively, as determined by FACS®. The B cells (105 cells/well) from chimeras were cultured in complete RPMI 1640 (RPMI 1640 medium supplemented with 10% FCS, 2-ME, penicillin, and streptomycin) with or without 25 μg/ml LPS, 0.5 μg/ml anti-CD40, 0.5 μg/ml anti-CD40 plus 200 U/ml IL-4, or 25 μg/ml LPS plus 0.5 μg/ml anti-CD40. After 48 h, they were pulsed with 0.2 μCi of [3H]thymidine (NEN Life Science Products) and cultured for a further 15 h. [3H]Thymidine incorporation was measured with a liquid scintillation counter, Top Count (Packard Instrument Co.).
Labeling Cells with 5-Bromo-2′-deoxyuridine.
Chimeric mice were fed with light-protected drinking water containing 1 mg/ml 5-bromo-2′-deoxyuridine (BrdU; Wako) for 4 d. BrdU-labeled cells were detected by flow cytometric analysis with FITC-conjugated anti-BrdU Ab, 3D4 (BD PharMingen). In brief, splenic cells were stained with PE-B220 and fixed with PBS containing 1% paraformaldehyde and 0.01% Tween 20 for 24 h at 4°C. Cells were washed and incubated for 1 h at 37°C in 40 mM Tris-HCl, pH 8.0, containing 10 mM NaCl, 6 mM MgCl2, and 50 Kuniz units of DNase I (Sigma-Aldrich). Then cells were washed and incubated with FITC–anti-BrdU or FITC-conjugated control Ab in the presence of 0.5% Tween 20. Stained cells were analyzed with a FACSCalibur™.
Electrophoretic Mobility Shift Assay.
Splenic B (CD43−) cells from IKKα1/+ and IKKα−/− mice were incubated with or without 25 μg/ml LPS or 0.5 μg/ml anti-CD40 for 1.5 h. Then, whole cell lysates were prepared, and NF-κB DNA binding activities were analyzed as described previously 33. For supershift analysis, Abs for each NF-κB component (Santa Cruz Biotechnology, Inc.) were incubated with the lysates before the addition of an NF-κB oligonucleotide probe.
Reverse Transcriptase PCR for A1.
Purified splenic B cells were incubated with or without 25 μg/ml LPS or 0.5 μg/ml anti-CD40 for 2 h. Extraction of total RNAs, reverse transcription, and PCR analysis were performed as described previously 31. Primers for A1 were as follows: sense primer, 5′-TCATGCATATCCACTCCCTGGCTGAGC-3′; and antisense primer, 5′-GTCCTGTCATCTGCAGAAAAGTCAGCC-3′.
Serum Ig Level and T Cell–dependent Immune Responses.
Serum Ig isotype concentrations of RAG2-deficient chimeras were analyzed by ELISA as described 34. Abs and standard Igs were purchased from Southern Biotechnology Associates, Inc. For T cell–dependent immune responses, mice were immunized with 100 μg/head of alum-precipitated chicken γ-globulin (CG) coupled to 4-hydroxy-3-nitro-phenylacetyl (NP) and bled at the indicated days. The serum titers of NP-specific IgM and IgG1 were determined by ELISA as described 34. Statistical analysis was performed with the unpaired Student's t test.
Histological Analysis of Splenic Sections.
Mice were killed 14 d after immunization with NP-CG, and the spleens were removed promptly. Each spleen was divided into two pieces, one piece for hematoxylin and eosin (HE) stain and the other for immunohistochemistry. For HE stain, spleen tissues were fixed in 10% buffered formalin, pH 7.2, and embedded in paraffin. Deparaffinized sections (4 μm thick) were then stained with HE. For immunohistochemistry, freshly dissected spleens were covered with Tissue-Tek OCT compound (Miles, Inc.) and quickly frozen in liquid nitrogen. Frozen sections (4 μm thick) were then fixed with ice cold acetone, and incubated in 3% H2O2 in 50% methanol for 30 min to inactivate internal peroxidase. After washing with PBS, the sections were incubated with normal goat serum or normal horse serum (Vector Laboratories) to block nonspecific binding of Abs, and subsequently with the following reagents: anti-B220 (BD PharMingen), anti-IgD (Southern Biotechnology Associates, Inc.), biotin-conjugated peanut agglutinin (PNA; Seikagaku kogyo), follicular dendritic cell (FDC)-M1 (reference 35; a gift from Dr. M.H. Kosco-Vilbois, Serono Pharmaceutical Research Institute, Geneva, Switzerland), biotinylated F4/80 (Serotec), MOMA-1 (Serotec), antisialoadhesin (Serotec), or rabbit anti–BST-1 serum (reference 36; a gift from Drs. T. Hirano and K. Ishihara, Osaka University). For negative controls, rabbit preimmune serum or isotype-matched rat IgGs were used. After washing with PBS, sections were further incubated with biotin-conjugated, goat anti–rabbit IgG (Vector Laboratories) or rabbit anti–rat Igs (Dako). Immunoreacted cells were then visualized by using a Vectastain ABC Elite kit (Vector Laboratories) and diaminobenzine tetrahydrochloride (Sigma-Aldrich). The sections were lightly counterstained with hematoxylin.
Results
Decrease of Mature B Cell Population in IKK−/− Chimeras.
IKKα-deficient mice die in an early neonatal period 8 9 10. To analyze roles of IKKα in hematopoietic cells, BM chimeras were established and analyzed for lymphocyte populations in various organs with flow cytometry (Fig. 1). In the peripheral blood (PB), B220+ cells in IKKα2/− chimeras significantly decreased compared with those in IKKα1/+ chimeras. Concomitant with decrease in the B cell population, the CD3+ T cell population increased in IKKα−/− chimeras (Fig. 1 A). Also in the spleen, decrease of B220+ and increase of CD3+ T cells were observed in IKKα2/− chimeras (Fig. 1 C). The results from IKKα1/− chimeras were similar to IKKα1/+ chimeras (data not shown). Mean total spleen cell numbers from 18 control (IKKα1/+ and +/−) and 19 −/− chimeras were 2.1 × 107 and 1.2 × 107, respectively. Therefore, the increase of CD3+ T cell population percentages is not because of an increase of its absolute numbers. Furthermore, CD4 versus CD8 staining of thymus and spleen revealed no significant differences between IKKα1/+ and IKKα−/− chimeras, indicating that T cell development proceeds normally in the absence of IKKα (data not shown).
Figure 1.
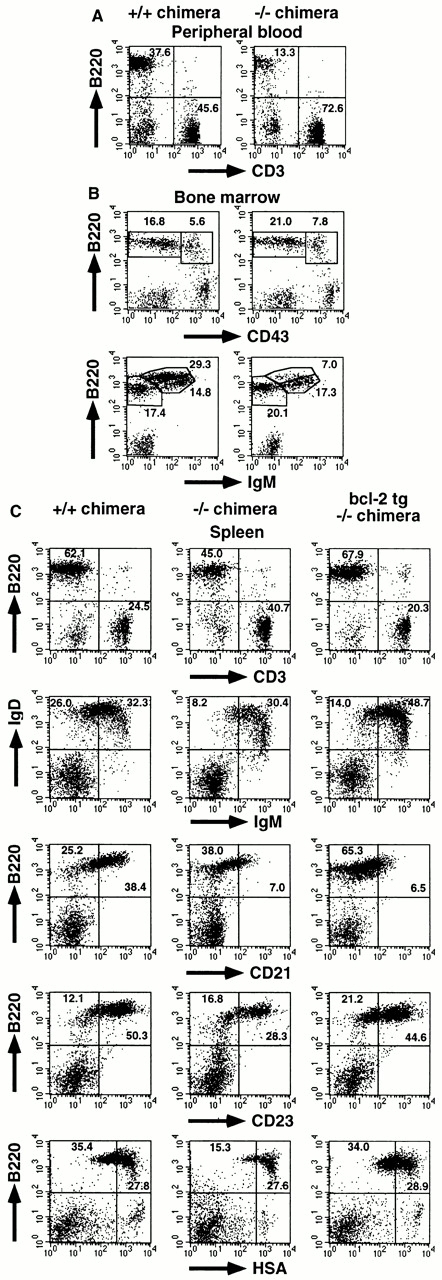
Mature B cell decrease in IKKα2/− RAG2-deficient B6 chimeras. Single cell suspensions from (A) PB, (B) BM, and (C) spleen were stained with the indicated Abs and analyzed using a FACSCalibur™ with CELLQuest™ software. The percentages of the quadrants or enclosed areas are indicated by numbers. For BM, triple color analysis was performed, and CD43 versus B220 and IgM versus B220 profiles are shown for IgM− and CD43− lymphoid cells, respectively. In C, data from IKKα2/− chimeras with transgene expression of bcl-2 are also shown. Four independent experiments were performed with similar results. One representative experiment is shown.
Next, we analyzed splenic B cell maturation status in chimeras (Fig. 1 C). Peripheral B cell development proceeds from immature IgMhighIgDhigh to mature IgMlowIgDhigh cells 37 38. The decrease of B cell numbers in IKKα2/− chimeras was more prominent in IgMlowIgDhigh cells than in IgMhighIgDhigh cells. Given the decrease of total spleen cell numbers, the IgMhighIgDhigh cell population in IKKα2/− chimeras was also reduced approximately twofold. B cell maturation is also characterized by the upregulation of CD21 and CD23 and the downregulation of heat stable antigen (HSA; reference 39). This marker analysis (Fig. 1 C), together with IgM versus IgD staining, clearly demonstrates that mature B cells were more severely decreased than immature B cells in the spleen of IKKα2/− chimeras.
We also analyzed early B cell development in the BM (Fig. 1 B). The population size of pro-B (CD43+ IgM−B220+), pre-B (CD43−IgM−B220+), and immature B (CD43−IgM+B220low) cells was comparable between IKKα1/+ and IKKα2/− chimeras. However, mature recirculating B (CD43−IgM+B220high) cells 40 significantly decreased in IKKα2/− chimeras (7.0%) compared with those in IKKα1/+ chimeras (29.3%).
Enhanced Cell Death of In Vitro IKKα2/− B Cells.
NF-κB activity has been shown to mediate antiapoptotic activity in various cells including B cells 41 42. We have examined survival of B lineage cells in both IKKα1/+ and IKKα2/− chimeras with annexin V binding activity. PB from the chimeras was cultured in vitro in the absence of mitogens for the indicated periods and analyzed for their annexin V binding activity with FACS® (Fig. 2A and Fig. B). Before culture (0 h), percentages of B220+annexin+ cells in PB were higher in IKKα2/− chimeras (3.3/7.1 + 3.3 = 31.7%) than in IKKα1/+ chimeras (4.3/4.3 + 26.2 = 14.1%) (Fig. 2A and Fig. B). B220+annexin+ cells reached >95% of the total B220+ cells in IKKα2/− chimeras after 24 h of culture, whereas they were 63.4 and 83.7% in IKKα1/+ chimeras after 24 and 48 h of culture, respectively. Likewise, survival of splenic B cells was severely impaired in IKKα2/− chimeras before and throughout the culture period (Fig. 2A and Fig. B).
Figure 2.

Impaired survival in vitro and increased turnover in vivo of IKKα2/− B cells. (A and B) PB and splenocytes from IKKα1/+ and IKKα2/− RAG2-deficient B6 chimeras were cultured in complete RPMI 1640 in the absence or presence of mitogens for the indicated periods and stained with FITC–annexin V and PE-B220. Percentages of annexin+ B cells were calculated by dividing percentages of B220+annexin+ cells by those of total B (B220+annexin+ and B220+annexin−) cells and shown as bar graphs in B. (C) Increased B cell turnover in IKKα2/− chimeras. The turnover of splenic B cells was determined by BrdU incorporation. Numbers represent percentages of the quadrants. Experiments were independently performed three times with similar results. One representative experiment is shown.
Mitogens such as LPS or anti-CD40 can induce B cell activation by inducing NF-κB activation. At 48 h, annexin+ B cell percentages of LPS-stimulated IKKα1/1 splenocytes (57.3%) were less than those of unstimulated ones (68.9%; Fig. 2 B). However, LPS could not decrease annexin+ B cell percentages in IKKα2/− chimeras. Furthermore, after 48 h of stimulation with anti-CD40, annexin+ B cell percentages in IKKα1/1 splenocytes decreased to 21.2%, while those in IKKα2/− splenocytes, although slightly reduced, remained at 71.3%. Thus, at any culture periods, with or without mitogens, cell death was significantly augmented in IKKα2/− B cells.
Increased Turnover of In Vivo IKKα2/− B Cells.
In vivo B cell turnover rate was evaluated in chimeras. BrdU-labeled B220+ cells were 10.9 and 8.6% in the spleens of IKKα1/+ and IKKα2/− chimeras, respectively (Fig. 2 C). Considering the decrease of B cell percentages in IKKα2/− chimeras, it can be reasonably assumed that about two times more percentages of B cells were labeled with BrdU in IKKα2/− chimeras than in IKKα1/+ chimeras. The results clearly suggest that B cell turnover in vivo is enhanced in the absence of IKKα.
Impaired Mitogenic Responses of IKKα2/− B Cells.
We have analyzed splenic B cell responses to LPS and anti-CD40 with thymidine incorporation (Fig. 3 A). The incorporation of IKKα2/− B cells stimulated with LPS or anti-CD40 was ∼5–10 times less than that of IKKα1/+ B cells. Simultaneous stimulation with LPS and anti-CD40 could enhance the proliferation of IKKα2/− B cells, but not to the same level as IKKα1/+ B cells (Fig. 3 A). Furthermore, blast formation by LPS or anti-CD40 was severely attenuated in IKKα2/− B cells (Fig. 3 B). In contrast, thymocytes and splenic T cells derived from IKKα2/− chimeras showed normal proliferative responses to T cell mitogens such as IL-2 plus anti-CD3 or IL-2 plus Con A (data not shown).
Figure 3.
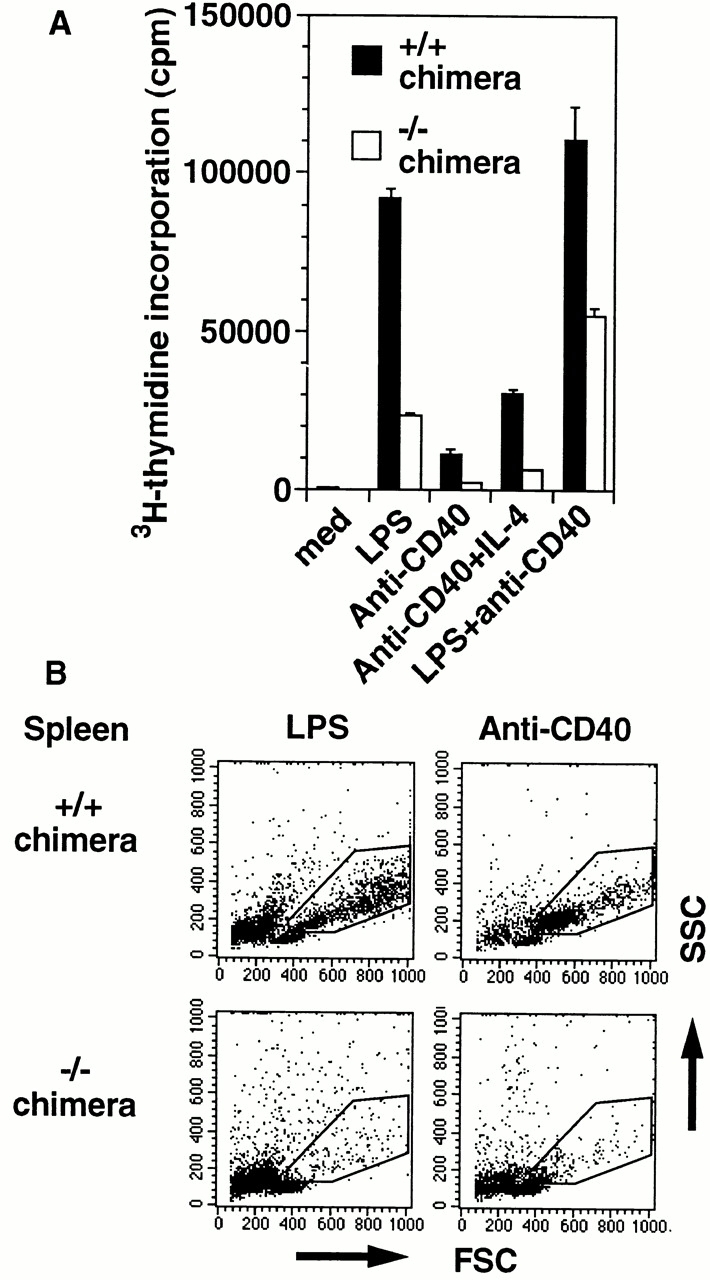
Impaired mitogenic responses of IKKα2/− B cells. (A) Splenic B cells from IKKα1/+ and IKKα2/− RAG2-deficient B6 chimeras were cultured in the absence or presence of mitogens for 72 h and analyzed for their [3H]thymidine incorporation. The data indicate means ± SD of triplicate samples of one representative experiment. (B) Splenocytes from IKKα1/+ and IKKα2/− RAG2-deficient B6 chimeras were cultured with LPS or anti-CD40 for 48 h. Forward scatter (FSC) and side scatter (SSC) of harvested cells were analyzed by FACS®. Blasts are indicated by enclosed areas. Experiments were independently performed three times with similar results. One representative experiment is shown.
Impaired NF-κB Activation in IKKα2/− B Cells.
We next analyzed NF-κB activity in IKKα1/+ and IKKα2/− splenic B cells with or without LPS or anti-CD40 (Fig. 4). Although the mitogens could enhance the NF-κB DNA binding activity in both IKKα1/+ and −/− B cells, the magnitude of the activity was decreased in IKKα2/− B cells (Fig. 4 A). Anti-CD40–induced NF-κB activation was more severely impaired than the LPS-induced one in IKKα2/− B cells.
Figure 4.

NF-κB DNA binding activity in IKKα1/+ and IKKα2/− B cells. (A) Splenic B cells were stimulated with or without 25 μg/ml LPS (L) or 0.5 μg/ml anti-CD40 40 for 1.5 h. The whole cell lysates were prepared and NF-κB activity was determined by electrophoretic mobility shift assay. (B) LPS-induced NF-κB complexes were analyzed by supershift analysis. Specific NF-κB complexes are indicated by arrowheads. *Nonspecific binding. Similar results were obtained from three independent experiments.
To determine the components of NF-κB complexes, we performed a supershift analysis by using specific Abs (Fig. 4 B). In IKKα1/+ B cells, anti-p50 supershifted nearly all the LPS-induced complexes, while anti-p65 did parts of them. Hence, the remaining complex in IKKα1/+ B cells with anti-p65 treatment seems to correspond mainly to p50/p50 homodimer. The results showed that LPS can mainly induce p50/p50 homodimer and p50/p65 heterodimer as NF-κB complexes. In addition, other components also contributed to NF-κB binding activity to some extent because anti-p52, –c-Rel, and -RelB Abs partially inhibited the binding. As observed in IKKα1/+ B cells, the detectable NF-κB complex in IKKα2/− B cells showed a similar supershift pattern, indicating that the composition of residual NF-κB complexes in IKKα2/− B cells is not grossly different from that in IKKα1/+ B cells.
Decreased Basal Level of A1 Expression in IKKα2/− B Cells.
NF-κB is involved in the expression of antiapoptotic genes. For example, c-Rel is essential for not only basal but also mitogen-induced expression of A1 43. A1 expression remains low during BM B cell development, but is upregulated 10-fold as cells maturate into a long-lived peripheral B cell stage, suggesting that constitutive A1 expression is critically involved in resting mature B cell survival 44. Therefore, we examined A1 gene expression in IKKα1/+ and IKKα2/− B cells with or without mitogens. LPS or anti-CD40 could enhance A1 gene expression not only in IKKα1/+ B cells but also in IKKα2/− B cells (Fig. 5). However, basal expression of A1 was lower in IKKα2/− B cells than in IKKα1/+ B cells.
Figure 5.

Decrease of basal A1 gene expression in IKKα2/− B cells. A1 expression was analyzed in IKKα1/+ or IKKα2/− B cells with or without LPS (L) or anti-CD40 40 stimulation through reverse transcription PCR analysis. Basal A1 gene expression was determined by amplification with 30 cycles. As controls, data for β-actin expression are shown. M, Φ×174/HaeIII digest marker.
Partial Rescue of Impaired B Cell Development in IKKα2/− Chimeras by Bcl-2 Expression.
To assess whether the prevention of IKKα2/− B cell apoptosis can restore peripheral B cell development, IKKα2/− chimeras expressing the bcl-2 transgene were established and analyzed by FACS® (Fig. 1 C). Total spleen cell numbers of bcl-2-tg-IKKα2/− chimeras (8.9 × 107, n = 5) were prominently increased compared with IKKα2/− chimeras. CD3 versus B220 staining revealed that Bcl-2 expression corrected the ratio of B to T cells in IKKα2/− chimeras. Next, B cell maturation status was analyzed with several markers. Although percentages of IgMlowIgDhigh and IgMhighIgDhigh cell populations increased, the ratio of IgMlowIgDhigh to IgMhighIgDhigh cell population in bcl-2-tg-IKKα2/− chimeras was not significantly different from IKKα2/− chimeras. Furthermore, in bcl-2-tg-IKKα2/− chimeras, CD21 expression on B220+ cells remained as low as IKKα2/− chimeras. However, CD23 upregulation and HSA downregulation were restored by Bcl-2 expression. Thus, bcl-2 transgene expression could enhance B lineage cell expansion, but only partially restore B cell maturation in the absence of IKKα.
Decreased Serum Ig Levels and Impaired T Cell–dependent Immune Responses in IKKα2/− Chimeras.
We further investigated immune functions of IKKα1/+ and IKKα2/− chimeras. Basal production of all Ig isotypes in IKKα2/− chimeras was reduced to 1/50 or less of those in IKKα1/+ chimeras (P < 0.0001, Fig. 6 A). Furthermore, T cell–dependent immune responses were severely impaired in IKKα2/− chimeras because Ag-specific IgM and IgG1 levels of IKKα2/− chimeras were <1/10 of IKKα1/+ chimeras (Fig. 6 B, anti-NP IgM at day 7, P < 0.001; anti-NP IgM at day 14 and IgG1 at day 7 and 14, P < 0.0001).
Figure 6.
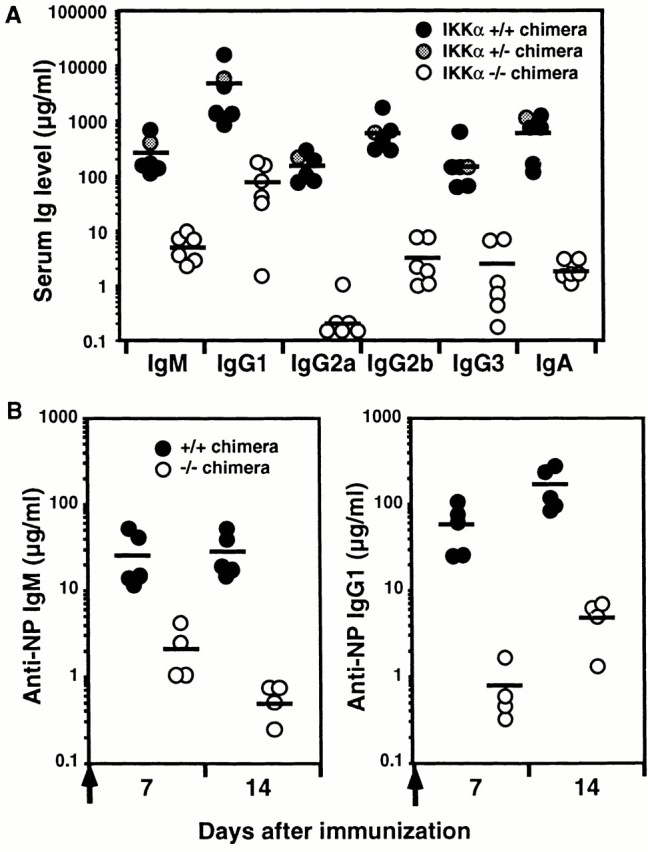
Decreased Ig production and impaired T cell–dependent immune responses in IKKα2/− chimeras. (A) Serum Ig isotype levels in unimmunized, IKKα1/+, IKKα1/−, and IKKα2/− RAG2-deficient B6 chimeras. Serum Ig titers were determined by isotype-specific ELISA. (B) Immune responses to the T cell–dependent Ag, NP-CG. IKKα1/+ and IKKα2/− RAG2-deficient B6 chimeras were immunized with alum-precipitated NP-CG and bled at the indicated days. NP-specific IgM and IgG1 titers were determined by ELISA. Bars indicate mean values.
Histological analysis was performed to evaluate the GC formation and splenic microarchitecture 14 d after immunization (Fig. 7). HE, B220, and IgD staining revealed the well-developed GC with clear B cell areas in control chimeras. In contrast, in IKKα2/− chimeras, poor GC formation was revealed with HE stain, although B220+ or IgD+ cells clearly formed B cell areas. Remarkably, PNA+ cell clusters, which represent GC B cells, were not detected at all in the spleen of IKKα2/− chimeras.
Figure 7.
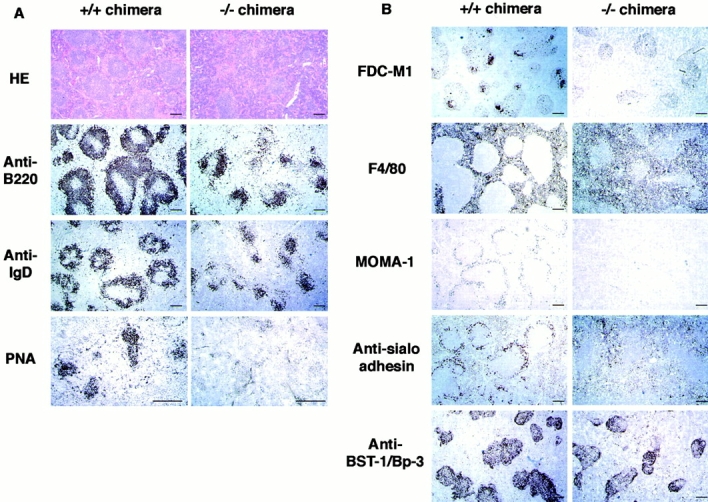
(A) Impaired GC formation in IKKα2/− chimeras. IKKα1/+ and IKKα2/− RAG2-deficient B6 chimeras were immunized with alum-precipitated NP-CG and killed 14 d after injection. Splenic sections were stained with HE or immunostained with B220, anti-IgD, or PNA. (B) Impairment of splenic microarchitecture in IKKα2/− chimeras. Frozen sections were stained with FDC-M1, F4/80, MOMA-1, or antisialoadhesin mAbs or rabbit anti–BST-1/Bp-3 antiserum. Scale bars, 200 μm.
To further investigate the alterations of the splenic microarchitecture in IKKα2/− chimeras, tissue sections were also stained with markers for FDCs or macrophages (Fig. 7 B). FDC clusters, defined by FDC-M1 35, were readily detected in IKKα1/+ chimeras but not in IKKα2/− chimeras. A stromal cell antigen, BST-1/Bp-3, is expressed on stromal cells in the T zone and follicles 45 46 47, which include precursors of FDCs 48. Expression patterns of BST-1/Bp-3 were equivalent in IKKα +/+ and IKKα2/− chimeras, suggesting that there are FDC precursors in IKKα2/− chimeras. Meanwhile, marginal metallophilic macrophages, identified by MOMA-1 or an antisialoadhesin Ab, were clearly detected in IKKα1/+ chimeras but not in IKKα2/− chimeras. Furthermore, F4/80+ red pulp macrophages in IKKα1/+ chimeras were clearly excluded into the red pulp, while some F4/80+ cells in IKKα2/− chimeras migrated into the white pulp. Collectively, these histological findings clearly indicate that IKKα is critical not only for GC formation but also for establishing the splenic microarchitecture.
Discussion
We have generated BM chimeras to elucidate in vivo roles of IKKα in lymphocytes. Although certain NF-κB subunit-deficient mice showed T cell defects in population size or proliferative responses 24 25 28 49, IKKα2/− chimeras did not, indicating that IKKα is not essential for T cell development or proliferation. This is also consistent with previous findings, including ours, that cytokine- or T cell receptor–induced NF-κB activation is dependent on IKKβ, but not on IKKα in T lineage cells 10 50. However, we cannot exclude the possibility that IKKα is involved in some T cell functions.
Our results clearly demonstrate that IKKα is involved in mature B cell development and function, but not in early B cell development. NF-κB components are developmentally regulated in B lineage cells 14 15. Gene targeting revealed that an individual NF-κB component plays its own critical roles in a B cell stage–specific manner. For example, BM chimeras transplanted with p50/p65 double knockout fetal liver cells lacked pro-B cells 27, indicating that the p50/p65 heterodimer is essential for early B lymphopoiesis. On the contrary, mutant mice lacking c-Rel showed normal BM B cell development, but manifested impairment of mitogenic responses, basal production of all Ig isotypes, and T cell–dependent immune responses 28. Although p50 is a major component for NF-κB throughout B cell development, p50-deficient mice showed impairment of mature B cell functions: low responses to LPS, reduction of some isotypes of serum Igs (IgG1, IgG2b, IgA, and IgE), and impairment of immune responses 19. Therefore, the phenotype of IKKα2/− chimeras was more similar to c-Rel– or p50-deficient mice than to p50/p65-deficient mice, in the respect that peripheral, not BM, B cell development is impaired.
Mutant mice lacking other NF-κB components also showed a similar phenotype to IKKα2/− chimeras. For example, targeting of p52 results in a decrease of B cells, low responses to B cell mitogens, and impaired GC formation 23 26. An IκB family member, Bcl-3, can function as a potent coactivator with p50/p50 or p52/p52 homodimers 51 52. Bcl-3–deficient mice also showed decrease of B cells and impairment of GC formation 53 54. However, GC formation was restored by the transfer of p52- or Bcl-3–deficient lymphocytes into irradiated RAG1-deficient mice 26. Thus, p52 and Bcl-3 play critical roles in non-BM stromal cells, while IKKα does in lymphocytes despite their similar phenotype.
Mutant mice established so far lacking a single NF-κB component showed an apparently less severe phenotype than IKKα2/− chimeras. For example, the B cell population is not decreased in c-Rel– or p50-deficient mice. In addition, not all isotypes of Ig production were impaired and GC formation was observed in p50-deficient mice 19 26 53. Double knockout mice manifested more severe phenotype than single knockout mice, exemplified by p50/p52 double knockout mice with severe mature B cell defects 25. Furthermore, tg mice harboring a dominant negative form of IκB, in which function of more than one NF-κB component is presumably attenuated, showed mature B cell decrease as in IKKα2/− chimeras 55. Given these findings, it can be assumed that IKKα is critically involved in the activation of several NF-κB components in B cells.
Histologically, B220+ or IgD+ staining indicates that IKKα expression in hematopoietic cells is dispensable for T–B compartmentalization. However, IKKα2/−chimeras showed loss of GC marker such as PNA or FDC-M1 with normal BST-1/Bp-3 expression. GC formation proceeds with FDC generation from FDC precursors expressing BST-1/Bp-3 in a mature B cell–dependent manner 48. Therefore, these histological findings are consistent with mature B cell decrease in IKKα2/− chimeras.
What brings about decrease of mature B cells in the absence of IKKα? It is possible that IKKα is critically involved in survival, mitogenic proliferation, or development in B cells. Cell turnover analysis in vivo and annexin staining in vitro (Fig. 2) clearly indicate enhanced cell death of IKKα2/− B cells. In addition, impaired mitogenic responses were also observed in IKKα2/− B cells (Fig. 3). Thus, IKKα should be critical not only for preventing B cell death but also for B cell mitogenic responses. Similar abnormalities were found in some NF-κB mutant mice. For example, p50−/− B cells manifested enhancement of apoptosis 22. Furthermore, B cell mitogenic responses were impaired in mutant mice lacking p50, c-Rel, or Rel-B 19 20 21 24 28.
We further addressed whether IKKα deficiency can lead to B cell developmental block. To rescue B cells from apoptosis, we have introduced bcl-2 transgene expression into the IKKα2/− background. Bcl-2 could restore the ratio of B to T cells, CD23 upregulation, and HSA downregulation, but neither differentiation into IgMlowIgDhigh cells nor CD21 upregulation in IKKα2/− chimeras. Partial restoration of IKKα2/− B cell development with bcl-2 transgene expression indicates that IKKα is critically involved in peripheral B cell development. It is also possible that the developmental defect caused by IKKα deficiency can make B cells susceptible to apoptosis and hyporesponsive to mitogens.
B cell receptor (BCR) signaling is critical for maintenance of the peripheral B cell population 56. Once B cells lack surface Ig, they die through an apoptotic pathway 56. Mature B cell reduction is also observed in mutant mice lacking BCR signaling molecules such as Bruton's tyrosine kinase (BTK; references 57, 58), B cell linker protein (BLNK/SLP-65; references 59, 60), phosphoinositide 3-kinase p85α 61 62, or phospholipase C-γ2 63 64. Notably, enhanced apoptosis of B lineage cells was also detected in the xid mice carrying the mutation in BTK 38. BTK is essential for coupling the BCR signal to NF-κB activation 65 66. In addition, BCR signaling can activate phosphoinositide 3-kinase and a serine/threonine kinase, Akt 67, which can induce phosphorylation of IKKα 68. These findings suggest the possibility that IKKα is involved in BCR signaling.
Several lines of evidences suggest that IKKβ, but not IKKα, is mainly involved in cytokine-induced NF-κB activation 8 9 10 11 12 13. This seems inconsistent with our finding that NF-κB activation by LPS or anti-CD40 was decreased in IKKα2/− B cells. However, activation is still inducible and sufficient for partial responses to LPS and anti-CD40 (Fig. 2 B and 3 A) and A1 gene induction (Fig. 5), indicating that IKKβ can still function without IKKα.
The inducible expression of A1 was also observed in p50-deficient B cells, which manifested enhanced apoptosis 22 43. In contrast, c-Rel–deficient B cells manifested impaired induction of A1 expression but no enhanced apoptosis 22 43. This might suggest that similar molecular defects, although unidentified, may underlie the augmented cell death of p50- and IKKα-deficient B cells.
In conclusion, this study strongly suggests that IKKα plays crucial roles in the survival, proliferative responses, and maturation of peripheral B cells. The defects caused by IKKα deficiency are most likely intrinsic to B cells, although the possibility cannot formally be excluded that dysfunction of BM-derived, non-B cells also contributes to the phenotype of IKKα2/− chimeras. It is unclear at present which contributes more to the observed B cell defects, disturbed maturation or dysfunction of already matured B cells. Although the molecular mechanism remains to be clarified yet, IKKα is a critical molecule for maintaining the mature B cell population.
Acknowledgments
We thank Ms. Akiko Maekawa and Nana Iwami for excellent technical assistance. We also thank Drs. Mitsuru Matsumoto, Takashi Nagasawa, and Yoshiko Murakami for helpful discussion. We are also grateful to Dr. Irving Weissman for the bcl-2-tg mice, Dr. Frederick W. Alt for the RAG2-deficient mice, Drs. Toshio Hirano and Katsuhiko Ishihara for the rabbit anti–BST-1 antiserum, Dr. Marie H. Kosco-Vilbois for the FDC-M1 Ab, and Ms. Claudia Uthoff-Hachenberg and Dr. Klaus Rajewsky for ELISA reagents.
This work was supported in part by grants from the Ministry of Education, Science, and Culture in Japan, Uehara Memorial Foundation, Novartis Foundation for the Promotion of Science, Kato Memorial Bioscience Foundation, Kowa Life Science Foundation, and Core Research for Evolutional Science and Technology (CREST) of Japan Science and Technology Corporation.
Footnotes
Abbreviations used in this paper: B6, C57BL/6; BCR, B cell receptor; BM, bone marrow; BrdU, 5-bromo-2′-deoxyuridine; BTK, Bruton's tyrosine kinase; CG, chicken γ-globulin; FDC, follicular dendritic cell; GC, germinal center; HE, hematoxylin and eosin; HSA, heat stable antigen; IKK, IκB kinase; NF, nuclear factor; NP, 4-hydroxy-3-nitro-phenylacetyl; PB, peripheral blood; PNA, peanut agglutinin; RAG, recombination activating gene; tg, transgenic.
References
- Baeuerle P.A., Baltimore D. NF-κBten years after. Cell. 1996;87:13–20. doi: 10.1016/s0092-8674(00)81318-5. [DOI] [PubMed] [Google Scholar]
- Barnes P.J., Karin M. Nuclear factor-κBa pivotal transcription factor in chronic inflammatory diseases. N. Engl. J. Med. 1997;336:1066–1071. doi: 10.1056/NEJM199704103361506. [DOI] [PubMed] [Google Scholar]
- Regnier C.H., Song H.Y., Gao X., Goeddel D.V., Cao Z., Rothe M. Identification and characterization of an IκB kinase. Cell. 1997;90:373–383. doi: 10.1016/s0092-8674(00)80344-x. [DOI] [PubMed] [Google Scholar]
- Zandi E., Rothwarf D.M., Delhase M., Hayakawa M., Karin M. The IκB kinase complex (IKK) contains two kinase subunits, IKKα and IKKβ, necessary for IκB phosphorylation and NF-κB activation. Cell. 1997;91:243–252. doi: 10.1016/s0092-8674(00)80406-7. [DOI] [PubMed] [Google Scholar]
- Woronicz J.D., Gao X., Cao Z., Rothe M., Goeddel D.V. IκB kinase-βNF-κB activation and complex formation with IκB kinase-α and NIK. Science. 1997;278:866–869. doi: 10.1126/science.278.5339.866. [DOI] [PubMed] [Google Scholar]
- Mercurio F., Zhu H., Murray B.W., Shevchenko A., Bennett B.L., Li J., Young D.B., Barbosa M., Mann M., Manning A., Rao A. IKK-1 and IKK-2cytokine-activated IκB kinases essential for NF-κB activation. Science. 1997;278:860–866. doi: 10.1126/science.278.5339.860. [DOI] [PubMed] [Google Scholar]
- DiDonato J.A., Hayakawa M., Rothwarf D.M., Zandi E., Karin M. A cytokine-responsive IκB kinase that activates the transcription factor NF-κB. Nature. 1997;388:548–554. doi: 10.1038/41493. [DOI] [PubMed] [Google Scholar]
- Li Q., Lu Q., Hwang J.Y., Buscher D., Lee K.F., Izpisua-Belmonte J.C., Verma I.M. IKK1-deficient mice exhibit abnormal development of skin and skeleton. Genes Dev. 1999;13:1322–1328. doi: 10.1101/gad.13.10.1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y., Baud V., Delhase M., Zhang P., Deerinck T., Ellisman M., Johnson R., Karin M. Abnormal morphogenesis but intact IKK activation in mice lacking the IKKα subunit of IκB kinase. Science. 1999;284:316–320. doi: 10.1126/science.284.5412.316. [DOI] [PubMed] [Google Scholar]
- Takeda K., Takeuchi O., Tsujimura T., Itami S., Adachi O., Kawai T., Sanjo H., Yoshikawa K., Terada N., Akira S. Limb and skin abnormalities in mice lacking IKKα. Science. 1999;284:313–316. doi: 10.1126/science.284.5412.313. [DOI] [PubMed] [Google Scholar]
- Li Q., Van Antwerp D., Mercurio F., Lee K.F., Verma I.M. Severe liver degeneration in mice lacking the IκB kinase 2 gene. Science. 1999;284:321–325. doi: 10.1126/science.284.5412.321. [DOI] [PubMed] [Google Scholar]
- Li Z.W., Chu W., Hu Y., Delhase M., Deerinck T., Ellisman M., Johnson R., Karin M. The IKKβ subunit of IκB kinase (IKK) is essential for nuclear factor κB activation and prevention of apoptosis. J. Exp. Med. 1999;189:1839–1845. doi: 10.1084/jem.189.11.1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka M., Fuentes M.E., Yamaguchi K., Durnin M.H., Dalrymple S.A., Hardy K.L., Goeddel D.V. Embryonic lethality, liver degeneration, and impaired NF-κB activation in IKK-β-deficient mice. Immunity. 1999;10:421–429. doi: 10.1016/s1074-7613(00)80042-4. [DOI] [PubMed] [Google Scholar]
- Miyamoto S., Schmitt M.J., Verma I.M. Qualitative changes in the subunit composition of κB-binding complexes during murine B-cell differentiation. Proc. Natl. Acad. Sci. USA. 1994;91:5056–5060. doi: 10.1073/pnas.91.11.5056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liou H.C., Sha W.C., Scott M.L., Baltimore D. Sequential induction of NF-κB/Rel family proteins during B-cell terminal differentiation. Mol. Cell. Biol. 1994;14:5349–5359. doi: 10.1128/mcb.14.8.5349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grumont R.J., Gerondakis S. The subunit composition of NF-κB complexes changes during B-cell development. Cell. Growth Differ. 1994;5:1321–1331. [PubMed] [Google Scholar]
- Francis D.A., Karras J.G., Ke X.Y., Sen R., Rothstein T.L. Induction of the transcription factors NF-κB, AP-1 and NF-AT during B cell stimulation through the CD40 receptor. Int. Immunol. 1995;7:151–161. doi: 10.1093/intimm/7.2.151. [DOI] [PubMed] [Google Scholar]
- Francis D.A., Sen R., Rice N., Rothstein T.L. Receptor-specific induction of NF-κB components in primary B cells. Int. Immunol. 1998;10:285–293. doi: 10.1093/intimm/10.3.285. [DOI] [PubMed] [Google Scholar]
- Sha W.C., Liou H.C., Tuomanen E.I., Baltimore D. Targeted disruption of the p50 subunit of NF-κB leads to multifocal defects in immune responses. Cell. 1995;80:321–330. doi: 10.1016/0092-8674(95)90415-8. [DOI] [PubMed] [Google Scholar]
- Snapper C.M., Zelazowski P., Rosas F.R., Kehry M.R., Tian M., Baltimore D., Sha W.C. B cells from p50/NF-κB knockout mice have selective defects in proliferation, differentiation, germ-line CH transcription, and Ig class switching. J. Immunol. 1996;156:183–191. [PubMed] [Google Scholar]
- Snapper C.M., Rosas F.R., Zelazowski P., Moorman M.A., Kehry M.R., Bravo R., Weih F. B cells lacking RelB are defective in proliferative responses, but undergo normal B cell maturation to Ig secretion and Ig class switching. J. Exp. Med. 1996;184:1537–1541. doi: 10.1084/jem.184.4.1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grumont R.J., Rourke I.J., O'Reilly L.A., Strasser A., Miyake K., Sha W., Gerondakis S. B lymphocytes differentially use the Rel and nuclear factor κB1 (NF-κB1) transcription factors to regulate cell cycle progression and apoptosis in quiescent and mitogen-activated cells. J. Exp. Med. 1998;187:663–674. doi: 10.1084/jem.187.5.663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caamano J.H., Rizzo C.A., Durham S.K., Barton D.S., Raventos-Suarez C., Snapper C.M., Bravo R. Nuclear factor (NF)-κB2 (p100/p52) is required for normal splenic microarchitecture and B cell–mediated immune responses. J. Exp. Med. 1998;187:185–196. doi: 10.1084/jem.187.2.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doi T.S., Takahashi T., Taguchi O., Azuma T., Obata Y. NF-κB RelA-deficient lymphocytesnormal development of T cells and B cells, impaired production of IgA and IgG1 and reduced proliferative responses. J. Exp. Med. 1997;185:953–961. doi: 10.1084/jem.185.5.953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzoso G., Carlson L., Xing L., Poljak L., Shores E.W., Brown K.D., Leonardi A., Tran T., Boyce B.F., Siebenlist U. Requirement for NF-κB in osteoclast and B-cell development. Genes Dev. 1997;11:3482–3496. doi: 10.1101/gad.11.24.3482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzoso G., Carlson L., Poljak L., Shores E.W., Epstein S., Leonardi A., Grinberg A., Tran T., Scharton-Kersten T., Anver M. Mice deficient in nuclear factor (NF)-κB/p52 present with defects in humoral responses, germinal center reactions, and splenic microarchitecture. J. Exp. Med. 1998;187:147–159. doi: 10.1084/jem.187.2.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horwitz B.H., Scott M.L., Cherry S.R., Bronson R.T., Baltimore D. Failure of lymphopoiesis after adoptive transfer of NF-κB-deficient fetal liver cells. Immunity. 1997;6:765–772. doi: 10.1016/s1074-7613(00)80451-3. [DOI] [PubMed] [Google Scholar]
- Kontgen F., Grumont R.J., Strasser A., Metcalf D., Li R., Tarlinton D., Gerondakis S. Mice lacking the c-rel proto-oncogene exhibit defects in lymphocyte proliferation, humoral immunity, and interleukin-2 expression. Genes Dev. 1995;9:1965–1977. doi: 10.1101/gad.9.16.1965. [DOI] [PubMed] [Google Scholar]
- Domen J., Gandy K.L., Weissman I.L. Systemic overexpression of BCL-2 in the hematopoietic system protects transgenic mice from the consequences of lethal irradiation. Blood. 1998;91:2272–2282. [PubMed] [Google Scholar]
- Shinkai Y., Rathbun G., Lam K.P., Oltz E.M., Stewart V., Mendelsohn M., Charron J., Datta M., Young F., Stall A.M., Alt F.W. RAG-2-deficient mice lack mature lymphocytes owing to inability to initiate V(D)J rearrangement. Cell. 1992;68:855–867. doi: 10.1016/0092-8674(92)90029-c. [DOI] [PubMed] [Google Scholar]
- Kaisho T., Tsutsui H., Tanaka T., Tsujimura T., Takeda K., Kawai T., Yoshida N., Nakanishi K., Akira S. Impairment of natural killer cytotoxic activity and interferon γ production in CCAAT/enhancer binding protein γ–deficient mice. J. Exp. Med. 1999;190:1573–1582. doi: 10.1084/jem.190.11.1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leitges M., Schmedt C., Guinamard R., Davoust J., Schaal S., Stabel S., Tarakhovsky A. Immunodeficiency in protein kinase Cβ-deficient mice. Science. 1996;273:788–791. doi: 10.1126/science.273.5276.788. [DOI] [PubMed] [Google Scholar]
- Adachi O., Kawai T., Takeda K., Matsumoto M., Tsutsui H., Sakagami M., Nakanishi K., Akira S. Targeted disruption of the MyD88 gene results in loss of IL-1- and IL-18-mediated function. Immunity. 1998;9:143–150. doi: 10.1016/s1074-7613(00)80596-8. [DOI] [PubMed] [Google Scholar]
- Kaisho T., Schwenk F., Rajewsky K. The roles of γ1 heavy chain membrane expression and cytoplasmic tail in IgG1 responses. Science. 1997;276:412–415. doi: 10.1126/science.276.5311.412. [DOI] [PubMed] [Google Scholar]
- Kosco M.H., Pflugfelder E., Gray D. Follicular dendritic cell-dependent adhesion and proliferation of B cells in vitro . J. Immunol. 1992;148:2331–2339. [PubMed] [Google Scholar]
- Itoh M., Ishihara K., Hiroi T., Lee B.O., Maeda H., Iijima H., Yanagita M., Kiyono H., Hirano T. Deletion of bone marrow stromal cell antigen-1 (CD157) gene impaired systemic thymus independent-2 antigen-induced IgG3 and mucosal TD antigen-elicited IgA responses. J. Immunol. 1998;161:3974–3983. [PubMed] [Google Scholar]
- Hardy R.R., Hayakawa K., Parks D.R., Herzenberg L.A. Demonstration of B-cell maturation in X-linked immunodeficient mice by simultaneous three-colour immunofluorescence. Nature. 1983;306:270–272. doi: 10.1038/306270a0. [DOI] [PubMed] [Google Scholar]
- Cariappa A., Kim T.J., Pillai S. Accelerated emigration of B lymphocytes in the Xid mouse. J. Immunol. 1999;162:4417–4423. [PubMed] [Google Scholar]
- Loder F., Mutschler B., Ray R.J., Paige C.J., Sideras P., Torres R., Lamers M.C., Carsetti R. B cell development in the spleen takes place in discrete steps and is determined by the quality of B cell receptor–derived signals. J. Exp. Med. 1999;190:75–89. doi: 10.1084/jem.190.1.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy R.R., Carmack C.E., Shinton S.A., Kemp J.D., Hayakawa K. Resolution and characterization of pro-B and pre-pro-B cell stages in normal mouse bone marrow. J. Exp. Med. 1991;173:1213–1225. doi: 10.1084/jem.173.5.1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerondakis S., Grossmann M., Nakamura Y., Pohl T., Grumont R. Genetic approaches in mice to understand Rel/NF-κB and IκB functiontransgenics and knockouts. Oncogene. 1999;18:6888–6895. doi: 10.1038/sj.onc.1203236. [DOI] [PubMed] [Google Scholar]
- Wu M., Lee H., Bellas R.E., Schauer S.L., Arsura M., Katz D., FitzGerald M.J., Rothstein T.L., Sherr D.H., Sonenshein G.E. Inhibition of NF-κB/Rel induces apoptosis of murine B cells. EMBO (Eur. Mol. Biol. Organ.) J. 1996;15:4682–4690. [PMC free article] [PubMed] [Google Scholar]
- Grumont R.J., Rourke I.J., Gerondakis S. Rel-dependent induction of A1 transcription is required to protect B cells from antigen receptor ligation-induced apoptosis. Genes Dev. 1999;13:400–411. doi: 10.1101/gad.13.4.400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomayko M.M., Cancro M.P. Long-lived B cells are distinguished by elevated expression of A1. J. Immunol. 1998;160:107–111. [PubMed] [Google Scholar]
- McNagny K.M., Bucy R.P., Cooper M.D. Reticular cells in peripheral lymphoid tissues express the phosphatidylinositol-linked BP-3 antigen. Eur. J. Immunol. 1991;21:509–515. doi: 10.1002/eji.1830210238. [DOI] [PubMed] [Google Scholar]
- Kaisho T., Ishikawa J., Oritani K., Inazawa J., Tomizawa H., Muraoka O., Ochi T., Hirano T. BST-1, a surface molecule of bone marrow stromal cell lines that facilitates pre-B-cell growth. Proc. Natl. Acad. Sci. USA. 1994;91:5325–5329. doi: 10.1073/pnas.91.12.5325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh M., Ishihara K., Tomizawa H., Tanaka H., Kobune Y., Ishikawa J., Kaisho T., Hirano T. Molecular cloning of murine BST-1 having homology with CD38 and Aplysia ADP-ribosyl cyclase. Biochem. Biophys. Res. Commun. 1994;203:1309–1317. doi: 10.1006/bbrc.1994.2325. [DOI] [PubMed] [Google Scholar]
- Yoshida K., Kaji M., Takahashi T., van den Berg T.K., Dijkstra C.D. Host origin of follicular dendritic cells induced in the spleen of SCID mice after transfer of allogeneic lymphocytes. Immunology. 1995;84:117–126. [PMC free article] [PubMed] [Google Scholar]
- Gerondakis S., Strasser A., Metcalf D., Grigoriadis G., Scheerlinck J.Y., Grumont R.J. Rel-deficient T cells exhibit defects in production of interleukin 3 and granulocyte-macrophage colony-stimulating factor. Proc. Natl. Acad. Sci. USA. 1996;93:3405–3409. doi: 10.1073/pnas.93.8.3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin X., O'Mahony A., Mu Y., Geleziunas R., Greene W.C. Protein kinase C-θ participates in NF-κB activation induced by CD3-CD28 costimulation through selective activation of IκB kinase β. Mol. Cell. Biol. 2000;20:2933–2940. doi: 10.1128/mcb.20.8.2933-2940.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caamano J.H., Perez P., Lira S.A., Bravo R. Constitutive expression of Bc1-3 in thymocytes increases the DNA binding of NF-κB1 (p50) homodimers in vivo . Mol. Cell. Biol. 1996;16:1342–1348. doi: 10.1128/mcb.16.4.1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolan G.P., Fujita T., Bhatia K., Huppi C., Liou H.C., Scott M.L., Baltimore D. The bcl-3 proto-oncogene encodes a nuclear IκB-like molecule that preferentially interacts with NF-κB p50 and p52 in a phosphorylation-dependent manner. Mol. Cell. Biol. 1993;13:3557–3566. doi: 10.1128/mcb.13.6.3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz E.M., Krimpenfort P., Berns A., Verma I.M. Immunological defects in mice with a targeted disruption in Bcl-3. Genes Dev. 1997;11:187–197. doi: 10.1101/gad.11.2.187. [DOI] [PubMed] [Google Scholar]
- Franzoso G., Carlson L., Scharton-Kersten T., Shores E.W., Epstein S., Grinberg A., Tran T., Shacter E., Leonardi A., Anver M. Critical roles for the Bcl-3 oncoprotein in T cell-mediated immunity, splenic microarchitecture, and germinal center reactions. Immunity. 1997;6:479–490. doi: 10.1016/s1074-7613(00)80291-5. [DOI] [PubMed] [Google Scholar]
- Bendall H.H., Sikes M.L., Ballard D.W., Oltz E.M. An intact NF-κB signaling pathway is required for maintenance of mature B cell subsets. Mol. Immunol. 1999;36:187–195. doi: 10.1016/s0161-5890(99)00031-0. [DOI] [PubMed] [Google Scholar]
- Lam K.P., Kuhn R., Rajewsky K. In vivo ablation of surface immunoglobulin on mature B cells by inducible gene targeting results in rapid cell death. Cell. 1997;90:1073–1083. doi: 10.1016/s0092-8674(00)80373-6. [DOI] [PubMed] [Google Scholar]
- Khan W.N., Alt F.W., Gerstein R.M., Malynn B.A., Larsson I., Rathbun G., Davidson L., Muller S., Kantor A.B., Herzenberg L.A. Defective B cell development and function in Btk-deficient mice. Immunity. 1995;3:283–299. doi: 10.1016/1074-7613(95)90114-0. [DOI] [PubMed] [Google Scholar]
- Kerner J.D., Appleby M.W., Mohr R.N., Chien S., Rawlings D.J., Maliszewski C.R., Witte O.N., Perlmutter R.M. Impaired expansion of mouse B cell progenitors lacking Btk. Immunity. 1995;3:301–312. doi: 10.1016/1074-7613(95)90115-9. [DOI] [PubMed] [Google Scholar]
- Pappu R., Cheng A.M., Li B., Gong Q., Chiu C., Griffin N., White M., Sleckman B.P., Chan A.C. Requirement for B cell linker protein (BLNK) in B cell development. Science. 1999;286:1949–1954. doi: 10.1126/science.286.5446.1949. [DOI] [PubMed] [Google Scholar]
- Jumaa H., Wollscheid B., Mitterer M., Wienands J., Reth M., Nielsen P.J. Abnormal development and function of B lymphocytes in mice deficient for the signaling adaptor protein SLP-65. Immunity. 1999;11:547–554. doi: 10.1016/s1074-7613(00)80130-2. [DOI] [PubMed] [Google Scholar]
- Suzuki H., Terauchi Y., Fujiwara M., Aizawa S., Yazaki Y., Kadowaki T., Koyasu S. Xid-like immunodeficiency in mice with disruption of the p85α subunit of phosphoinositide 3-kinase. Science. 1999;283:390–392. doi: 10.1126/science.283.5400.390. [DOI] [PubMed] [Google Scholar]
- Fruman D.A., Snapper S.B., Yballe C.M., Davidson L., Yu J.Y., Alt F.W., Cantley L.C. Impaired B cell development and proliferation in absence of phosphoinositide 3-kinase p85α. Science. 1999;283:393–397. doi: 10.1126/science.283.5400.393. [DOI] [PubMed] [Google Scholar]
- Wang D., Feng J., Wen R., Marine J.C., Sangster M.Y., Parganas E., Hoffmeyer A., Jackson C.W., Cleveland J.L., Murray P.J. Phospholipase Cγ2 is essential in the functions of B cell and several Fc receptors. Immunity. 2000;13:25–35. doi: 10.1016/s1074-7613(00)00005-4. [DOI] [PubMed] [Google Scholar]
- Hashimoto A., Takeda K., Inaba M., Sekimata M., Kaisho T., Ikehara S., Homma Y., Akira S., Kurosaki T. Cutting edgeessential role of phospholipase C-γ2 in B cell development and function. J. Immunol. 2000;165:1738–1742. doi: 10.4049/jimmunol.165.4.1738. [DOI] [PubMed] [Google Scholar]
- Bajpai U.D., Zhang K., Teutsch M., Sen R., Wortis H.H. Bruton's tyrosine kinase links the B cell receptor to nuclear factor κB activation. J. Exp. Med. 2000;191:1735–1744. doi: 10.1084/jem.191.10.1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petro J.B., Rahman S.M., Ballard D.W., Khan W.N. Bruton's tyrosine kinase is required for activation of IκB kinase and nuclear factor κB in response to B cell receptor engagement. J. Exp. Med. 2000;191:1745–1754. doi: 10.1084/jem.191.10.1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold M.R., Scheid M.P., Santos L., Dang-Lawson M., Roth R.A., Matsuuchi L., Duronio V., Krebs D.L. The B cell antigen receptor activates the Akt (protein kinase B)/glycogen synthase kinase-3 signaling pathway via phosphatidylinositol 3-kinase. J. Immunol. 1999;163:1894–1905. [PubMed] [Google Scholar]
- Ozes O.N., Mayo L.D., Gustin J.A., Pfeffer S.R., Pfeffer L.M., Donner D.B. NF-κB activation by tumour necrosis factor requires the Akt serine-threonine kinase. Nature. 1999;401:82–85. doi: 10.1038/43466. [DOI] [PubMed] [Google Scholar]