

Abstract
The promyelocytic leukemia retinoic acid receptor α (PMLRARα) chimeric protein is associated with acute promyelocytic leukemia (APL). PMLRARα transgenic mice develop leukemia only after several months, suggesting that PMLRARα does not by itself confer a fully malignant phenotype. Suppression of apoptosis can have a central role in tumorigenesis; therefore, we assessed whether BCL-2 influenced the ability of PMLRARα to initiate leukemia. Evaluation of preleukemic animals showed that whereas PMLRARα alone modestly altered neutrophil maturation, the combination of PMLRARα and BCL-2 caused a marked accumulation of immature myeloid cells in bone marrow. Leukemias developed more rapidly in mice coexpressing PMLRARα and BCL-2 than in mice expressing PMLRARα alone, and all mice expressing both transgenes succumbed to leukemia by 7 mo. Although both preleukemic, doubly transgenic mice and leukemic animals had abundant promyelocytes in the bone marrow, only leukemic mice exhibited thrombocytopenia and dissemination of immature cells. Recurrent gain of chromosomes 7, 8, 10, and 15 and recurrent loss of chromosome 2 were identified in the leukemias. These chromosomal changes may be responsible for the suppression of normal hematopoiesis and dissemination characteristic of the acute leukemias. Our results indicate that genetic changes that inhibit apoptosis can cooperate with PMLRARα to initiate APL.
Keywords: leukemia, myeloid/leukemia, promyelocytic, acute/leukopoiesis/PML protein/receptors, retinoic acid
Introduction
Many genetic alterations have been associated with acute myeloid leukemia (AML) 1 2. Nevertheless, our understanding of the combinations of changes sufficient to fully transform myeloid cells remains limited. The t(15;17)(q22;q11.2) chromosomal translocation is closely associated with human acute promyelocytic leukemia (APL; reference 3). This translocation fuses the gene encoding promyelocytic leukemia (PML), a protein that limits proliferation and induces apoptosis, with the gene encoding the retinoic acid receptor α (RARα), a hormone receptor that represses transcription in the absence of retinoic acid and activates transcription in response to this ligand. A PMLRARα fusion protein is expressed in almost all cases of APL, and this protein can initiate acute leukemias with promyelocytic features when expressed in mice 4 5 6.
PMLRARα is thought to contribute to leukemogenesis primarily by inhibiting neutrophilic differentiation. Retinoids can induce differentiation of myeloid and other cell types 7. PMLRARα inhibits differentiation of myeloid cell lines 8 9 10 11, and all-trans retinoic acid (tRA) binding to PMLRARα results in transcriptional activation and neutrophilic differentiation 3 12 13 14. Nevertheless, mice expressing PMLRARα exhibited only mild abnormalities before the appearance of acute leukemia (which arises with limited penetrance only after several months; references 4–6), suggesting that additional genetic changes cooperate with PMLRARα to block differentiation.
PMLRARα may also contribute to leukemogenesis by enhancing the survival of immature myeloid cells. Whereas PMLRARα induces apoptosis of nonhematopoietic and some hematopoietic cell lines 15, PMLRARα confers increased resistance to apoptosis in myeloid cell lines in which its expression is tolerated 8 16. PMLRARα also enhances survival of primary myeloid CFU in response to proapoptotic stimuli 17 and hematopoietic progenitor cells after growth factor deprivation 18. PMLRARα-mediated suppression of apoptosis might influence the ability of this fusion protein to initiate leukemia.
BCL-2 protects cells from apoptosis by inhibiting the activation of caspases necessary to execute a cell death program 19. In follicular lymphoma, BCL-2 is overexpressed as a result of a t(11;14)(q32;q21) chromosomal translocation. BCL-2 expression has also been observed in AMLs, including leukemic cells of some APL patients, and such expression has been associated with poor prognosis 20 21 22 23 24 25 26. Thus, BCL-2, or related proteins, may play a role in the pathogenesis of a subset of APL and might influence the response of patients to therapy.
Transgenic mice expressing PMLRARα under the control of the MRP8 promoter were bred with MRP8-BCL-2 transgenic mice to assess whether BCL-2 could cooperate with PMLRARα to initiate leukemia. We observed that coexpression of BCL-2 and PMLRARα caused a marked accumulation of immature myeloid cells. Furthermore, BCL-2 accelerated the development of acute leukemia in mice that express PMLRARα.
Materials and Methods
Mice.
Mice were bred and maintained at the University of California at San Francisco and their care was in accordance with University of California at San Francisco guidelines. MRP8-PMLRARα transgenic mice 4 were bred with MRP8-BCL-2 transgenic mice 27 to generate the litters of control, singly transgenic, and doubly transgenic mice used for experiments.
Preparation of Tissues for Analysis.
Blood was obtained from anesthetized animals by venipuncture of the retroorbital venous plexus. Bone marrow was obtained by flushing HBSS through mouse long bones. Blood smears and bone marrow smears were prepared according to standard hematological techniques. Sternums were decalcified for 2–3 h (formic acid 11%, formaldehyde 8%). Sternums, livers, spleens, and kidneys were fixed overnight in buffered formalin and embedded in paraffin before sectioning.
Peripheral Blood Counts and Bone Marrow Differential Counts. Blood counts of nonleukemic, preleukemic, and some leukemic mice were obtained on automated hematology analyzers running veterinary software (Cell-Dyne3500, Abbott; Hemavet 850, CDC Technologies). Blood counts of leukemias that arose early in the study were obtained on a Technicon H-3 automated hematology analyzer running the standard software used for clinical analysis. Peripheral blood differential white blood cell counts (total of 200 cells each) and bone marrow differential counts (total of 400 cells each) were performed on stained smears as described 28.
Analysis of Proliferation.
Bone marrow cells were fixed in ice-cold 70% ethanol for 30 min. After two washes in PBS, cells were resuspended in PBS containing 50 μg/ml propidium iodide (Boehringer) and 10 μg/ml RNase (Sigma-Aldrich). After a 30-min incubation, cells were analyzed using a FACScan™ flow cytometer (Becton Dickinson) and cell cycle distribution of 10,000 events was assessed using Modfit software (Becton Dickinson).
Analysis of Apoptosis.
Immediately after harvest, bone marrow cells were washed twice in PBS with 2% heat-inactivated fetal bovine serum (wash buffer), and cells were then stained with FITC-conjugated antibodies to CD18 or an isotype control (BD PharMingen) for 20 min on ice. Then, cells were washed and stained for 30 min in the dark, on ice, with 20 μg/ml 7-amino actinomycin D (7-AAD; Calbiochem). After being washed twice, cells were resuspended in wash buffer containing 10 μg/ml actinomycin D (Calbiochem). Analysis was carried out using a FACScan™ flow cytometer within 1 h of staining. 50,000 events were analyzed using CELLQuest™ software (Becton Dickinson). CD18+ cells were gated and three populations were identified: 7-AADlow live nonapoptotic cells; 7-AADmoderate apoptotic cells; and 7-AADbright dead cells 29.
Methylcellulose Cultures of Bone Marrow.
As described previously 28, bone marrow cells were cultured in duplicate in 35-mm petri dishes in Methocult M3230 methylcellulose medium (StemCell Technologies Inc.) supplemented with either 50 U/ml of G-CSF (Boehringer) or 20 ng/ml GM-CSF (StemCell Technologies Inc.). 1-ml cultures contained 50,000 viable bone marrow cells. Cultures were examined at 7 d. Colonies were counted on an inverted light microscope. Duplicate plates were averaged. Cells were harvested, pelleted, washed once in HBSS, and counted before preparation of cytospins. Differential cell counts (total of 200 cells each) were performed on stained cytospins to assess percentages and calculate absolute numbers of neutrophilic cells. To assess the effects of growth factor deprivation, cells were placed into methylcellulose medium with or without GM-CSF and incubated for 24 h before plating matched cultures in the presence of GM-CSF. Results were expressed as a percentage (CFU of growth factor–deprived culture/CFU of matched culture without growth factor deprivation × 100).
Transplantation of Bone Marrow.
Essentially as described previously 4, total bone marrow isolated from tibias and femurs of 4–6-wk-old mice was divided for intravenous injection into 6–10 recipient mice (∼1–3 × 106 nucleated marrow cells/recipient). 6–12-wk-old FVB/N mice were prepared for transplantation by cesium irradiation totaling 10 Gy, divided into two doses 3–6 h apart. The results shown in Fig. 3 include five recipients of PMLRARα/BCL-2 doubly transgenic marrow that was 0.75 FVB/N strain, 0.25 BA strain; six recipients of matched PMLRARα singly transgenic marrow (0.75 FVB/N, 0.25 BA); eight recipients of PMLRARα/BCL-2 doubly transgenic marrow (0.98 FVB/N, 0.02 BA); and 21 recipients of PMLRARα singly transgenic marrow (1.0 FVB/N). The difference in survival of recipients of doubly transgenic and singly transgenic marrow was statistically significant whether the data were analyzed as a whole or as subgroups.
Figure 3.

BCL-2 cooperates with PMLRARα to initiate leukemia. Lethally irradiated nontransgenic FVB/N mice were reconstituted with bone marrow from PMLRARα or PMLRARα/BCL-2 transgenic mice. Kaplan-Meier curves are shown. All PMLRARα/BCL-2 mice died of leukemia. Mortality among PMLRARα mice from causes other than leukemia was censored at the date of death. PMLRARα/BCL-2, n = 13; PMLRARα, n = 27; P < 0.00001.
Comparative Genomic Hybridization.
Comparative genomic hybridization was performed as described previously 30 31 at the Molecular Cytogenetics Core Facility, Comprehensive Cancer Center, University of California at San Francisco.
Retinoic Acid/Arsenic Treatment.
As described previously, mice were treated by subcutaneous implantation of a 21-d release pellet containing 5 mg tRA or placebo (Innovative Research of America; reference 4), and/or by daily intraperitoneal injection of As2O3 (arsenic) at a dose of 5 μg/g body wt 32.
Statistical Analysis.
The software program Statistica (v.4.1 for Macintosh) was used to prepare Kaplan-Meier curves and to compare survival using the Wilcoxon test. Other statistical analyses were performed with Excel (v.5.0/95) using the Student's t test, two-tailed distribution, and unequal variance. Comparisons are with control animals unless otherwise noted.
Results
BCL-2 Cooperates with PMLRARα to Impair Neutrophil Maturation and Expand Myeloid Progenitors.
Previously, we developed a mouse model of APL by expressing PMLRARα in the neutrophilic cells of transgenic mice 4. PMLRARα had a modest effect on neutrophil differentiation but did not initially arrest maturation. Some of the PMLRARα transgenic mice went on to develop acute leukemia with promyelocytic features. The latency and limited penetrance of the leukemic phenotype indicated that in this mouse model, additional events were required to cooperate with PMLRARα in the genesis of acute leukemia. In our effort to identify genetic changes that can cooperate with PMLRARα to induce leukemia, we considered previous studies indicating that PMLRARα induces apoptosis of many cell lines 15 and evidence that apoptosis can be a critical regulator of malignant transformation in vivo 33.
To assess the hypothesis that suppression of apoptosis cooperates with PMLRARα in leukemogenesis, we crossed MRP8-PMLRARα transgenic mice with MRP8-BCL-2 transgenic mice 27. Previous studies of MRP8-BCL-2 transgenic mice indicated that (a) the BCL-2 transgene inhibited apoptosis of neutrophils but not their engulfment by macrophages 27, (b) the transgene inhibited apoptosis of monocytes as well as rescued macrophages and partially reversed osteopetrosis in op/op mice 34, (c) the transgene inhibited Fas-induced apoptosis of myeloid progenitors 35, and (d) in the context of FAS l pr/lpr mice, the BCL-2 transgene contributed to the development of a lethal proliferation of myeloid cells 35. These results showed that the BCL-2 transgene was able to suppress apoptosis of myeloid cells and contribute to myeloid neoplasms.
MRP8-BCL-2 transgenic mice were mated with MRP8-PMLRARα mice to examine the effects of the combination of transgenes on hematopoiesis. In the peripheral blood, total leukocyte counts and hemoglobin levels of 3–5-wk-old transgenic mice were normal (Fig. 1 A). Singly transgenic mice had normal platelet counts, whereas those of doubly transgenic mice were elevated. PMLRARα transgenic mice had normal peripheral blood neutrophil counts, but mice expressing BCL-2 alone or in combination with PMLRARα were neutropenic (Fig. 1 B). Decreased neutrophil counts were previously observed in MRP8-BCL-2 transgenic mice 34. In the bone marrow, both PMLRARα and BCL-2 singly transgenic mice exhibited a shift from mature neutrophilic cells to immature neutrophilic cells with a small increase in promyelocytes. Doubly transgenic mice had very few neutrophilic cells beyond the promyelocyte stage (Fig. 1 C). Whereas the bone marrow of singly transgenic mice looked modestly different from control mice (Fig. 2A–C), doubly transgenic bone marrow was filled with immature cells and morphologically resembled acute leukemia (Fig. 2 D). However, unlike leukemic animals, the organs of these doubly transgenic mice had a near normal appearance (Fig. 2E–J).
Figure 1.



BCL-2 cooperates with PMLRARα to impair neutrophil production in vivo. (A) Peripheral blood. White blood cell count (WBC, 1,000/μL), hemoglobin (HGB, g/dL), and platelet count (PLT, 100,000/μL) are shown. Littermate controls, n = 6; PMLRARα, n = 5; BCL-2, n = 4; and PMLRARα/BCL-2, n = 6. Platelet count was increased in PMLRARα/BCL-2 mice (P = 0.002). (B) Peripheral blood. Absolute cell counts (1,000/μL). Littermate controls, n = 6; PMLRARα, n = 4; BCL-2, n = 4; and PMLRARα/BCL-2, n = 6. Neutrophils were decreased in BCL-2 and PMLRARα/BCL-2 mice (P = 0.01). Monocytes were increased in PMLRARα/BCL-2 mice (P = 0.02). (C) Bone marrow. Percentages of nucleated cells were derived from 400 cell differential counts of Wright's Giemsa-stained bone marrow smears. Blast+Pro, blasts and promyelocytes; Imm Neut, neutrophilic myelocytes and metamyelocytes; Mat Neut, neutrophilic band, mature ring, and polymorphonuclear forms; Lymph, lymphocytes; Eosin, eosinophils. Littermate controls, n = 6; PMLRARα, n = 4; BCL-2, n = 4; PMLRARα/BCL-2, n = 6. In PMLRARα mice, blasts plus promyelocytes were increased (P = 0.01). In BCL-2 mice, blasts plus promyelocytes were increased (P = 0.02), immature neutrophils were increased (P = 0.04), and mature neutrophils were decreased (P = 0.02). In PMLRARα/BCL-2 mice, blasts plus promyelocytes were increased (P = 0.001), immature neutrophils were decreased (P = 0.003), mature neutrophils were decreased (P = 0.0004), and lymphocytes were decreased (P = 0.002). Graphs depict arithmetic means ± SD.
Figure 2.
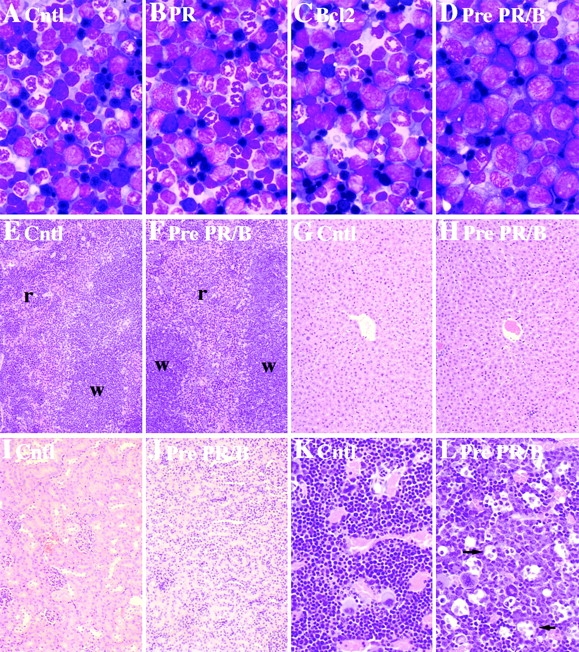
PMLRARα and BCL-2 transgenes cooperate to arrest neutrophil differentiation. (A–D) Bone marrow morphology: (A) Control (Cntl); (B) preleukemic PMLRARα (PR); (C) BCL-2; (D) preleukemic PMLRARα/BCL2 (Pre PR/B). Wright's Giemsa stain; original magnifications: ×330. Immature cells are not disseminated in preleukemic doubly transgenic mice. (E–F) Spleen. Red pulp (r); white pulp (w). (G–H) Liver. (I–J) Kidney. (E, G, and I) Control. (F, H, and J) Preleukemic PMLRARα/BCL-2. Hematoxylin and eosin; original magnifications: ×66. Tingible body macrophages are increased in preleukemic PMLRARα/BCL-2 mice. (K and L) Bone marrow histology. (K) Control. (L) Preleukemic PMLRARα/BCL-2. Arrows indicate selected macrophages. Hematoxylin and eosin; original magnifications: ×165.
We also assessed proliferation and apoptosis in the transgenic mice (Table ). PMLRARα did not discernibly alter the number of proliferating or apoptotic cells detected in bone marrow. BCL-2 increased the number of proliferating cells, and the combination of transgenes increased both proliferation and apoptotic cell numbers. Bone marrow histology provided additional evidence of increased cell turnover in doubly transgenic bone marrow; compared with control, increased numbers of tingible body macrophages were present (Fig. 2K and Fig. L). The increased proliferation in doubly transgenic mice is consonant with the presence of increased promyelocytes because promyelocytes, unlike mature neutrophils, are mitotically active. The increase in apoptotic cells likely reflects the fact that in some settings, such as growth factor deprivation, BCL-2 delays but does not prevent cell death 36. The combination of BCL-2 and PMLRARα caused an accumulation of numerous immature myeloid cells in the bone marrow. These cells could proliferate, but very few differentiated into mature neutrophils. BCL-2 appeared unable to block indefinitely the death of the resulting expanded promyelocyte population.
Table 1.
Bone Marrow Proliferation and Apoptosis in Nonleukemic Animals
| Transgenic mice | Proliferation | Apoptosis |
|---|---|---|
| Control | 24.6 ± 3.4 | 3.02 ± 1.08 |
| PMLRARα | 23.7 ± 2.1 NS | 3.07 ± 1.31 NS |
| BCL-2 | 31.0 ± 3.1 P = 0.03 | 5.98 ± 2.87 NS |
| PMLRARα/BCL-2 | 36.9 ± 1.3 P = 0.003 | 10.11 ± 1.27 P = 0.0002 |
Bone marrows of mice were harvested and analyzed without prior culture to assess the effects of the transgenes on proliferation and apoptosis in vivo. Data are shown as arithmetic means ± SD. For each group, n = 4. P values for comparisons with control are shown. NS, not significant.
Methylcellulose cultures were performed to examine the effects of the transgenes on myeloid colony formation in vitro (Table ). PMLRARα did not alter the number of colonies formed in GM-CSF or G-CSF. In contrast, BCL-2 resulted in increased colonies, an effect that was statistically significant for doubly transgenic bone marrow. The effect of growth factor deprivation on CFU was also assessed for singly transgenic mice. Matched GM-CSF cultures of control, PMLRARα, and BCL-2 bone marrow cells were plated with or without 24 h of growth factor deprivation. Both PMLRARα and BCL-2 resulted in a trend towards enhanced survival (control 61 ± 7%, n = 6; PMLRARα 77 ± 17%, n = 6, P = 0.07; BCL-2 95 ± 14%, n = 4, P = 0.01). With regard to differentiation, PMLRARα, BCL-2, and PMLRARα/BCL-2 transgenic bone marrows gave rise to a decreased percentage of neutrophils, accompanied by increased immature and monocytic cells. Of note, although the combination of BCL-2 and PMLRARα caused profound neutropenia in vivo, neutrophil maturation was not completely blocked in bone marrow cultures. In GM-CSF, absolute numbers of neutrophils in BCL-2 and PMLRARα/BCL-2 cultures were normal because increased colony numbers offset the relative decrease in neutrophil maturation. However, in G-CSF absolute numbers of neutrophils were decreased. These results suggest that PMLRARα and BCL-2 impeded neutrophil differentiation of myeloid precursors. Altogether, our observations of doubly transgenic animals showed that BCL-2 expanded myeloid progenitors, allowed increased numbers of cells to proliferate, and enhanced the ability of PMLRARα to inhibit neutrophil differentiation.
Table 2.
Methylcellulose Cultures of Bone Marrow of Nonleukemic Animals
| Colonies | Immature cells | Monocytes/ macrophages | Neutrophilic cells | Total cells | Neutrophilic cells | |
|---|---|---|---|---|---|---|
| n/ml | % | % | % | 1,000/ml | 1,000/ml | |
| GM-CSF | ||||||
| Control | 96 ± 25 | 9.7 ± 4.6 | 53.6 ± 2.9 | 33.8 ± 3.4 | 149 ± 39 | 51.1 ± 16.8 |
| PMLRARα | 106 ± 16 | 9.3 ± 2.3 | 76.4 ± 7.1, P = 0.001 | 10.8 ± 6.5,P < 0.001 | 192 ± 72 | 17.4 ± 6.3,P = 0.008 |
| BCL-2 | 179 ± 78 | 13.0 ± 1.4 | 61.7 ± 10.6 | 19.3 ± 7.8,P = 0.03 | 323 ± 182 | 74.7 ± 61.1 |
| PMLRARα/BCL-2 | 354 ± 147,P = 0.007 | 12.4 ± 4.8 | 77.6 ± 6.4, P < 0.001 | 7.7 ± 5.6,P < 0.001 | 732 ± 428, P = 0.02 | 45.7 ± 41.2 |
| G-CSF | ||||||
| Control | 40.0 ± 18 | 22.1 ± 9.4 | 37.3 ± 10.9 | 40.4 ± 17.5 | 16.5 ± 8.4 | 5.7 ± 2.2 |
| PMLRARα | 36.0 ± 10 | 30.7 ± 12 | 50.5 ± 11.8 | 17 ± 5.5,P = 0.04 | 7.7 ± 7.5 | 1.1 ± 0.8,P = 0.007 |
| BCL-2 | 70.0 ± 20 | 32.6 ± 13.1 | 57.5 ± 21.3 | 9.3 ± 9.1,P = 0.01 | 21.8 ± 8.3 | 1.8 ± 1.3,P = 0.01 |
| PMLRARα/BCL-2 | 114.0 ± 45, P = 0.009 | 40.0 ± 26 | 55.8 ± 25.4 | 3.5 ± 2.7,P = 0.009 | 51.7 ± 39.7 | 2.1 ± 3,P = 0.05 |
Bone marrows of mice were harvested and cultured in duplicate at 50,000 cells/ml culture in the presence of either GM-CSF or G-CSF. On day 7, colonies/plate were counted, cells were harvested, pooled from duplicate plates, and counted. Immature cells, monocytes/macrophages, and neutrophilic cells were identified among 200 cells counted on Wright's Giemsa-stained cytospins of pooled cells. Data are shown as arithmetic means ± SD. For each group, n = 4–6. P values (excluding values >0.05) for comparisons with control are shown.
BCL-2 Cooperates with PMLRARα to Initiate Leukemia.
The experiments described above were performed on matched 3–5-wk-old littermates. We noted that mice that inherited both transgenes were smaller than littermates at 3 wk of age, had the skin abnormalities we have seen in our PMLRARα transgenics 4, and died between 4 and 6 wk of age. To assess the degree to which these deaths were due solely to a hematopoietic disorder or in part to nonhematopoietic effects of the transgenes, we harvested bone marrow from doubly transgenic mice and used this marrow to reconstitute lethally irradiated nontransgenic animals. Marrow harvested from PMLRARα singly transgenic mice was similarly transplanted. Transplantation of either PMLRARα transgenic or PMLRARα/BCL-2 doubly transgenic bone marrow rescued recipient mice from lethal irradiation. Recipients remained healthy for 3 mo or more after transplantation, suggesting that the early deaths of untransplanted doubly transgenic mice were, at least in part, caused by nonhematopoietic effects of the transgenes.
Mice reconstituted with PMLRARα or PMLRARα/BCL-2 bone marrow were observed for the development of leukemia. Leukemias developed more rapidly in mice coexpressing PMLRARα and BCL-2 than in mice expressing PMLRARα alone (median time to leukemia 127 vs. 257 d; Fig. 3). All mice reconstituted with doubly transgenic bone marrow succumbed to leukemia by 196 d of age. Similar to the leukemias that arose from singly transgenic PMLRARα mice, leukemias arising from PMLRARα/BCL-2 doubly transgenic mice were characterized by anemia and thrombocytopenia, and by bone marrow that was effaced by cells at the blast-promyelocyte stage of differentiation (Fig. 4 and Fig. 5 A). Although there was a trend towards increased peripheral white blood cell counts in PMLRARα/BCL-2 leukemias, this trend was not statistically significant (P > 0.05).
Figure 4.


PMLRARα and PMLRARα/BCL-2 leukemic mice are anemic, thrombocytopenic, and have bone marrow that is filled with promyelocytes. (A) Peripheral blood. Results are displayed as in the legend to Fig. 1 A. Adult controls, n = 7; PMLRARα leukemia, n = 10; PMLRARα/BCL-2 leukemia, n = 10. White blood cell count (WBC) was not significantly different among the groups. Hemoglobin (HGB) and platelet count (PLT) were reduced in leukemic mice (P < 0.00001). (B) Bone marrow. Results are displayed as in the legend to Fig. 1 C. Adult controls, n = 7; PMLRARα leukemia, n = 6; PMLRARα/BCL-2 leukemia, n = 6. All values were significantly different in leukemic mice compared with controls (P < 0.00001 to P = 0.002), but were not significantly different between PMLRARα and PMLRARα/BCL-2 leukemias.
Figure 5.
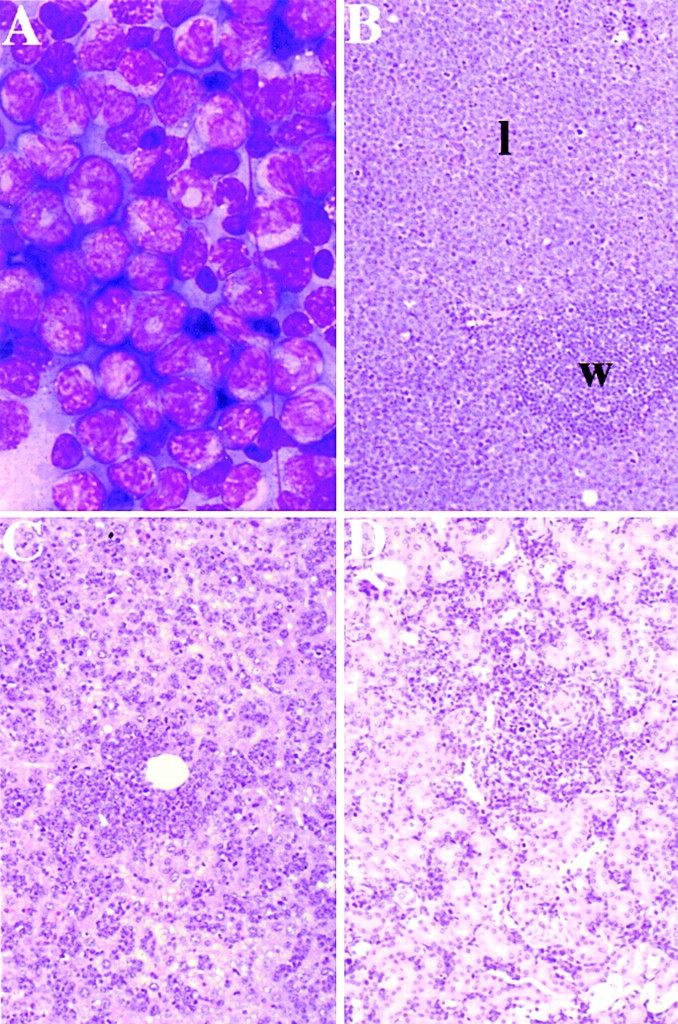
Leukemic transformation is characterized by dissemination of immature cells. Leukemic PMLRARα/BCL-2. (A) Bone marrow morphology. Wright's Giemsa stain; original magnification: ×330. (B) Spleen. l, leukemic infiltrate. (C) Liver. (D) Kidney. (B–D) Hematoxylin and eosin; original magnifications: ×66.
Dissemination Appears to Be a Central Characteristic of AML.
PMLRARα/BCL-2 transgenic mice had bone marrow that morphologically resembled acute leukemia (Fig. 2 D and 5 A), yet mice whose bone marrow expressed both transgenes did not become ill for a period of 3–7 mo. Therefore, we sought to identify differences between preleukemic and overtly leukemic, doubly transgenic animals. Bone marrow differential counts showed very low numbers of maturing neutrophils in preleukemic and leukemic animals (Fig. 1 C, PMLRARα/BCL-2; Fig. 4 B). However, in the leukemias suppression of normal hematopoiesis was evident: there was a marked decrease of nucleated erythroid cells in the bone marrow, and in contrast to preleukemic animals, the leukemic mice were anemic and thrombocytopenic (Fig. 1 A and 4 A). Furthermore, in the leukemic mice, immature neutrophilic cells were disseminated. Small numbers of promyelocytes were present in the peripheral blood, and the spleens, livers, and kidneys in leukemic mice were enlarged and contained infiltrates of leukemic cells (Fig. 5B–D). The existence of a prolonged preleukemic state in PMLRARα/BCL-2 mice suggests that the suppression of normal hematopoiesis and dissemination of leukemic cells observed in AML are not a direct consequence of arrested differentiation.
Acquired Chromosomal Abnormalities Are Common in Murine APL.
Preleukemic, doubly transgenic mice initially produced adequate red blood cells and platelets and did not become ill for 3–7 mo. These findings indicated that the combination of BCL-2 and PMLRARα was sufficient to initiate but not complete leukemic transformation. Therefore, we sought to identify additional genetic changes in our leukemias. Comparative genomic hybridization was performed on 30 murine APLs, including 20 PMLRARα leukemias and 10 PMLRARα/BCL-2 leukemias. Chromosomal copy number abnormalities were observed in 17/20 PMLRARα leukemias and 9/10 PMLRARα/BCL-2 leukemias. Gain of chromosomes 7, 8, 10, and 15 and loss of chromosome 2 occurred in three or more samples including both PMLRARα and PMLRARα/BCL2 leukemias (Fig. 6). Losses of the X chromosome from female samples and the Y chromosome from male samples were also observed. These results identify chromosomal regions whose loss or gain may complete the process of leukemic transformation initiated by PMLRARα.
Figure 6.

Murine APL is characterized by karyotypic abnormalities. Results of comparative genomic hybridization of 30 APLs (11 female, 19 male) are shown, including 20 PMLRARα and 10 PMLRARα/BCL-2 leukemias. Ideograms of mouse chromosomes are numbered. Each bar to the left of chromosomes indicates a region lost in one leukemic sample. Each bar to the right of chromosomes indicates a region gained in one sample. Each wide bar represents chromosomal gain in 10 samples. Losses of the X chromosome were seen in female samples.
Murine APL Expressing BCL-2 Responds to Retinoic Acid and Arsenic Trioxide.
We assessed whether the expression of BCL-2 influenced the ability of tRA therapy to cause differentiation and prolong survival of mice with APL. Nontransgenic mice received intravenous injections of 5 × 106 spleen cells from leukemia 2995A4, which expresses both PMLRARα and BCL-2 transgenes. In one experiment, 32 d after the injection of leukemic cells, three mice were treated with placebo and three with tRA. The placebo-treated mice died on days 3 and 5, and five died after initiation of therapy, whereas tRA-treated mice died on days 17, 25, and 33. The difference in survival was significant (P = 0.04). In a second experiment, groups of six mice were treated 14 d after the injection of leukemic cells; again, tRA prolonged survival (Fig. 7 A). The prolongation in survival we observed (median 42 d, range 11–73) was comparable to the increase in survival seen in recipients of a PMLRARα leukemia that was similarly treated (median 41 d, range 33–175; reference 32). tRA induced neutrophilic differentiation of leukemic blasts coexpressing PMLRARα and BCL-2 (Fig. 7 B), but did not abrogate expression of the transgenes as assessed by Western blot analysis (data not shown).
Figure 7.
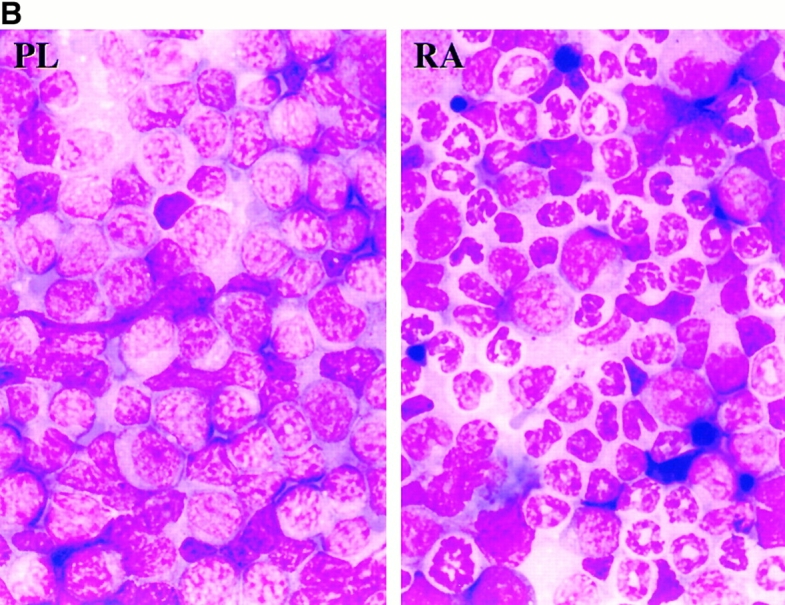
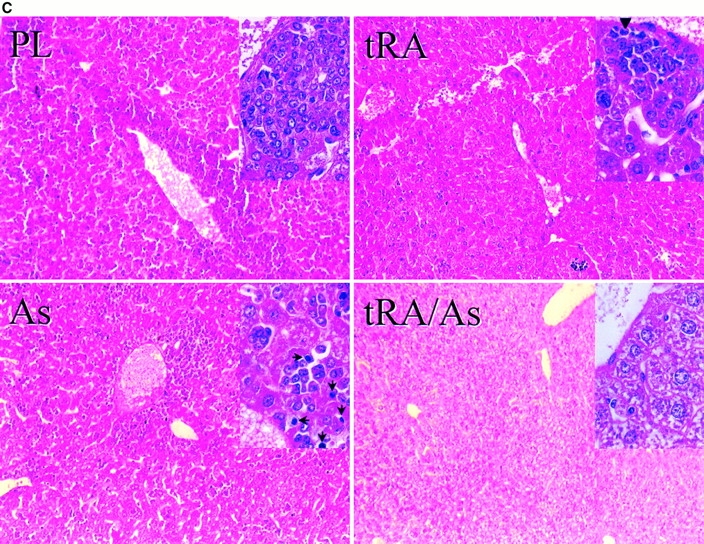

APL expressing PMLRARα and BCL-2 is responsive to therapy. (A and B) Unirradiated nontransgenic FVB/N mice received 5 × 106 PMLRARα/BCL-2 leukemic cells by intravenous injection. (A) 14 d after injection of leukemic cells, 5 mg placebo (n = 6) or tRA (n = 6) pellets (21-d release) were implanted into mice. Kaplan-Meier curves are shown. P = 0.02. (B) Bone marrow morphology. Recipients of PMLRARα/BCL-2 leukemic cells were treated with placebo or tRA when ill. PL, placebo-treated; RA, tRA-treated, day 11. Wright's Giemsa stain; original magnification: ×455. (C) FVB/N mice received 107 PMLRARα/BCL-2 leukemic cells. 15 d after injection, mice were treated with tRA and/or arsenic for 4 d. Representative sections of livers are shown. PL, placebo: a leukemic infiltrate is present in the periportal and parenchymal regions of the liver; inset shows immature myeloid cells; tRA, retinoic acid: leukemic cells are much reduced; inset shows hepatocytes accompanied by smaller residual leukemic cells, arrowhead indicates a maturing neutrophil. As, arsenic; leukemic infiltrate present; arrows (inset) indicate condensed nuclei of apoptotic cells. tRA/As, retinoic acid/arsenic: leukemic cells are not seen. Hematoxylin and eosin: original magnifications: ×125; inset ×625.
We also examined whether arsenic and tRA were an effective combination therapy for APL expressing BCL-2. In three experiments, nontransgenic mice received intravenous injections of 107 leukemic 2995A4 cells. After 15 d, mice were treated with tRA and/or arsenic for 4 d, two mice per group, and then killed. Similar to what was previously observed for murine APL that does not express BCL-2 32, arsenic-induced apoptosis and some differentiation, and the combination of arsenic and tRA, caused rapid regression of the leukemia. 4 d of therapy decreased spleen size (control, 0.57, 0.46 g; tRA, 0.21, 0.20 g; arsenic, 0.40, 0.30 g; tRA/arsenic: 0.10, 0.09 g; representative data from one of three experiments). In the liver, tRA caused regression but not the disappearance of disease, and the addition of arsenic eliminated the visible leukemic cell population (Fig. 7 C). These results suggest that APL cells expressing BCL-2 remain sensitive to arsenic plus tRA combination therapy.
Discussion
Expression of BCL-2 in mice that also express the APL-associated PMLRARα fusion protein caused a marked accumulation of immature myeloid cells and accelerated the development of leukemia. Progression from preleukemia to acute leukemia was characterized by suppression of normal hematopoiesis and dissemination of leukemic cells. Additional genetic changes accompanied this transition from abnormal neutrophil maturation to lethal disease.
A Role for Suppression of Apoptosis in APL Pathogenesis.
The possible role of apoptosis in APL pathogenesis has been unclear. Whereas PMLRARα induces cell death in many cell lines, it also inhibits apoptosis of some myeloid cell lines and primary myeloid cells. We anticipated that BCL-2 might cooperate with PMLRARα to initiate leukemia. On the other hand, given the ability of PMLRARα itself to block apoptosis, it was possible that BCL-2 would have little impact on leukemogenesis. We observed that the antiapoptotic protein BCL-2 did cooperate with PMLRARα to initiate leukemia, indicating that genetic changes that suppress apoptosis can cooperate with the PMLRARα fusion in APL.
The MRP8-BCL-2 transgene has been demonstrated to increase survival of myeloid cells (27, 34, 35, and our results). We observed increased CFU and young myeloid forms in bone marrows of BCL-2 and doubly transgenic mice. It is likely that this increase is in part attributable to the ability of BCL-2 to enhance survival of immature myeloid cells. BCL-2 makes cells less dependent on external survival signals. BCL-2 did not, however, permit unlimited survival of immature cells. We speculate that the preleukemic, doubly transgenic cells remained responsive to extracellular signals that limited their expansion to hematopoietic microenvironments. Nevertheless, it appears that by expanding the population of immature myeloid cells in PMLRARα transgenic mice, BCL-2 made it inevitable that additional leukemogenic mutations would accumulate. Expression of BCL-2 therefore promotes promyelocyte transformation but is not sufficient to complete this process. Interestingly, whereas BCL-2 may facilitate the acquisition of genetic changes that induce acute leukemia in PMLRARα transgenic mice, it does not appear to alter the type of chromosomal changes that occur.
Cytogenetic Changes in Murine APL.
The t(15;17) translocation is present as an isolated karyotypic abnormality in the majority of APL patients at presentation. However, additional karyotypic abnormalities are observed in 24–39% of untreated APL patients 37 38 39 40. The fact that a substantial minority of new APL cases have additional cytogenetic changes provides evidence that human APL results from a combination of acquired mutations. The most common additional abnormality is trisomy 8, which is seen in ∼17% of patients at presentation. Other recurrent aberrations include loss of chromosome 9q and isochromosome of the long arm of the derivative chromosome 17. The particular genes through which these additional cytogenetic changes may contribute to leukemia have not been identified.
Some of the changes seen in murine APL correspond to karyotypic abnormalities in human APL. Murine chromosomes 8 and 15 contain regions homologous to human chromosome 8. The area of loss we noted on mouse chromosome 2 contains genes that in humans are located on human chromosome 9q. In the broader context of attempts to understand leukemia pathogenesis in mice, the gain of chromosome 15 and the loss of chromosome 2 have been observed previously. Gibbons et al. 41 observed a common gain of chromosome 15 in B/myeloid leukemias that arose in irradiated Eμ-BCL-2 mice. The authors suggested that the presence of BCL-2 permitted cells damaged by radiation to tolerate chromosomal damage, and the subsequently damaged cells gave rise to the observed leukemias. In our experiments, BCL-2 could have similarly tolerized early myeloid cells to spontaneous genetic changes including a pathogenic gain of chromosome 15. Loss of murine chromosome 2 has been observed in radiation-induced myeloid leukemias in mice (for a review, see reference 42). The region of murine chromosome 2 identified in these leukemias was lost in all four APLs in which we observed copy number changes on chromosome 2. Of interest, another group studying the effects of PMLRARα, under the control of a different promoter in a different strain of mice, has also identified recurrent gain of chromosome 15 and recurrent loss of chromosomes 2, X, and Y in their leukemias 43. The chromosome copy number abnormalities observed in murine APLs may facilitate identification of genetic changes that cooperate with PMLRARα to cause acute leukemia.
BCL-2 Effects on Neutrophil Differentiation.
Although PMLRARα can inhibit differentiation, its effects on neutrophilic differentiation of primary bone marrow cells were modest and most apparent in vitro (Table ). In vivo, BCL-2 cooperated with PMLRARα to block maturation at the promyelocyte stage. Given the central role of BCL-2 family members in regulating cell death, the inhibition of neutrophil differentiation in PMLRARα transgenic mice brought by coexpression of BCL-2 is likely an indirect effect of BCL-2. Enhanced survival of neutrophil precursors may abrogate signals within the bone marrow that normally stimulate promyelocytes to differentiate into mature neutrophils. The finding that mature erythroid cells stimulate the death of erythroid progenitors 44 raises the possibility that interactions between mature and immature myeloid cells play a role in neutrophil maturation. Such interactions could be disrupted by enhanced survival of immature elements. On the other hand, our data are consistent with the speculative idea that BCL-2 may directly influence neutrophil maturation. Our finding that BCL-2 transgenic marrow gave rise to relatively decreased neutrophils in vitro might reflect enhanced production of monocytic cells, but, alternatively, this decrease could reflect a cell autonomous effect of BCL-2 on neutrophil differentiation. In previous studies, BCL-2 did not block tRA-induced differentiation of HL-60 cells 45, but BCL-2 did delay neutrophilic maturation of 32DCl3 cells in response to G-CSF 46. Another finding suggesting that, in some settings, BCL-2 family members might directly influence neutrophil differentiation is the observation that tRA-mediated differentiation of APL cells is accompanied by caspase activation 13. BCL-2 family members might influence differentiation through an activity that is distinct from their effects on cell survival. Such a possibility has precedent: BCL-2 has an effect on proliferation separable from its effect on survival 47.
Multistep Pathogenesis for APL.
The pathogenesis of human APL may parallel that of murine APL. The acquisition of the t(15;17)(q22,q11.2) with resulting expression of PMLRARα could initiate leukemia. Increased expression of BCL-2, or other genetic changes that enhance survival and impair differentiation of the initiated cells, may occur in some patients and facilitate the acquisition of genetic changes that result in acute leukemia. Further study of human APLs could inform our understanding of the role of BCL-2–like proteins in the human disease. Although BCL-2 expression was not seen in one study of APL cases 21, subsequent reports indicated that BCL-2 is expressed in most APLs 22 23 24 25 48. Elucidating whether this BCL-2 expression is because of a genetic lesion in the BCL-2 gene or a downstream result of other lesions should be of value, as should assessing the expression of other BCL-2 family members in APL.
It is notable that the t(15;17) translocation is likely to have effects that are more pronounced than those caused by PMLRARα alone. Similar to BCL-2 expression, loss of Pml, a genetic event that suppresses apoptosis, facilitated the development of acute leukemia in CathepsinG-PMLRARα transgenic mice 49. The reciprocal fusion protein, RARαPML, also increased the penetrance of leukemia in mice expressing PMLRARα 50. Given the fact that the incidence of t(15;17)-associated myeloid leukemia does not increase with age 51, we favor a model in which the translocation initiates the disease and additional required genetic changes may accumulate in a relatively brief period of time.
BCL-2 Does Not Block the Response to APL Therapy.
Downregulation of BCL-2 accompanies the differentiation of APL cells that is induced by tRA 52, and this downregulation may be important for some effects of tRA 13. We found that overexpression of BCL-2 did not block the differentiative effect of tRA. Similarly, BCL-2 might have blocked the therapeutic effect of arsenic because arsenic is thought to induce remissions primarily by stimulating apoptosis of the APL blasts 53 54 55. The fact that murine APL overexpressing BCL-2 still responded to arsenic suggests that arsenic can overcome a BCL-2–mediated delay in apoptosis. This result is also consistent with the hypothesis that differentiative effects of arsenic contribute to its therapeutic value. Although we studied the response of a single transplantable PMLRARα/BCL-2 leukemia to tRA and arsenic and, therefore, have not addressed possible heterogeneity among such leukemias, our results nevertheless indicate that BCL-2 expression can be associated with an intact response. Thus, human APLs that overexpress BCL-2 or have acquired analogous changes may remain responsive to the novel APL therapies, tRA and arsenic.
Is Dissemination of Myeloid Leukemia Cells the Equivalent of Metastasis of Solid Tumors?
Discussions of AML pathogenesis often focus on the inhibition of differentiation characteristic of the disease. The existence of a preleukemic state in PMLRARα/BCL-2 transgenic mice characterized by maturation that is nearly arrested suggests that additional features of AML, including suppression of normal hematopoiesis and dissemination of leukemic cells, are not the direct result of inhibited differentiation. AML may result from two fundamental abnormalities: (a) a relative inhibition of differentiation that is accompanied by (b) a relative autonomy that allows the leukemic cells to survive and proliferate outside of their normal microenvironment. The idea that survival and proliferation outside of the normal microenvironment are central to AML parallels the concept that for solid tumors, invasion and metastasis define malignancy. In our APL model, although BCL-2 enhances cell survival, other genetic lesions are still required to fully overcome normal growth controls.
Acknowledgments
The authors thank Anne Janin and Hugues de Thé for their assistance with arsenic studies; Lois Koren-Serxner, Gayatry Mohapatra, and Meijuan Zhou for their assistance with mice and DNA preparation; Bob Flandermeyer, Gregg Magrane, and Fred Waldman of the University of California at San Francisco Comprehensive Cancer Center Molecular Cytogenetics Core Facility for comparative genomic hybridization; Daniel Stites and Frank McCormick for their continuing support; and H. Jeffrey Lawrence and Daphne Haas-Kogan for critical comments on the manuscript.
This work was supported by grants CA4338 and CA75986 from the National Institutes of Health and funds from the G.W. Hooper Research Foundation. S.C. Kogan is a recipient of a Burroughs Wellcome Fund Career Award and is an Edward Mallinckrodt Jr. Foundation Scholar.
Footnotes
Abbreviations used in this paper: 7-AAD, 7-amino actinomycin D; AML, acute myeloid leukemia; APL, acute promyelocytic leukemia; PML, promyelocytic leukemia; RARα, retinoic acid receptor α; tRA, all-trans retinoic acid.
References
- Cline M.J. The molecular basis of leukemia. N. Engl. J. Med. 1994;330:328–336. doi: 10.1056/NEJM199402033300507. [DOI] [PubMed] [Google Scholar]
- Look A.T. Oncogenic transcription factors in the human acute leukemias. Science. 1997;278:1059–1064. doi: 10.1126/science.278.5340.1059. [DOI] [PubMed] [Google Scholar]
- Melnick A., Licht J.D. Deconstructing a diseaseRARα, its fusion partners, and their roles in the pathogenesis of acute promyelocytic leukemia. Blood. 1999;93:3167–3215. [PubMed] [Google Scholar]
- Brown D., Kogan S., Lagasse E., Weissman I., Alcalay M., Pelicci P.G., Atwater S., Bishop J.M. A PMLRARα transgene initiates murine acute promyelocytic leukemia. Proc. Natl. Acad. Sci. USA. 1997;94:2551–2556. doi: 10.1073/pnas.94.6.2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grisolano J.L., Wesselschmidt R.L., Pelicci P.G., Ley T.J. Altered myeloid development and acute leukemia in transgenic mice expressing PML-RARα under control of cathepsin G regulatory sequences. Blood. 1997;89:376–387. [PubMed] [Google Scholar]
- He L.Z., Tribioli C., Rivi R., Peruzzi D., Pelicci P.G., Soares V., Cattoretti G., Pandolfi P.P. Acute leukemia with promyelocytic features in PML/RARα transgenic mice. Proc. Natl. Acad. Sci. USA. 1997;94:5302–5307. doi: 10.1073/pnas.94.10.5302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sporn, M.B., A.B. Roberts, and D.S. Goodman, editors. 1994. The Retinoids: Biology, Chemistry, and Medicine. 2nd ed. Raven Press, New York. 679 pp.
- Grignani F., Ferrucci P.F., Testa U., Talamo G., Fagioli M., Alcalay M., Mencarelli A., Grignani F., Peschle C., Nicoletti I., Pelicci P.G. The acute promyelocytic leukemia-specific PML-RARα fusion protein inhibits differentiation and promotes survival of myeloid precursor cells. Cell. 1993;74:423–431. doi: 10.1016/0092-8674(93)80044-f. [DOI] [PubMed] [Google Scholar]
- Rousselot P., Hardas B., Patel A., Guidez F., Gaken J., Castaigne S., Dejean A., de The H., Degos L., Farzaneh F., Chomienne C. The PML-RARα gene product of the t(15;17) translocation inhibits retinoic acid-induced granulocytic differentiation and mediated transactivation in human myeloid cells. Oncogene. 1994;9:545–551. [PubMed] [Google Scholar]
- Grignani F., Testa U., Fagioli M., Barberi T., Masciulli R., Mariani G., Peschle C., Pelicci P.G. Promyelocytic leukemia-specific PML-retinoic acid α receptor fusion protein interferes with erythroid differentiation of human erythroleukemia K562 cells. Cancer Res. 1995;55:440–443. [PubMed] [Google Scholar]
- Testa U., Grignani F., Hassan H.J., Rogaia D., Masciulli R., Gelmetti V., Guerriero R., Macioce G., Liberatore C., Barberi T. Terminal megakaryocytic differentiation of TF-1 cells is induced by phorbol esters and thrombopoietin and is blocked by expression of PML/RARα fusion protein. Leukemia. 1998;12:563–570. doi: 10.1038/sj.leu.2400967. [DOI] [PubMed] [Google Scholar]
- Shao W., Benedetti L., Lamph W.W., Nervi C., Miller W.H., Jr. A retinoid-resistant acute promyelocytic leukemia subclone expresses a dominant negative PML-RARα mutation. Blood. 1997;89:4282–4289. [PubMed] [Google Scholar]
- Nervi C., Ferrara F.F., Fanelli M., Rippo M.R., Tomassini B., Ferrucci P.F., Ruthardt M., Gelmetti V., Gambacorti-Passerini C., Diverio D. Caspases mediate retinoic acid-induced degradation of the acute promyelocytic leukemia PML/RARα fusion protein. Blood. 1998;92:2244–2251. [PubMed] [Google Scholar]
- Kogan S.C., Hong S.H., Shultz D.B., Privalsky M.L., Bishop J.M. Leukemia initiated by PMLRARαthe PML domain plays a critical role while retinoic acid-mediated transactivation is dispensable. Blood. 2000;95:1541–1550. [PubMed] [Google Scholar]
- Ferrucci P.F., Grignani F., Pearson M., Fagioli M., Nicoletti I., Pelicci P.G. Cell death induction by the acute promyelocytic leukemia-specific PML/RARα fusion protein. Proc. Natl. Acad. Sci. USA. 1997;94:10901–10906. doi: 10.1073/pnas.94.20.10901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu S., Consoli U., Hanania E.G., Zu Z., Claxton D.F., Andreeff M., Deisseroth A.B. PML/RARα, a fusion protein in acute promyelocytic leukemia, prevents growth factor withdrawal-induced apoptosis in TF-1 cells. Clin. Cancer Res. 1995;1:583–590. [PubMed] [Google Scholar]
- Wang Z.G., Ruggero D., Ronchetti S., Zhong S., Gaboli M., Rivi R., Pandolfi P.P. PML is essential for multiple apoptotic pathways. Nat. Genet. 1998;20:266–272. doi: 10.1038/3073. [DOI] [PubMed] [Google Scholar]
- Grignani F., Valtieri M., Gabbianelli M., Gelmetti V., Botta R., Luchetti L., Masella B., Morsilli O., Pelosi E., Samoggia P. PML/RARα fusion protein expression in normal human hematopoietic progenitors dictates myeloid commitment and the promyelocytic phenotype. Blood. 2000;96:1531–1537. [PubMed] [Google Scholar]
- Gross A., McDonnell J.M., Korsmeyer S.J. BCL-2 family members and the mitochondria in apoptosis. Genes Dev. 1999;13:1899–1911. doi: 10.1101/gad.13.15.1899. [DOI] [PubMed] [Google Scholar]
- Karakas T., Maurer U., Weidmann E., Miething C.C., Hoelzer D., Bergmann L. High expression of bcl-2 mRNA as a determinant of poor prognosis in acute myeloid leukemia. Ann. Oncol. 1998;9:159–165. doi: 10.1023/a:1008255511404. [DOI] [PubMed] [Google Scholar]
- Campos L., Rouault J.P., Sabido O., Oriol P., Roubi N., Vasselon C., Archimbaud E., Magaud J.P., Guyotat D. High expression of bcl-2 protein in acute myeloid leukemia cells is associated with poor response to chemotherapy. Blood. 1993;81:3091–3096. [PubMed] [Google Scholar]
- Bensi L., Longo R., Vecchi A., Messora C., Garagnani L., Bernardi S., Tamassia M.G., Sacchi S. Bcl-2 oncoprotein expression in acute myeloid leukemia. Haematologica. 1995;80:98–102. [PubMed] [Google Scholar]
- Porwit-MacDonald A., Ivory K., Wilkinson S., Wheatley K., Wong L., Janossy G. Bcl-2 protein expression in normal human bone marrow precursors and in acute myelogenous leukemia. Leukemia. 1995;9:1191–1198. [PubMed] [Google Scholar]
- DiGiuseppe J.A., LeBeau P., Augenbraun J., Borowitz M.J. Multiparameter flow-cytometric analysis of bcl-2 and Fas expression in normal and neoplastic hematopoiesis. Am. J. Clin. Pathol. 1996;106:345–351. doi: 10.1093/ajcp/106.3.345. [DOI] [PubMed] [Google Scholar]
- Molica S., Mannella A., Dattilo A., Levato D., Iuliano F., Peta A., Consarino C., Magro S. Differential expression of BCL-2 oncoprotein and Fas antigen on normal peripheral blood and leukemic bone marrow cells. A flow cytometric analysis. Haematologica. 1996;81:302–309. [PubMed] [Google Scholar]
- Seiter K., Feldman E.J., Dorota Halicka H., Deptala A., Traganos F., Burke H.B., Hoang A., Goff H., Pozzuoli M., Kancherla R. Clinical and laboratory evaluation of all-trans retinoic acid modulation of chemotherapy in patients with acute myelogenous leukaemia. Br. J. Haematol. 2000;108:40–47. doi: 10.1046/j.1365-2141.2000.01804.x. [DOI] [PubMed] [Google Scholar]
- Lagasse E., Weissman I.L. Bcl-2 inhibits apoptosis of neutrophils but not their engulfment by macrophages. J. Exp. Med. 1994;179:1047–1052. doi: 10.1084/jem.179.3.1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kogan S.C., Lagasse E., Atwater S., Bae S.C., Weissman I., Ito Y., Bishop J.M. The PEBP2βMYH11 fusion created by Inv(16)(p13;q22) in myeloid leukemia impairs neutrophil maturation and contributes to granulocytic dysplasia. Proc. Natl. Acad. Sci. USA. 1998;95:11863–11868. doi: 10.1073/pnas.95.20.11863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid I., Uittenbogaart C.H., Keld B., Giorgi J.V. A rapid method for measuring apoptosis and dual-color immunofluorescence by single laser flow cytometry. J. Immunol. Methods. 1994;170:145–157. doi: 10.1016/0022-1759(94)90390-5. [DOI] [PubMed] [Google Scholar]
- Mohapatra G., Kim D.H., Feuerstein B.G. Detection of multiple gains and losses of genetic material in ten glioma cell lines by comparative genomic hybridization. Genes Chromosomes Cancer. 1995;13:86–93. doi: 10.1002/gcc.2870130203. [DOI] [PubMed] [Google Scholar]
- Weiss W.A., Aldape K., Mohapatra G., Feuerstein B.G., Bishop J.M. Targeted expression of MYCN causes neuroblastoma in transgenic mice. EMBO (Eur. Mol. Biol. Organ.) J. 1997;16:2985–2995. doi: 10.1093/emboj/16.11.2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lallemand-Breitenbach V., Guillemin M.C., Janin A., Daniel M.T., Degos L., Kogan S.C., Bishop J.M., de The H. Retinoic acid and arsenic synergize to eradicate leukemic cells in a mouse model of acute promyelocytic leukemia. J. Exp. Med. 1999;189:1043–1052. doi: 10.1084/jem.189.7.1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symonds H., Krall L., Remington L., Saenz-Robles M., Lowe S., Jacks T., Van Dyke T. p53-dependent apoptosis suppresses tumor growth and progression in vivo. Cell. 1994;78:703–711. doi: 10.1016/0092-8674(94)90534-7. [DOI] [PubMed] [Google Scholar]
- Lagasse E., Weissman I.L. Enforced expression of Bcl-2 in monocytes rescues macrophages and partially reverses osteopetrosis in op/op mice. Cell. 1997;89:1021–1031. doi: 10.1016/s0092-8674(00)80290-1. [DOI] [PubMed] [Google Scholar]
- Traver D., Akashi K., Weissman I.L., Lagasse E. Mice defective in two apoptosis pathways in the myeloid lineage develop acute myeloblastic leukemia. Immunity. 1998;9:47–57. doi: 10.1016/s1074-7613(00)80587-7. [DOI] [PubMed] [Google Scholar]
- Nuñez G., London L., Hockenbery D., Alexander M., McKearn J.P., Korsmeyer S.J. Deregulated Bcl-2 gene expression selectively prolongs survival of growth factor-deprived hemopoietic cell lines. J. Immunol. 1990;144:3602–3610. [PubMed] [Google Scholar]
- Berger R., Le Coniat M., Derre J., Vecchione D., Jonveaux P. Cytogenetic studies in acute promyelocytic leukemiaa survey of secondary chromosomal abnormalities. Genes Chromosomes Cancer. 1991;3:332–337. doi: 10.1002/gcc.2870030503. [DOI] [PubMed] [Google Scholar]
- Schoch C., Haase D., Haferlach T., Freund M., Link H., Lengfelder E., Loffler H., Buchner T., Fonatsch C. Incidence and implication of additional chromosome aberrations in acute promyelocytic leukaemia with translocation t(15;17)(q22;q21)a report on 50 patients. Br. J. Haematol. 1996;94:493–500. doi: 10.1046/j.1365-2141.1996.d01-1829.x. [DOI] [PubMed] [Google Scholar]
- Hiorns L.R., Swansbury G.J., Mehta J., Min T., Dainton M.G., Treleaven J., Powles R.L., Catovsky D. Additional chromosome abnormalities confer worse prognosis in acute promyelocytic leukaemia. Br. J. Haematol. 1997;96:314–321. doi: 10.1046/j.1365-2141.1997.d01-2037.x. [DOI] [PubMed] [Google Scholar]
- Slack J.L., Arthur D.C., Lawrence D., Mrózek K., Mayer R.J., Davey F.R., Tantravahi R., Pettenati M.J., Bigner S., Carroll A.J. Secondary cytogenetic changes in acute promyelocytic leukemia—prognostic importance in patients treated with chemotherapy alone and association with the intron 3 breakpoint of the PML genea cancer and leukemia group B study. J. Clin. Oncol. 1997;15:1786–1795. doi: 10.1200/JCO.1997.15.5.1786. [DOI] [PubMed] [Google Scholar]
- Gibbons D.L., MacDonald D., McCarthy K.P., Cleary H.J., Plumb M., Wright E.G., Greaves M.F. An E mu-BCL-2 transgene facilitates leukaemogenesis by ionizing radiation. Oncogene. 1999;18:3870–3877. doi: 10.1038/sj.onc.1202721. [DOI] [PubMed] [Google Scholar]
- Silver A., Moody J., Dunford R., Clark D., Ganz S., Bulman R., Bouffler S., Finnon P., Meijne E., Huiskamp R., Cox R. Molecular mapping of chromosome 2 deletions in murine radiation-induced AML localizes a putative tumor suppressor gene to a 1.0 cM region homologous to human chromosome segment 11p11-12. Genes Chromosomes Cancer. 1999;24:95–104. [PubMed] [Google Scholar]
- Zimonjic D.B., Pollock J.L., Westervelt P., Popescu N.C., Ley T.J. Acquired, nonrandom chromosomal abnormalities associated with the development of acute promyelocytic leukemia in transgenic mice. Proc. Natl. Acad. Sci. USA. 2000;97:13306–13311. doi: 10.1073/pnas.97.24.13306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Maria R., Testa U., Luchetti L., Zeuner A., Stassi G., Pelosi E., Riccioni R., Felli N., Samoggia P., Peschle C. Apoptotic role of Fas/Fas ligand system in the regulation of erythropoiesis. Blood. 1999;93:796–803. [PubMed] [Google Scholar]
- Park J.R., Robertson K., Hickstein D.D., Tsai S., Hockenbery D.M., Collins S.J. Dysregulated bcl-2 expression inhibits apoptosis but not differentiation of retinoic acid-induced HL-60 granulocytes. Blood. 1994;84:440–445. [PubMed] [Google Scholar]
- Kohzaki H., Ito K., Huang G., Wee H.J., Murakami Y., Ito Y. Block of granulocytic differentiation of 32Dcl3 cells by AML1/ETO(MTG8) but not by highly expressed Bcl-2. Oncogene. 1999;18:4055–4062. doi: 10.1038/sj.onc.1202735. [DOI] [PubMed] [Google Scholar]
- Vairo G., Innes K.M., Adams J.M. Bcl-2 has a cell cycle inhibitory function separable from its enhancement of cell survival. Oncogene. 1996;13:1511–1519. [PubMed] [Google Scholar]
- Wuchter C., Karawajew L., Ruppert V., Büchner T., Schoch C., Haferlach T., Ratei R., Dörken B., Ludwig W.D. Clinical significance of CD95, Bcl-2 and Bax expression and CD95 function in adult de novo acute myeloid leukemia in context of P-glycoprotein function, maturation stage, and cytogenetics. Leukemia. 1999;13:1943–1953. doi: 10.1038/sj.leu.2401605. [DOI] [PubMed] [Google Scholar]
- Rego E.M., Wang Z.G., Peruzzi D., He L.Z., Cordon-Cardo C., Pandolfi P.P. Role of promyelocytic leukemia (PML) in tumor suppression. J. Exp. Med. 2001;193:521–529. doi: 10.1084/jem.193.4.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollock J.L., Westervelt P., Kurichety A.K., Pelicci P.G., Grisolano J.L., Ley T.J. A bcr-3 isoform of RARα-PML potentiates the development of PML-RARα-driven acute promyelocytic leukemia. Proc. Natl. Acad. Sci. USA. 1999;96:15103–15108. doi: 10.1073/pnas.96.26.15103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vickers M., Jackson G., Taylor P. The incidence of acute promyelocytic leukemia appears constant over most of a human lifespan, implying only one rate limiting mutation. Leukemia. 2000;14:722–726. doi: 10.1038/sj.leu.2401722. [DOI] [PubMed] [Google Scholar]
- Bocchia M., Xu Q., Wesley U., Xu Y., Korontsvit T., Loganzo F., Albino A.P., Scheinberg D.A. Modulation of p53, WAF1/p21 and BCL-2 expression during retinoic acid-induced differentiation of NB4 promyelocytic cells. Leuk. Res. 1997;21:439–447. doi: 10.1016/s0145-2126(96)00085-9. [DOI] [PubMed] [Google Scholar]
- Chen G.Q., Zhu J., Shi X.G., Ni J.H., Zhong H.J., Si G.Y., Jin X.L., Tang W., Li X.S., Xong S.M. In vitro studies on cellular and molecular mechanisms of arsenic trioxide (As2O3) in the treatment of acute promyelocytic leukemiaAs2O3 induces NB4 cell apoptosis with downregulation of Bcl-2 expression and modulation of PML-RARα/PML proteins. Blood. 1996;88:1052–1061. [PubMed] [Google Scholar]
- Chen G.Q., Shi X.G., Tang W., Xiong S.M., Zhu J., Cai X., Han Z.G., Ni J.H., Shi G.Y., Jia P.M. Use of arsenic trioxide (As2O3) in the treatment of acute promyelocytic leukemia (APL). I. As2O3 exerts dose-dependent dual effects on APL cells. Blood. 1997;89:3345–3353. [PubMed] [Google Scholar]
- Soignet S.L., Maslak P., Wang Z.G., Jhanwar S., Calleja E., Dardashti L.J., Corso D., DeBlasio A., Gabrilove J., Scheinberg D.A. Complete remission after treatment of acute promyelocytic leukemia with arsenic trioxide. N. Engl. J. Med. 1998;339:1341–1348. doi: 10.1056/NEJM199811053391901. [DOI] [PubMed] [Google Scholar]