

Abstract
We have shown previously that neutralizing antibodies (nAbs) are important contributors to the long-term immune control of lymphocytic choriomeningitis virus infection, particularly if cytotoxic T cell responses are low or absent. Nevertheless, virus escape from the nAb response due to mutations within the surface glycoprotein gene may subsequently allow the virus to persist. Here we show that most of the antibody-escape viral mutants retain their immunogenicity. We present evidence that the failure of the infected host to mount effective humoral responses against emerging neutralization-escape mutants correlates with the rapid loss of CD4+ T cell responsiveness during the establishment of viral persistence. Similar mechanisms may contribute to the persistence of some human pathogens such as hepatitis B and C viruses, and human immunodeficiency virus.
Keywords: persistent infection, lymphocytic choriomeningitis virus, T helper cells, humoral responses, viral evasion
Introduction
Strong CD8+ CTL responses characterize the initial immunosurveillance of infections with poorly or noncytopathic viruses such as hepatitis B and C viruses, HIV, and the murine RNA virus lymphocytic choriomeningitis virus (LCMV 1 2 3). Nevertheless, this initial clearance of viremia by CD8+ CTLs does not always prevent the establishment of a chronic infection. Besides CTLs, neutralizing Abs (nAbs) in association with other noncytolytic factors (IFN-γ, TNF-α, and chemokines) play a crucial role in controlling persisting virus infection 4 5 6 7 8. However, virus escape from the nAb response during chronic infections does occur and may contribute to viral persistence 9. We have recently demonstrated in a model system that low CTL activity during LCMV infection facilitates the detection of neutralization-escape virus variants 10. Escape was due to single point mutations within the genes coding for the envelope glycoprotein (GP)-1. The ensuing amino acid changes affect the conformation of the neutralizing epitope 11.
The following questions have been further addressed in this study. (a) Are the envelope GPs of nAb-escape LCMV variants less immunogenic? (b) How quickly and efficiently are nAbs generated against emerging escape variants? (c) Do variants evolve that are in general neutralization resistant? (d) Is the impairment of nAb responses due to deletion or anergy of virus-specific T helper cells?
We studied the nAb response during a long-term infection of CD8−/− mice with LCMV, strain WE 12. Due to the absence of CTLs in these mice, augmented virus production occurs, which is transiently controlled by polyclonal nAbs in the blood and only to a limited extent in solid organs 10. The results indicate that the broadening of nAb responses against emerging neutralization-resistant virus variants in this high viremia model infection was mainly limited by a decrease and eventual loss of virus-specific CD4+ T cell responses. Changes in replication properties or a decreased immunogenicity of some virus variants may additionally contribute to virus persistence.
Materials and Methods
Mice and Viruses.
CD8−/− mice 13, LCMV-GP61-80–specific CD4+ TCR transgenic (tg) SMARTA mice 14, and control C57BL/6 (B6) mice were obtained from the Institut für Labortierkunde (University of Zürich, Zürich, Switzerland). In certain experiments, B6 mice were in vivo depleted of CD8+ T cells by treatment with a tested anti-CD8 monoclonal Ab on days 3 and 1 preceding the infection, as described 10. The degree of depletion was always >95% in blood and spleen. All animals were kept under specific pathogen-free conditions.
LCMV strain WE originally was obtained from F. Lehmann-Grube (Heinrich Pette Institut, Hamburg, Germany) and propagated on L929 cells. Mice were infected with 2 × 106 PFU of LCMV-WE intravenously. LCMV titers in blood or virus titers of stock solutions were determined with an immunological focus assay 15.
LCMV nAb–escape variants were isolated from the blood of CD8−/− mice (120 and 240 d after infection), grown on MC57 cells for 48 h, and subsequently plaque purified two times in vitro as described 16. For de novo infections, mice were immunized with 2 × 104 PFU of selected virus variants intravenously.
Vesicular stomatitis virus (VSV) Indiana (VSV-IND; Mudd-Summers isolate) was originally obtained from Dr. D. Kolakovsky (University of Geneva, Geneva, Switzerland) and was grown on BHK21 cells. Mice were infected with 2 × 106 PFU of VSV-IND intravenously.
Neutralizing Activity.
Neutralizing activity against LCMV was measured in a focus reduction assay 15. The neutralizing titer was defined as the dilution causing half-maximal reduction of plaques of LCMV when compared with the same amount of virus incubated with control sera from uninfected mice. VSV neutralization assay was performed as described 17. The highest dilution of serum that reduced the number of plaques by 50% was taken as the titer. To determine IgG Ab titers, undiluted serum was pretreated with 0.1 M β-mercaptoethanol.
Adoptive Transfers.
Spleen cell suspensions were prepared from naive SMARTA tg mice previously in vivo depleted of CD8+ T cells by treatment with anti-CD8 monoclonal Abs. 5 × 105 SMARTA splenocytes were transferred intravenously into recipient B6 or CD8−/− mice, 1 d before infection with 2 × 106 PFU of LCMV-WE. Both SMARTA and CD8−/− mice have a pure B6 background. Clonal expansion and disappearance of transferred tg cells in the blood of recipients were monitored by FACS® analysis using TCR Vα2 and Vβ8.3 monoclonal Abs.
Intracellular IFN-γ Staining.
Splenocytes (106 cells/well in 96-well plates) were stimulated in vitro with media or with the immunodominant LCMV-specific peptide GP61-80 (1 μg/ml). Brefeldin A (Sigma-Aldrich) was added after 1 h of culture. After a 5-h stimulation, the cells were harvested, washed once in PBS containing 2% FCS, and stained with PE-conjugated anti-CD4 monoclonal Abs (Caltag) for 30 min at 4°C. After washing of unbound Ab, the cells were fixed with PBS/4% paraformaldehyde for 10 min and then permeabilized with PBS/0.1% Saponin (Sigma-Aldrich). For intracellular staining, FITC-conjugated anti–IFN-γ monoclonal Abs were used and incubated with the cells for 30 min at 4°C. After washing twice with PBS/0.1% Saponin, the cells were resuspended in PBS containing 2% FCS and analyzed using a FACScan™ (Becton Dickinson). To control for nonspecific intracellular staining, parallel samples of stimulated and permeabilized CD4+ T cells were stained with FITC-conjugated isotype-matched monoclonal Abs of irrelevant specificity, which did not result in any staining signal.
Molecular Analysis.
Total RNA of MC57 cells infected either with LCMV-WE wild-type (wt) or with variant virus isolates for 48 h at an initial multiplicity of infection of 0.01 was extracted by using RNeasy kit (QIAGEN). Reverse transcription PCR was performed using the LCMV-GP1–specific primers R1 (5′-1037TCG TAG CAT GTC ACA GAA CTC TTC1014-3′) for reverse transcription and the primer pairs 001/R1 and 001/RC1 (001, 5′-1CGC ACC GGG GAT CCT AGG CTT21-3′; RC1, 5′-965GAG CTC TGC AGC AAG GAT CAT CC942-3′) for Hot Start PCR amplification. PCR products were sequenced by automated Taq cycle sequencing (Taq Dye Deoxy Terminator Cycle Sequencing kit; Applied Biosystems; Bio-Rad Laboratories) using the primers 001 and RC1.
Results
NAb Responses against Emerging nAb-escape Virus Mutants.
Virus titers as well as nAb titers were sequentially determined in the blood of five LCMV-WE–infected CD8−/− mice (animals M7–M11) for up to 240 d. As reported previously 10, nAb-mediated control of viremia, attained within 50–60 d after infection, was only transient and virus reappeared in the blood 2–4 wk after initial control (Fig. 1). This occurred despite the presence of relatively high titers of nAbs (Fig. 2, filled circles). Viremia was not controlled at later time points in CD8−/− mice (Fig. 1), suggesting that induction of new nAb responses against emerging neutralization-resistant virus variants had failed. To assess long-term virus-specific humoral and T helper responses in these mice, we next characterized the virus variants emerging in vivo. Virus was isolated from the blood of mice M7–M11 after the recrudescence of viremia (day 120). Sequence analysis of the gene encoding the envelope GP1 of LCMV isolates revealed amino acid alterations of the predominant viral clone (WE-M7 to WE-M11) within the bulk virus isolated from each animal (at least 5 out of 8–10 independent clones; Table ). One to three base pair exchanges per GP1 gene were identified, leading to amino acid substitutions within the three regions of GP1 that have been shown to correlate with virus escape from the nAb response 10. These mutations affected the efficiency of variant virus neutralization by polyclonal hyperimmune serum (pooled from B6 mice immunized with LCMV-WE) and by LCMV-WE-GP1–specific mAb (data not shown).
Figure 1.

Transient control of viremia in CD8−/− mice. CD8−/− mice (animals M7 to M11; •) and control B6 mice (○) were infected with 2 × 106 PFU of LCMV-WE-wt intravenously, and sequential blood samples were analyzed for virus titers. Data shown are the mean for five mice ± SEM.
Figure 2.

NAb responses against immunizing LCMV-WE-wt and emerging LCMV variants in five individual CD8−/− mice. (A–E) Five CD8−/− mice (animals M7–M11) were infected with 2 × 106 PFU of LCMV-WE-wt. Sequential serum samples were tested for neutralizing (Neutr.) activity against LCMV-WE-wt (•) and against the predominant LCMV variant (WE-M7 to WE-M11; ○) isolated from the corresponding mouse after viral reemergence on day 120 after infection (see characterization of variants in Table ). α, anti.
Table 1.
LCMV-WE nAb-escape Variants Contain Amino Acid–changing Point Mutations within the Sequence Coding for the Envelope Protein GP1 (Amino Acids 59–262)
| Amino acid substitution at indicated position | |||||||
|---|---|---|---|---|---|---|---|
| Day | Virus | 122 | 177 | 182 | 211 | 212 | |
| Mouse | 0 | LCMV-WE-wt | Phe | Pro | Ser | Ala | Gly |
| M7 | 120 | WE-M7 | Arg | Asp | |||
| 240 | Arg | Asp | |||||
| M8 | 120 | WE-M8 | Asn | ||||
| 240 | Ser | Asn | |||||
| M9 | 120 | WE-M9 | Ser | Asp | |||
| 240 | Ser | Asp | |||||
| M10 | 120 | WE-M10 | Ser | Leu | Thr | ||
| M11 | 120 | WE-M11 | Asn | Thr | |||
Virus was isolated from the blood of five CD8−/− mice (M7 to M11) 120 and 240 d after infection with 2 × 106 PFU of LCMV-WE and double plaque purified. Positions of alterations in deduced amino acid residues of the envelope GP1 of the predominant viral clone (at least 5 out of 8–10 clones) from each animal are shown.
We next performed autologous serum neutralization assays with virus variants WE-M7 to WE-M11 and with LCMV-WE-wt (Fig. 2). The strong initial nAb response against the immunizing virus (LCMV-WE-wt) peaked around day 75 and declined very slowly. In contrast, low or no neutralizing activity was detected against virus escape variants at all time points tested. Hence, these variants have indeed escaped the original nAb response, and failed to induce specific nAbs over a period of 120 d (Fig. 2). The failure of the variants to induce an effective nAb response could have several reasons. Theoretically, we cannot exclude that novel nAbs are generated, as this could be masked by outgrowth of newly evolving escape mutants. We therefore sequenced day 240 isolates from the blood of three test animals (M7, M8, and M9). The GP1 sequence of the predominant clone (at least 5/8 isolates) is shown in Table . No sequence changes were observed when viral isolates derived on day 120 and 240 after infection were compared in mice M7 and M9, respectively. In mouse M8, the predominant virus clone had one additional amino acid–changing mutation at position 122 on day 240, compared with day 120. Nevertheless, a clone with this genotype was already present in the viral quasispecies on day 120 (1/8 clones; data not shown). Overall, the escape mutants were relatively stable, which suggests the absence of a specific newly induced immune selection pressure. Selection of predominant virus variants within the quasispecies at late time points, as seen in animal M8, might therefore be influenced by viral fitness 18.
Loss of CD4+ T Cell Responsiveness Precedes the Emergence of nAb-escape Virus Mutants.
Failure of the hosts to elicit new nAb responses against emerging nAb-escape virus variants could be due to insufficient CD4+ T helper responses at late time points. This could be the consequence of CD4+ T cell unresponsiveness induced during establishment of persistent infection 19, T helper epitope variation, or a decrease of infected CD4+ T cells.
LCMV-specific CD4+ T cell responses were compared between LCMV-infected CD8−/− mice, which show a high and sustained viremia, particularly in solid organs, and control B6 mice, which rapidly control the virus (10; Fig. 1). We monitored the number of splenic CD4+ T cells specific for the LCMV-immunodominant epitope GP61–80 and expressing intracellular IFN-γ after antigenic stimulation at different time points after infection (Fig. 3 A). In agreement with previous studies 20 21 22, this response peaked at day 9 in control B6 mice (∼3% of CD4+ T cells) and decreased to 0.4% by day 50. This percentage was stable in the memory phase (up to 240 d). By contrast, only 0.6% of CD4+ T cells stained positive for intracellular IFN-γ at the peak of the response in CD8−/− mice. This percentage rapidly dropped to background levels by day 20. In CD8−/− test animals M7 to M11, only background levels of virus-specific CD4+ T helper activity were detected on day 240 (Fig. 3 A), irrespective of the ability of the mice to produce low or high nAb titers against the immunizing LCMV-WE-wt virus (Fig. 2). We were not able to monitor an additional T helper response against the nucleoprotein NP309–328 epitope because on day 8 after infection, frequencies of specific CD4+ T cells were below background levels in LCMV-WE–infected CD8−/− mice. To ascertain that the responses measured in the in vitro assays were not affected by the absence of CD8+ T cells, we performed the following control experiment. LCMV-WE–immune B6 mice (70 d after infection) were either depleted of CD8+ T cells or left untreated. We then evaluated the number of splenic LCMV-specific, IFN-γ–expressing CD4+ T cells. Similar percentages of GP61-80–specific CD4+ T cells were measured in the spleens of mice of the two groups (data not shown). As indicated by sequence analysis, failure to detect GP61-80–specific CD4+ T cells in CD8−/− mice was not due to epitope variation (Table , and data not shown).
Figure 3.

Induction of specific CD4+ T cell unresponsiveness in LCMV-infected CD8−/− mice. B6 mice (triangles) and CD8−/− mice (circles) were infected with 2 × 106 PFU LCMV-WE intravenously. (A) Splenocytes from the indicated days after infection were stimulated in vitro with the immunodominant LCMV class II–restricted epitope (GP61–80; filled symbols) or with no peptide (open symbols), and the percentage of peptide-specific CD4+ T cells expressing intracellular IFN-γ was then assessed. (B) 5 × 105 splenocytes from in vivo CD8-depleted, LCMV-GP61-80–specific CD4+ TCR tg SMARTA mice (B6 background) were adoptively transferred intravenously to recipient B6 mice that were either infected with LCMV-WE 1 d later (filled symbols) or were not infected (open symbols). The percentage of tg CD4+ T cells (TCR Vα2+; Vβ8.3+) in the blood was then sequentially determined by FACS® analysis. Data shown are the mean for four mice ± SEM.
As mentioned above, several factors could account for the low levels of CD4+ T cells in the high viremia CD8−/− model system. Loss of total CD4+ T cell population due to infection and/or immunopathology, specific unresponsiveness of the LCMV-specific T helper subset, but also a deficiency in generating the initial response, have to be considered. To specifically study the effects of persistent high viremia on CD4+ T cell expansion, we performed adoptive transfer experiments with LCMV GP61–80 epitope–specific splenocytes from in vivo CD8-depleted SMARTA TCR tg mice. This approach allowed for monitoring of the transferred LCMV-specific T helper cells via the tg TCR Vα and Vβ chains (Fig. 3 B). Compared with B6 mice, clonal expansion of the transferred tg CD4+ T cells was compromised in LCMV-infected CD8−/− mice. Moreover, we found that these cells disappeared more rapidly from the blood of recipient LCMV-infected CD8−/− mice. The tg CD4+ T cells were still present in high numbers on day 16 in the blood of LCMV-infected B6 mice and were still above background levels by about day 40. No obvious differences were seen in uninfected B6 and CD8−/− mice, suggesting that the effects seen were a consequence of high level viral replication.
To investigate whether the reduction of LCMV-specific T helper cells was due to an overall decline of the CD4+ T cell population in the high viremia model, we assessed the absolute number of splenic CD4+ T cells by flow cytometry before and after LCMV infection. No depletion of the general CD4+ T cell pool was detected after infection of CD8−/− mice with LCMV-WE (Fig. 4).
Figure 4.

The CD4+ T cell population is not depleted during LCMV infection. B6 mice and CD8−/− mice were infected with 2 × 106 PFU of LCMV-WE. Splenocytes at various time points after infection were stained for CD4. Numbers of CD4+ T cells were determined by flow cytometry. Data shown are the mean of three mice per group ± SEM.
To test whether loss of CD4+ T cells during high viral replication was restricted to LCMV-specific cells or whether unrelated, non-LCMV responses were also affected, we evaluated CD4 T cell help against VSV. VSV induces a very early neutralizing IgM Ab response that is largely T cell help independent; the subsequent IgM switch to IgG is in contrast strictly dependent on functional CD4+ T cell help 23. CD8−/− and B6 mice, 240 d after LCMV infection, and uninfected control animals were immunized with 2 × 106 PFU of VSV-IND intravenously. Anti-VSV IgM and IgG Ab titers were assessed in sequential serum samples (Fig. 5A and Fig. B). LCMV-infected CD8−/− mice were able to switch their anti-VSV IgM Ab responses to IgG, proving that functional VSV-specific CD4+ T cell help was present (Fig. 5 B). Hence, the time-dependent induction of T helper unresponsiveness in CD8−/− mice was LCMV specific.
Figure 5.
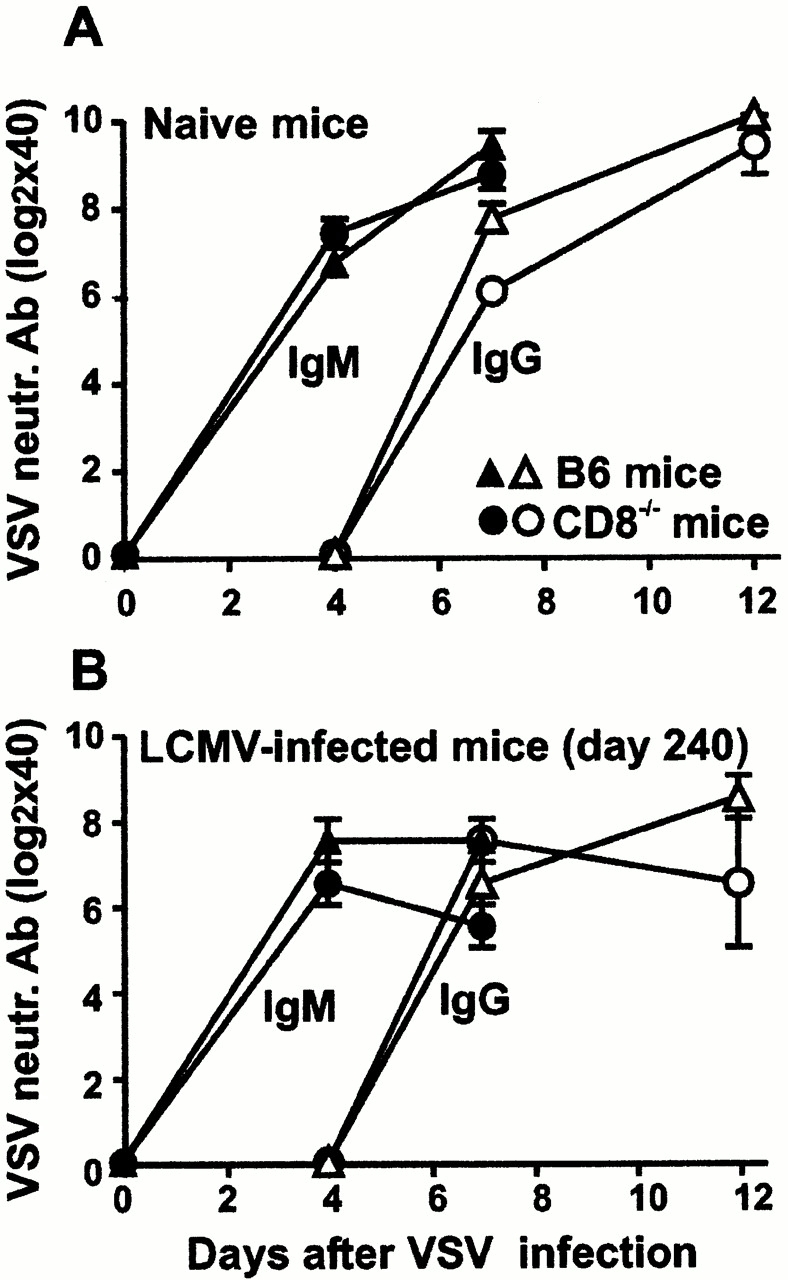
Functional VSV-specific T cell help in LCMV-WE infected CD8−/− mice. B6 mice (triangles) and CD8−/− mice (circles) infected with 2 × 106 PFU of LCMV-WE 240 d previously (B), as well as naive control animals (A), were immunized with 2 × 106 PFU of VSV-IND. Sequential serum samples were obtained for quantitation of VSV-neutralizing (neutr.) IgM Ab titers (filled symbols) or IgG Ab titers (open symbols), distinguished from IgM by reduction with 0.1 M of β-mercaptoethanol. The results are given as the mean ± SEM of three to four mice per group.
Immunogenicity of nAb-escape Virus Variants.
In addition to insufficient CD4 T help, nAb responses against escape viruses might be low as a consequence of decreased immunogenicity of their envelope proteins. To study the immunogenicity of nAb-escape variants in vivo, we have chosen to use the model infection of mice depleted in vivo of CD8+ T cells by treatment with anti-CD8 monoclonal Abs. In contrast to CD8−/− mice, earlier virus control through the antiviral activity of reappearing CTLs takes place in this model 10. Therefore, due to the lower viremia, less Ab is masked by excess antigen. Escape mutants were tested in infections with 2 × 104 PFU, a dose available for all variants. Two isolates (WE-M10 and WE-M11) persisted for longer periods in CD8-depleted mice, whereas WE-M7, WE-M8, and WE-M9 were eliminated with similar kinetics as LCMV-WE-wt (Fig. 6 A). In contrast to our previous study 10, in which neutralization-resistant variants that showed enhanced persistence induced lower autologous nAb titers, all variants studied here raised autologous nAb responses similar to LCMV-WE-wt (Fig. 7A–E). Interestingly, heterologous nAb titers against LCMV-WE-wt in mice infected with variant viruses were at least as high as autologous titers, demonstrating a broad neutralizing response in these mice (Fig. 7A–E).
Figure 6.

In vivo replication of nAb-escape variants. CD8-depleted B6 mice (A) and CD8−/− mice (B) were infected with 2 × 104 PFU of LCMV-WE-wt or of the indicated nAb-escape LCMV variant (for CD8−/− mice only variant WE-M7). Sequential blood samples were obtained for quantitation of virus titers. Data shown are the mean of three to four mice per group ± SEM. The experiment shown in A was performed twice with similar results.
Figure 7.

Immunogenicity of LCMV-WE nAb-escape variants. (A–E) Sequential blood samples from CD8-depleted mice infected with 2 × 104 PFU of five nAb-escape variants WE-M7 (A), WE-M8 (B), WE-M9 (C), WE-M10 (D), and WE-M11 (E) were obtained for quantitation of nAb titers against the immunizing virus (•) and of nAb titers against LCMV-WE-wt (○). The dotted lines represent the autologous nAb response after infection of CD8-depleted mice with an equivalent dose of LCMV-WE-wt. The experiment was repeated in CD8−/− mice for variant WE-M7 (F). Data shown are the mean for three mice per group ± SEM. Neutr., neutralizing.
To assess whether differences in immunogenicity are dependent on the animal model used, we performed de novo infection experiments with 2 × 104 PFU of WE-M7 isolate and LCMV-WE-wt in CD8−/− mice. Infection with the variant strain was cleared with similar kinetics (within 60 d) as wt virus in these animals (Fig. 6 B). Moreover, both variant and wt virus induced an equally strong neutralization response (Fig. 7 F). Of note is that the nAb response in CD8−/− mice was, for both wt and escape virus, lower than in CD8-depleted mice.
Discussion
We have previously introduced a LCMV infection model in CD8-deficient mice that is characterized by a high persistent viremia, and that only transiently is controlled by nAb, as neutralization-resistant variants are generated very rapidly 10. This model of prolonged absence of CD8+ T cells is reflected in several virus diseases in which low or absent CTL activity during establishment of persistent infections occurs. CTL responses may exhaust or become unresponsive after overwhelming infection with LCMV 24 25, may physically or functionally disappear during HIV-1 infection 26 27 28, and become scarce once infection with hepatitis C virus is established 3. In addition, viral escape from CTL responses through selection of mutations in the relevant epitopes has been documented 29 30 31 32, and may even occur early in the course of infection 33 34.
As documented here, several factors may allow the persistence of emerging neutralization-resistant variants. A weaker immunogenicity of the escape variants seems not to be a general phenomenon, as most of the variants were able to induce autologous nAb responses after de novo infections similar to wt LCMV-WE. However, we observed an asymmetric pattern of cross-reactivity between neutralizing responses induced by wt and escape virus isolates. Whereas the original wt strain induced a response against itself but not against the emerging escape variants, the mutant viruses were able to induce nAbs that inhibited both wt and escape viruses. Thus, the variant viruses reveal via nAbs a recapitulation of their evolution and exhibit a new sort of coevolutionarily directed connectivity that one might call “archetypical”. In some aspects, this recall of genetic history is reminiscent of findings during influenza virus infections in the context of preexisting immune memory: after infection with an escape mutant after antigenic drift, influenza-immune individuals will generate higher Ab titers against the influenza surface hemagglutinin experienced during the original infection than against the mutated hemagglutinin of the drifted variant. This has been called original antigenic sin 35 36.
Other factors may contribute to the long-term persistence of nAb-resistant viruses, depending on the variant analyzed. Some variants may have better intrinsic replication capacities in vivo. Alternatively, changes in cell or tissue tropism, as well as possible differences in resistance to interferons, may account for the observed enhanced persistence. All these alterations may be the consequence of some of the documented mutations in the GP1 gene, as demonstrated at several occasions for different LCMV strains 37 38 39. In addition, mutations in other genes cannot be excluded, particularly on the L strand which encodes the viral polymerase and which was not sequenced in this study.
However, the most important mechanism leading to the persistence of emerging nAb-escape variants is the induction of specific CD4+ T cell unresponsiveness, as demonstrated here. The absence of an adequate T helper response at the moment of viral escape impaired the induction of new nAb responses against the virus mutants and allowed them to persist. Interestingly, the primary nAb response in CD8−/− mice after infection with LCMV-WE was very potent, despite the fact that the CD4+ T cell expansion was much lower than in normal mice and the helper response was rapidly lost. Several factors may account for this finding. First, virus-specific B cells, infected through the LCMV-GP1–recognizing receptor, are not killed in the absence of CTL responses 40. Second, the antigen load is much higher in CD8−/− mice. On the other hand, memory B cell responses may be less dependent on functional T help 41, explaining how high nAb titers can be maintained in CD8−/− mice against the immunizing LCMV-WE-wt despite vanishing T helper responses.
The CD4+ T cell dysfunction in CD8−/− mice does not affect the overall CD4+ T cell population and is only confined to the LCMV-specific compartment. Furthermore, as exemplified by the adoptive transfer of tg SMARTA splenocytes into LCMV-infected CD8−/− mice, LCMV-specific CD4+ T cell numbers remain equal to levels in uninfected control animals for at least 3 wk, indicating that the cells might undergo a phase of functional unresponsiveness before being physically deleted.
It has to be emphasized that CD8−/− mice are able to mount vigorous CD4+ T cell responses when infected with a low dose of a slowly replicating LCMV strain (LCMV-Armstrong; up to 14% at day 9; reference 21). An influence of elevated virus titers on the fate of T helper responses is indicated by the fact that a similar low percentage of LCMV-specific CD4+ T cells was reached during infection with the clone 13 strain of LCMV 21, which is able to establish persistent infections in immunocompetent mice 37. The high viral replication level achieved in CD8−/− mice (but not B6 mice) infected with LCMV-WE is comparable to the overwhelming infection of immunocompetent hosts by the rapidly replicating Docile strain of LCMV, where virus-specific T helper cell loss has also been shown 19. As CD8+ T cells may also contribute positively to T helper cell responsiveness via bystander factors 42, their absence in CD8−/− mice might also impact on the loss of specific CD4+ T cells.
Decrease of virus-specific T helper cells seems to be a critical evolutionary phenomenon which allows the survival of the host by preventing lethal immunopathology during persisting noncytopathic virus infection 19. The molecular mechanisms involved in the loss of T cell function remain unclear. As hypothesized for the described exhaustion of CTLs 24 43, high antigenic load after wide viral spread may induce a general activation of all available virus-specific CD4+ T cells. These cells then would die of activation-induced apoptosis preceded by a period of anergy. Replenishment of virus-specific T helper cells would not be possible as a consequence of negative T cell selection in the infected thymus 44. Other contributing factors like interleukin starvation have been postulated, but their involvement has not been conclusively demonstrated.
The relevance of this in vivo model of inactivation of specific CD4+ T cells is based on increasing evidence for a role of T helper cells in the immune control of noncytopathic viral infections, by supporting not only nAb responses, but also CTLs 45. Studies of LCMV-infected mice deficient in CD4+ T cells either by in vivo treatment with anti-CD4 monoclonal Ab or by gene targeting have demonstrated a loss of long-term functional CTL responses and the enhancement of viral persistence in the absence of T helper cells 4 5 25 46 47 48 49. Low CD4+ T cell responses have also been shown to correlate with high viral load 50 51 and with low CTL responses 52 during HIV infection. Similarly, hepatitis C virus–specific CD4+ T cells seem to be essential not only for initial viral clearance, but also for long-term viral control 53. With regard to the impact of early viral load on subsequent impairment of T helper responses, it has been recently shown that early suppression of HIV-1 by highly active antiretroviral therapy may preserve CD4+ and CD8+ T cell function and may be associated with immune control 54 55. The preserved T helper cell function may allow multiple rounds of nAb-escape and may therefore lead to the observed broadly cross-reactive nAb responses in HIV-infected long-term nonprogressors 56.
Together with earlier observations demonstrating an influence of nAb-producing B cells on LCMV long-term control 4 5 10 57, this study provides further evidence for the interactions between cellular and humoral immune responses for efficient virus control. However, an adequate balance between the two arms of the acquired immune system is needed in order to avoid virus escape due to virus variant selection through a unilateral immune response.
Acknowledgments
We thank Dr. Alexandra Trkola for helpful discussions and critical reading of the manuscript and Edit Horvath for expert technical assistance.
This work was supported by Swiss National Foundation Grants 31.50900.97 and 31.50884.97, and the Kanton of Zurich.
Footnotes
A. Ciurea and L. Hunziker contributed equally to this work.
P. Klenerman's present address is Nuffield Department of Medicine, John Radcliffe Hospital, Oxford OX3 9DU, UK.
Abbreviations used in this paper: GP, glycoprotein; LCMV, lymphocytic choriomeningitis virus; nAb, neutralizing Ab; tg, transgenic; VSV, vesicular stomatitis virus; VSV-IND, VSV Indiana; wt, wild-type.
References
- Kagi D., Ledermann B., Bürki K., Seiler P., Odermatt B., Olsen K.J., Podack E.R., Zinkernagel R.M., Hengartner H. Cytotoxicity mediated by T cells and natural killer cells is greatly impaired in perforin-deficient mice. Nature. 1994;369:31–37. doi: 10.1038/369031a0. [DOI] [PubMed] [Google Scholar]
- Schmitz J.E., Kuroda M.J., Santra S., Sasseville V.G., Simon M.A., Lifton M.A., Racz P., Tenner-Racz K., Dalesandro M., Scallon B.J. Control of viremia in simian immunodeficiency virus infection by CD8+ lymphocytes. Science. 1999;283:857–860. doi: 10.1126/science.283.5403.857. [DOI] [PubMed] [Google Scholar]
- Lechner F., Wong D.K., Dunbar P.R., Chapman R., Chung R.T., Dohrenwend P., Robbins G., Phillips R., Klenerman P., Walker B.D. Analysis of successful immune responses in persons infected with hepatitis C virus. J. Exp. Med. 2000;191:1499–1512. doi: 10.1084/jem.191.9.1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Planz O., Ehl S., Furrer E., Horvath E., Brundler M.A., Hengartner H., Zinkernagel R.M. A critical role for neutralizing-antibody-producing B cells, CD4+ T cells, and interferons in persistent and acute infections of mice with lymphocytic choriomeningitis virusimplications for adoptive immunotherapy of virus carriers. Proc. Natl. Acad. Sci. USA. 1997;94:6874–6879. doi: 10.1073/pnas.94.13.6874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomsen A.R., Johansen J., Marker O., Christensen J.P. Exhaustion of CTL memory and recrudescence of viremia in lymphocytic choriomeningitis virus-infected MHC class II- deficient mice and B cell- deficient mice. J. Immunol. 1996;157:3074–3080. [PubMed] [Google Scholar]
- Guidotti L.G., Borrow P., Brown A., McClary H., Koch R., Chisari F.V. Noncytopathic clearance of lymphocytic choriomeningitis virus from the hepatocyte. J. Exp. Med. 1999;189:1555–1564. doi: 10.1084/jem.189.10.1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guidotti L.G., Rochford R., Chung J., Shapiro M., Purcell R., Chisari F.V. Viral clearance without destruction of infected cells during acute HBV infection. Science. 1999;284:825–829. doi: 10.1126/science.284.5415.825. [DOI] [PubMed] [Google Scholar]
- Wagner L., Yang O.O., Garcia-Zepeda E.A., Ge Y., Kalams S.A., Walker B.D., Pasternack M.S., Luster A.D. Beta-chemokines are released from HIV-1-specific cytolytic T-cell granules complexed to proteoglycans. Nature. 1998;391:908–911. doi: 10.1038/36129. [DOI] [PubMed] [Google Scholar]
- Parren P.W.H.I., Moore J.P., Burton D.R., Sattentau Q.J. The neutralizing antibody response to HIV-1viral evasion and escape from humoral immunity. AIDS. 1999;13:S137–S162. [PubMed] [Google Scholar]
- Ciurea A., Klenerman P., Hunziker L., Horvath E., Senn B.M., Ochsenbein A.F., Hengartner H., Zinkernagel R.M. Viral persistence in vivo through selection of neutralizing antibody-escape variants. Proc. Natl. Acad. Sci. USA. 2000;97:2749–2754. doi: 10.1073/pnas.040558797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parekh B.S., Buchmeier M.J. Proteins of lymphocytic choriomeningitis virusantigenic topography of the viral glycoproteins. Virology. 1986;153:168–178. doi: 10.1016/0042-6822(86)90020-6. [DOI] [PubMed] [Google Scholar]
- Ciurea A., Klenerman P., Hunziker L., Horvath E., Odermatt B., Ochsenbein A.F., Hengartner H., Zinkernagel R.M. Persistence of lymphocytic choriomeningitis virus at very low levels in immune mice. Proc. Natl. Acad. Sci. USA. 1999;96:11964–11969. doi: 10.1073/pnas.96.21.11964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fung-Leung W.P., Schilham M.W., Rahemtulla A., Kundig T.M., Vollenweider M., Potter J., Van Ewijk W., Mak T.W. CD8 is needed for development of cytotoxic T cells but not helper T cells. Cell. 1991;65:443–449. doi: 10.1016/0092-8674(91)90462-8. [DOI] [PubMed] [Google Scholar]
- Oxenius A., Bachmann M.F., Zinkernagel R.M., Hengartner H. Virus-specific MHC class II-restricted TCR-transgenic miceeffects on humoral and cellular immune responses after viral infection. Eur. J. Immunol. 1998;28:390–400. doi: 10.1002/(SICI)1521-4141(199801)28:01<390::AID-IMMU390>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- Battegay M., Cooper S., Althage A., Baenziger J., Hengartner H., Zinkernagel R.M. Quantification of lymphocytic choriomeningitis virus with an immunological focus assay in 24- or 96-well plates. J. Virol. Methods. 1991;33:191–198. doi: 10.1016/0166-0934(91)90018-u. [DOI] [PubMed] [Google Scholar]
- Seiler P., Senn B.M., Brundler M.A., Zinkernagel R.M., Hengartner H., Kalinke U. In vivo selection of neutralization-resistant virus variants but no evidence of B cell tolerance in lymphocytic choriomeningitis virus carrier mice expressing a transgenic virus-neutralizing antibody. J. Immunol. 1999;162:4536–4541. [PubMed] [Google Scholar]
- Ochsenbein A.F., Pinschewer D.D., Odermatt B., Ciurea A., Hengartner H., Zinkernagel R.M. Correlation of T cell independence of antibody responses with antigen dose reaching secondary lymphoid organsimplications for splenectomized patients and vaccine design. J. Immunol. 2000;164:6296–6302. doi: 10.4049/jimmunol.164.12.6296. [DOI] [PubMed] [Google Scholar]
- Domingo E., Holland J.J. RNA virus mutations and fitness for survival. Annu. Rev. Microbiol. 1997;51:151–178. doi: 10.1146/annurev.micro.51.1.151. [DOI] [PubMed] [Google Scholar]
- Oxenius A., Zinkernagel R.M., Hengartner H. Comparison of activation versus induction of unresponsiveness of virus-specific CD4+ and CD8+ T cells upon acute versus persistent viral infection. Immunity. 1998;9:449–457. doi: 10.1016/s1074-7613(00)80628-7. [DOI] [PubMed] [Google Scholar]
- Varga S.M., Welsh R.M. Detection of a high frequency of virus-specific CD4+ T cells during acute infection with lymphocytic choriomeningitis virus. J. Immunol. 1998;161:3215–3218. [PubMed] [Google Scholar]
- Varga S.M., Welsh R.M. High frequency of virus-specific interleukin-2-producing CD4+ T cells and Th1 dominance during lymphocytic choriomeningitis virus infection. J. Virol. 2000;74:4429–4432. doi: 10.1128/jvi.74.9.4429-4432.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamperschroer C., Quinn D.G. Quantification of epitope-specific MHC class-II-restricted T cells following lymphocytic choriomeningitis virus infection. Cell. Immunol. 1999;193:134–146. doi: 10.1006/cimm.1999.1458. [DOI] [PubMed] [Google Scholar]
- Charan S., Zinkernagel R.M. Antibody mediated suppression of secondary IgM response in nude mice against vesicular stomatitis virus. J. Immunol. 1986;136:3057–3061. [PubMed] [Google Scholar]
- Moskophidis D., Lechner F., Pircher H., Zinkernagel R.M. Virus persistence in acutely infected immunocompetent mice by exhaustion of antiviral cytotoxic effector T cells. Nature. 1993;362:758–761. doi: 10.1038/362758a0. [DOI] [PubMed] [Google Scholar]
- Zajac A.J., Blattman J.N., Murali-Krishna K., Sourdive D.J., Suresh M., Altman J.D., Ahmed R. Viral immune evasion due to persistence of activated T cells without effector function. J. Exp. Med. 1998;188:2205–2213. doi: 10.1084/jem.188.12.2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantaleo G., Soudeyns H., Demarest J.F., Vaccarezza M., Graziosi C., Paolucci S., Daucher M., Cohen O.J., Denis F., Biddison W.E. Evidence for rapid disappearance of initially expanded HIV-specific CD8+ T cell clones during primary HIV infection. Proc. Natl. Acad. Sci. USA. 1997;94:9848–9853. doi: 10.1073/pnas.94.18.9848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein M.R., van Baalen C.A., Holwerda A.M., Kerkhof G.S., Bende R.J., Keet I.P., Eeftinck-Schattenkerk J.K., Osterhaus A.D., Schuitemaker H., Miedema F. Kinetics of Gag-specific cytotoxic T lymphocyte responses during the clinical course of HIV-1 infectiona longitudinal analysis of rapid progressors and long-term asymptomatics. J. Exp. Med. 1995;181:1365–1372. doi: 10.1084/jem.181.4.1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appay V., Nixon D.F., Donahoe S.M., Gillespie G.M., Dong T., King A., Ogg G.S., Spiegel H.M., Conlon C., Spina C.A. HIV-specific CD8+ T cells produce antiviral cytokines but are impaired in cytolytic function. J. Exp. Med. 2000;192:63–75. doi: 10.1084/jem.192.1.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pircher H., Moskophidis D., Rohrer U., Bürki K., Hengartner H., Zinkernagel R.M. Viral escape by selection of cytotoxic T cell-resistant virus variants in vivo. Nature. 1990;346:629–633. doi: 10.1038/346629a0. [DOI] [PubMed] [Google Scholar]
- Phillips R.E., Rowland-Jones S., Nixon D.F., Gotch F.M., Edwards J.P., Ogunlesi A.O., McMichael A.J. Human immunodeficiency virus genetic variation that can escape cytotoxic T cell recognition. Nature. 1991;354:453–459. doi: 10.1038/354453a0. [DOI] [PubMed] [Google Scholar]
- Evans D.T., O'Connor D.H., Jing P., Dzuris J.L., Sidney J., Da Silva J., Allen T.M., Horton H., Venham J.E., Rudersdorf R.A. Virus-specific cytotoxic T-lymphocyte responses select for amino-acid variation in simian immunodeficiency virus Env and Nef. Nat. Med. 1999;5:1270–1276. doi: 10.1038/15224. [DOI] [PubMed] [Google Scholar]
- Weiner A., Erickson A.L., Kansopon J., Crawford K., Muchmore E., Hughes A.L., Houghton M., Walker C.M. Persistent hepatitis C virus infection in a chimpanzee is associated with emergence of a cytotoxic T lymphocyte escape variant. Proc. Natl. Acad. Sci. USA. 1995;92:2755–2759. doi: 10.1073/pnas.92.7.2755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borrow P., Lewicki H., Wei X., Horwitz M.S., Peffer N., Meyers H., Nelson J.A., Gairin J.E., Hahn B.H., Oldstone M.B., Shaw G.M. Antiviral pressure exerted by HIV-1-specific cytotoxic T lymphocytes (CTLs) during primary infection demonstrated by rapid selection of CTL escape virus. Nat. Med. 1997;3:205–211. doi: 10.1038/nm0297-205. [DOI] [PubMed] [Google Scholar]
- Allen T.M., O'Connor D.H., Jing P., Dzuris J.L., Mothe B.R., Vogel T.U., Dunphy E., Liebl M.E., Emerson C., Wilson N. Tat-specific cytotoxic T lymphocytes select for SIV escape variants during resolution of primary viraemia. Nature. 2000;407:386–390. doi: 10.1038/35030124. [DOI] [PubMed] [Google Scholar]
- Fazekas de St., Groth S., Webster R.G. Disquisitions on original antigenic sin. IEvidence in man. J. Exp. Med. 1966;140:355–360. doi: 10.1084/jem.124.3.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fazekas de St., Groth S., Webster R.G. Disquisitions on original antigenic sin. IIProof in lower creatures. J. Exp. Med. 1966;124:347–361. doi: 10.1084/jem.124.3.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matloubian M., Kolhekar S.R., Somasundaram T., Ahmed R. Molecular determinants of macrophage tropism and viral persistenceimportance of single amino acid changes in the polymerase and glycoprotein of lymphocytic choriomeningitis virus. J. Virol. 1993;67:7340–7349. doi: 10.1128/jvi.67.12.7340-7349.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moskophidis D., Battegay M., van den Broek M.F., Laine E., Hoffmann Rohrer U., Zinkernagel R.M. Role of virus and host variables in virus persistence or immunopathological disease caused by a non-cytolytic virus. J. Gen. Virol. 1995;76:381–391. doi: 10.1099/0022-1317-76-2-381. [DOI] [PubMed] [Google Scholar]
- Moskophidis D., Battegay M., Bründler M.-A., Laine E., Gresser I., Zinkernagel R.M. Resistance of lymphocytic choriomeningitis virus to alpha/beta interferon and to gamma interferon. J. Virol. 1994;68:1951–1955. doi: 10.1128/jvi.68.3.1951-1955.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Planz O., Seiler P., Hengartner H., Zinkernagel R.M. Specific cytotoxic T cells eliminate cells producing neutralizing antibodies. Nature. 1996;382:726–729. doi: 10.1038/382726a0. [DOI] [PubMed] [Google Scholar]
- Vieira P., Rajewsky K. Persistence of memory B cells in mice deprived of T cell help. Int. Immunol. 1990;2:487–494. doi: 10.1093/intimm/2.6.487. [DOI] [PubMed] [Google Scholar]
- Ruedl C., Kopf M., Bachmann M.F. CD8+ T cells mediate CD40-independent maturation of dendritic cells in vivo. J. Exp. Med. 1999;189:1875–1884. doi: 10.1084/jem.189.12.1875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallimore A., Glithero A., Godkin A., Tissot A.C., Pluckthun A., Elliott T., Hengartner H., Zinkernagel R.M. Induction and exhaustion of lymphocytic choriomeningitis virus-specific cytotoxic T lymphocytes visualized using soluble tetrameric major histocompatibility complex class I-peptide complexes. J. Exp. Med. 1998;187:1383–1393. doi: 10.1084/jem.187.9.1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King C.C., Jamieson B.D., Reddy K., Bali N., Concepcion R.J., Ahmed R. Viral infection of the thymus. J. Virol. 1992;66:3155–3160. doi: 10.1128/jvi.66.5.3155-3160.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalams S.A., Walker B.D. The critical need for CD4 help in maintaining effective cytotoxic T lymphocyte responses. J. Exp. Med. 1998;188:2199–2204. doi: 10.1084/jem.188.12.2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battegay M., Moskophidis D., Rahemtulla A., Hengartner H., Mak T.W., Zinkernagel R.M. Enhanced establishment of a virus carrier state in adult CD4+ T-cell-deficient mice. J. Virol. 1994;68:4700–4704. doi: 10.1128/jvi.68.7.4700-4704.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen J.P., Marker O., Thomsen A.R. The role of CD4+ T cells in cell-mediated immunity to LCMVstudies in MHC class I and class II deficient mice. Scand. J. Immunol. 1994;40:373–382. doi: 10.1111/j.1365-3083.1994.tb03477.x. [DOI] [PubMed] [Google Scholar]
- Matloubian M., Concepcion R.J., Ahmed R. CD4+ T cells are required to sustain CD8+ cytotoxic T-cell responses during chronic viral infection. J. Virol. 1994;68:8056–8063. doi: 10.1128/jvi.68.12.8056-8063.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Herrath M.G., Yokoyama M., Dockter J., Oldstone M.B., Whitton J.L. CD4-deficient mice have reduced levels of memory cytotoxic T lymphocytes after immunization and show diminished resistance to subsequent virus challenge. J. Virol. 1996;70:1072–1079. doi: 10.1128/jvi.70.2.1072-1079.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg E.S., Billingsley J.M., Caliendo A.M., Boswell S.L., Sax P.E., Kalams S.A., Walker B.D. Vigorous HIV-1-specific CD4+ T cell responses associated with control of viremia. Science. 1997;278:1447–1450. doi: 10.1126/science.278.5342.1447. [DOI] [PubMed] [Google Scholar]
- Pitcher C.J., Quittner C., Peterson D.M., Connors M., Koup M., Maino V.C., Picker L.J. HIV-1-specific CD4+ T cells are detectable in most individuals with active HIV-1 infection, but decline with prolonged viral suppression. Nat. Med. 1999;5:518–525. doi: 10.1038/8400. [DOI] [PubMed] [Google Scholar]
- Kalams S.A., Buchbinder S.P., Rosenberg E.S., Billingsley J.M., Colbert D.S., Jones N.G., Shea A.K., Trocha A.K., Walker B.D. Association between virus-specific cytotoxic T-lymphocyte and helper responses in human immunodeficiency virus type 1 infection. J. Virol. 1999;73:6715–6720. doi: 10.1128/jvi.73.8.6715-6720.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerlach J.T., Diepolder H.M., Jung M.C., Gruener N.H., Schraut W.W., Zachoval R., Hoffmann R., Schirren C.A., Santantonio T., Pape G.R. Recurrence of hepatitis C virus after loss of virus-specific CD4+ T-cell response in acute hepatitis C. Gastroenterology. 1999;117:933–941. doi: 10.1016/s0016-5085(99)70353-7. [DOI] [PubMed] [Google Scholar]
- Oxenius A., Price D.A., Easterbrook P.J., O'Callaghan C.A., Kelleher A.D., Whelan J.A., Sontag G., Sewell A.K., Phillips R.E. Early highly active antiretroviral therapy for acute HIV-1 infection preserves immune function of CD8+ and CD4+ T lymphocytes. Proc. Natl. Acad. Sci. USA. 2000;97:3382–3387. doi: 10.1073/pnas.97.7.3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg E.S., Altfeld M., Poon S.H., Phillips M.N., Wilkes B.M., Eldridge R.L., Robbins G.K., D'Aquila R.T., Goulder P.J.R., Walker B.D. Immune control of HIV-1 after early treatment of acute infection. Nature. 2000;407:523–526. doi: 10.1038/35035103. [DOI] [PubMed] [Google Scholar]
- Bradney A.P., Scheer S., Crawford J.M., Buchbinder S.P., Montefiori D.C. Neutralization escape in human immunodeficiency virus type 1-infected long-term nonprogressors. J. Infect. Dis. 1999;179:1264–1267. doi: 10.1086/314711. [DOI] [PubMed] [Google Scholar]
- Baldridge J.R., McGraw T.S., Paoletti A., Buchmeier M.J. Antibody prevents the establishment of persistent arenavirus infection in synergy with endogenous T cells. J. Virol. 1997;71:755–758. doi: 10.1128/jvi.71.1.755-758.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]