

Abstract
We eluted peptides from class I molecules of HLA-A2.1+ breast adenocarcinoma and loaded reverse phase high-performance liquid chromatography (HPLC) fractions onto dendritic cells to prime naive CD8+ T cells. Fractions that supported growth of tumor-specific cytotoxic T lymphocytes were analyzed by nano-HPLC micro-ESI tandem mass spectrometry. Six HLA-A2.1-binding peptides, four 9-mers (P1-P4) differing in the COOH-terminal residue, and two 10-mers (P5 and P6) with an additional COOH-terminal alanine, were identified in one fraction. Peptide sequences were homologous to cyclin B1. We primed CD8+ T cells from another HLA-A2.1+ healthy donor with synthetic peptides and generated P4-specific responses. We also detected memory T cells specific for one or more of these peptides in patients with breast cancer and squamous cell carcinomas of the head and neck (SCCHN). T cells from one patient, restimulated once in vitro, could kill the tumor cell line from which the peptides were derived. Immunohistochemical analysis of tumor lines and tissue sections showed cyclin B1 overexpression and aberrant localization in the cytoplasm instead of the nucleus. Sequencing genomic DNA and cDNA corresponding to P1–P6 region showed that differences in COOH-terminal residues were not due to either DNA mutations or errors in transcription, suggesting a high error rate in translation of cyclin B1 protein in tumors.
Keywords: dendritic cells, in vitro priming, tandem mass spectrometry, CTL, ELISPOT
Introduction
For tumor-specific immunotherapy to be successful, tumor antigens must be identified that are effective in inducing strong immune responses in the host. A number of methods have been described to identify tumor antigens, including transfection of recombinant tumor cDNA libraries and HLA molecules into target cells (“genetic approach”) (1, 2), elution of peptides from the binding cleft of tumor HLA molecules (“peptide-elution approach”) (3, 4), deduction of peptide sequences from known oncogenes or tumor-associated proteins using known HLA-anchor motifs (“reverse immunology approach”) (5, 6), and more recently, serological analysis of recombinant tumor cDNA expression libraries (SEREX) (7). These methods have led to the discovery of a number of tumor antigens, primarily in melanomas (8). These tumor antigens have been classified into five categories: (a) cancer testis antigens, such as MAGE (9, 10), BAGE (11), GAGE (12), and more recently, NY-ESO-1 (13); (b) differentiation antigens, such as gp100 (14), Melan-A/Mart-1 (2, 15), and tyrosinase (1); (c) mutated antigens, such as cdk4 (16), β-catenin (17), and cdc27 (18); (d) overexpressed and/or ubiquitous antigens, such as PRAME (19), and p53 (20), and (e) viral antigens, such as human papillomavirus (HPV) (21) and EBV (22). Very few tumor-specific antigens have been identified in epithelial tumors, the best known of which are CEA (23), PSA (24), Her-2/Neu (25), and MUC1 (26).
We recently described a novel method for tumor antigen discovery whereby we primed naive CD8+ T cells from healthy donors to in vitro generated dendritic cells (DCs)* loaded with peptides eluted from HLA class I molecules of a breast cancer cell line (27). We identified several fractions that supported growth of tumor-specific CTLs, suggesting that these fractions contained peptides that could potentially be tumor antigens. In this paper we report that one of these fractions contained peptides derived from cyclin B1, a cell cycle checkpoint protein that regulates the transition from G2 to M phase (28). Staining of the tumor cell line with anti-cyclin B1 antibody revealed that this protein was greatly overexpressed and found mostly in the cytoplasm rather than in its normal location, the nucleus. We were able to prime naive CD8+ T cells from another healthy donor using synthetic peptides and detect T cell responses to one of the peptides using the IFN-γ enzyme-linked immunospot (ELISPOT) assay. We also detected secondary T cell responses to one or more of these peptides in breast cancer patients and patients with squamous cell carcinomas of the head and neck (SCCHN). Where we were able to evaluate, these T cell responses correlated with the overexpression of cyclin B1 protein in the patients' tumors. In the fresh tumor samples, like in the cell lines, the overexpressed protein was found predominantly in the cytoplasm instead of the nucleus. We propose that this aberrant overexpression of cyclin B1 in the cytoplasm leads to overabundance of cyclin B1–derived peptides in tumor HLA class I molecules, which can then be recognized by T cells as tumor antigens.
Materials and Methods
Cells and Tumor Cell Lines
MS-A2 is an HLA-A*0201 transfected tumor cell line derived from a breast adenocarcinoma cell line MS, characterized in our laboratory (27, 29). MS-A2/CD80 is the MS-A2 cell line retrovirally transduced with the CD80 gene obtained from Corixa Corporation. The lung tumor cell line, 201T, was obtained from Dr. Jill Siegfried, University of Pittsburgh School of Medicine. The head and neck tumor cell line, PCI-13, was also established by us (30). T cells, DCs, and macrophages were derived from the peripheral blood of HLA-A*0201+ healthy donors and cancer patients under an Institutional Review Board approved protocol and with signed informed consent.
Peptide Synthesis
All peptides used in this study were synthesized with F-moc chemistry using the 432A Synergy Peptide Synthesizer (Applied Biosystems). Peptides were purified by reverse phase (RP)-HPLC to greater than 85% purity, dissolved in DMSO, and frozen until further use.
Antibodies
MA2.1, a mouse anti–human HLA-A2.1 antibody, was produced by the MA2.1 hybridoma; W6/32, a mouse anti–human MHC class I antibody, was produced by the W6/32 hybridoma; both were obtained from the American Type Culture Collection. GNS-1, a mouse anti–human cyclin B1 antibody, was purchased from BD PharMingen.
Mass Spectrometry Data Acquisition
Active first-dimension HPLC fractions were screened for peptide content on a home-built Fourier transform mass spectrometer (FTMS), equipped with a nano-flow high performance liquid chromatography micro-electrospray ionization (nano-HPLC micro-ESI) interface (31). Nano-HPLC columns were constructed from 50 μm internal diameter fused silica capillaries and packed with an 8-cm bed length of 5-μm diameter reversed phase beads. An integrated microESI emitter tip (∼2 μm diameter) was located a few mm from the column bed. Typically, ∼0.75 μl (corresponding to ∼2.3 × 108 cell equivalents or ∼1.5%) of an active, first-dimension HPLC fraction was loaded onto a column and eluted directly into the mass spectrometer with a linear, 17-min gradient of 0–70% acetonitrile in 0.1% acetic acid. Full scan mass spectra, over a mass-to-charge (m/z) range 300 ≤ m/z ≤ 2500, were acquired at a rate of ∼1 scan/second.
Mass Spectrometry/Mass Spectrometry Data Acquisition
Mass spectrometry/mass spectrometry (MS/MS) data were acquired on a Finnigan LCQ quadrupole ion trap mass spectrometer (Finnigan Corp.), equipped with a nano-HPLC micro-ESI source as described above. Typically, ∼1.5 μl (corresponding to ∼4.5 × 108 cell equivalents or ∼3%) of an active, first-dimension HPLC fraction was loaded onto a column eluted directly into the mass spectrometer with a linear, 30 min gradient of 0–30% acetonitrile in 0.1% acetic acid. Data-dependent spectral acquisition (32) was performed as follows: A full scan mass spectrum (MS) was acquired over the range 300 ≤ m/z ≤ 2,000. The instrument control computer then selected the top five most abundant ion species in the MS scan for subsequent MS/MS analysis over the next five scans. After acquiring MS/MS data on a particular ion species, its corresponding m/z value was excluded from consideration by the instrument control computer for a period equal to the observed chromatographic peak width (∼1.5 min for the data herein). This data acquisition procedure minimized redundancy and allowed MS/MS analysis on peptide species whose abundances spanned a wide dynamic range. After acquisition, tandem mass spectral data were searched using SEQUEST (33), an algorithm that matches uninterpreted MS/MS spectra to theoretical spectra for peptides generated from user-specified databases. All data herein were searched against nonredundant (nr) protein databases compiled at the National Center for Biotechnology Information (NCBI). In addition, manual (e.g., de novo) interpretation of MS/MS spectra was performed. Peptide sequence information obtained in this manner was compared with sequences in the nr (nonredundant) protein database using the MS-TAG algorithm (34). Candidate peptide sequences were subsequently confirmed by comparison of their MS/MS spectra acquired for synthetic analogs.
HLA Class I Stabilization Assays
Peptide-induced stabilization of HLA-A2.1 molecules on T2 cells was done as described previously (35). 2 × 105 T2 cells were incubated with 20 μg/ml of the indicated synthetic peptides in 3 μg/ml human B2m (Calbiochem) for 18–20 h at room temperature. The cells were then stained with the HLA-A2.1–specific antibody, MA2.1, for 45 min, washed with FACS® buffer (PBS, 5% FBS, and 0.01% sodium azide), and stained with a secondary FITC-conjugated anti-IgG antibody (Biosource International). The cells were fixed in 4% formaldehyde before flow cytometry analysis. The negative control consisted of T2 cells without peptide. The positive control consisted of T2 cells loaded with the Flu matrix peptide, GILGFVFTL. Flow cytometric analysis was done using a FACScan™ flow cytometer (Becton Dickinson). Experimental results are depicted as X-Fold increase = mean fluorescent intensity of T2 cells loaded with peptide/mean fluorescent intensity of T2 cells with no peptide. An X-Fold increase of >1 indicates that the peptide binds to HLA-A2.1.
IFN-γ ELISPOT Assays
IFN-γ ELISPOT assays were done as described previously (36). Briefly, nitrocellulose plates (Millipore) were coated with the anti–IFN-γ capture mAb 1-D1K (MabTech) overnight at 4°C. For assays using DCs as APCs, DCs were loaded with 10 μg of the indicated peptides for 2–6 h, and mixed with autologous T cells at a DC/T ratio of 1:10 for 20 h at 37°C. For assays using autologous PBMCs as APCs, PBMCs were irradiated at 3,000 Rads, loaded with 10 μg of peptides for 4 h, and mixed with autologous T cells at an APC/T ratio of 1:5 for 40 h at 37°C. The T cells were seeded at 3 × 103–105 cells/well. All assays were done in serum-free AIM-V medium (GIBCO BRL). The plates were then washed in PBS plus 0.1% Tween and stained with anti–IFN-γ mAb 7-B6–1 (Mabtech) for 2 h at 37°C. The plates were washed, and the avidin-peroxidase complex (Vectastain ABC Kit; Vector Laboratories) was added to the plates for 1 h. The plates were then developed using AEC (Sigma-Aldrich) substrate, and spots were quantified microscopically with an inverted phase-contrast microscope (Carl Zeiss, Inc.) along with a computer-assisted image analysis system (KS ELISPOT). For HLA class I blocking experiments, the W6/32 antibody was added to the APCs for 30–45 min before incubation with T cells.
T Cell Cultures
Priming Naive CD8+ T Cells from a Healthy Donor to Cyclin B1 Peptides In Vitro
Naive CD8+ T cells and in vitro generated DCs were purified as described previously (27). 2 × 104 DCs were loaded overnight with 10 μg/well of peptide in 96-well U-bottom plates (Falcon) and mixed with 2 × 105 autologous naive CD8+ T cells the next day in the presence of 2 ng/ml IL-1β (R&D Systems), 20 U/ml IL-2 (DuPont), and 10 U/ml IL-4 (Schering Plough). Depending on growth kinetics, T cells were fed every 3–4 d with 10 U/ml IL-2 and 5 U/ml IL-4. T cells were restimulated every 7–10 d using peptide-loaded autologous macrophages.
Stimulating CD8+ T Cells from Breast Cancer and SCCHN Patients with Cyclin B1 Peptides In Vitro
PBMCs from cancer patients were X-irradiated at 3,000 Rads, loaded with 10 μg of the indicated peptides for 2–4 h, and mixed with autologous PBMCs in the presence of 20 U/ml IL-2 (DuPont). The T cell cultures were fed every 3–4 d with 10 U/ml IL-2, and restimulated every 10–12 d, if necessary, using peptide-loaded autologous irradiated PBMCs. All T cell cultures were grown in RPMI medium (ICN Biomedicals) supplemented with 10% human AB sera (Gemini Products), L-glutamine, and penicillin/streptomycin (Life Technologies).
Cytotoxicity Assays
Target cells were labeled with 50 μCi of Na2 51CrO4 (Amersham Pharmacia Biotech) for 90 min at 37°C. The labeled cells were then washed three times and plated at 103 cells/well in a 96-well V-bottom plate (Costar) with various numbers of effector T cells. In addition, a 50-fold excess of unlabeled K562 (5 × 104) cells was added to the wells for 30 min before the addition of T cells to minimize the detection of lymphokine-activated killer (LAK) activity in the assay. The plates were briefly centrifuged and incubated for 4 h at 37°C. All determinations were done in triplicate. Supernatants were harvested using a Skatron harvesting process (Skatron Instruments) and counted on a Cobra II series auto gamma counting system (Packard Instrument Co.). Maximum release was obtained by adding 50 μl of 1% Triton X-100 to the labeled target cells. Spontaneous release was obtained by incubating the labeled cells in the absence of T cells. Percent specific lysis was calculated from the following formula: % specific lysis = 100 × (experimental release − spontaneous release)/(maximum release − spontaneous release).
Immunohistochemical Staining of Cyclin B1 in Tumor Cell Lines and in Tumor Sections
For tumor cell lines, the cells were left to adhere overnight on poly-lysine charged slides (Fisher Scientific) in the presence of RPMI plus 10% FBS (Cellgro®; Media Tech, Inc.). The cells were then fixed for 15 min on ice with either 2% Triton-X or 50% Formalin/50% acetone, blocked with serum, and stained with the anti-cyclin B1 antibody, GNS-1 (BD PharMingen). The avidin-biotin peroxidase method was then applied according to manufacturer's instructions using the Vectastain ABC Elite™ staining kit (Vector Laboratories). For SCCHN sections, formalin-fixed, paraffin-embedded tumor tissues were sectioned (3–5 μm), air-dried overnight at 37°C, deparaffinized and dehydrated, and stained with the anti–human cyclin B1 antibody. The avidin-biotin peroxidase method was applied as above according to the instructions supplied by the manufacturer (Dako).
PCR Amplification of the Cyclin B1 Gene Fragment
Genomic DNA from two tumor cell lines (PCI-13, MS-A2) and from PBLs of two normal donors was extracted, ethanol precipitated, and first amplified with PCR primers P1 (5′-GTGTGCCCAAGAAGATGC-3′) and P3 (5′-AGTTGGTGTCCATTCACCATT-3′) flanking the region encoding peptides P1-P6 (nucleotide region 1,056–1,085 in HeLa cyclin B1 cDNA: GenBank/EMBL/DDBJ accession number M25753) under the following PCR conditions: initial denaturation step for 3 min at 94°C, followed by 35 cycles (30 s at 94°C, 1 min 55°C, 30 s at 72°C, and an elongation step for 7 min at 72°C. The PCR products were gel purified and a second nested-PCR reaction was performed using primers P3 (described above) and P4 (5′-ATGCTGCAGCTGGTTGGTGTC-3′) to ensure that the region of interest in the cyclin B1 gene was actually being amplified. The PCR products were then sequenced.
Cloning of Cyclin B1 cDNA Fragment Amplified by PCR
Total cellular RNA was extracted with Trizol™ following the manufacturer's protocol (GIBCO BRL), and reverse-transcribed into cDNA using the GeneAmp® RNA PCR Kit (PerkinElmer). Total RNA from normal lung tissue was purchased from Ambion. PCR amplification was done using primers P1 and P3 (described above), and the PCR products were cloned into the pcDNA3.1/V5-His-TOPO® plasmid vector using the TA Cloning® Kit (Invitrogen). Transformants were screened for inserts using PCR primers P3 and P4, and plasmids isolated from positive clones were sequenced.
DNA Sequence Analysis
DNA sequencing was done at the University of Pittsburgh DNA Sequencing Facility using the dideoxy-chain termination method (PerkinElmer) with the specific cyclin B1 oligonucleotides P3 and P4 as primers.
Results
Identification of Tumor-derived Peptides Stimulatory to T Cells
Peptides were acid-extracted from immunoaffinity purified HLA class I molecules of an HLA-A*0201 epithelial tumor cell line MS-A2, fractionated by RP-HPLC, and loaded onto DCs from a healthy donor to prime in vitro autologous naive CD8+ T cells as described previously (27). One RP-HPLC fraction whose peptides supported the growth of tumor-specific CTLs (27) was further analyzed by nano-HPLC micro ESI tandem mass spectrometry. Analysis of the resulting MS/MS data yielded six candidate peptide sequences that corresponded to the mass range expected for HLA class I–associated peptides (700–1,300 D). The abundances of these candidates represented the majority of total ion current observed in the mass range of 700–1,300 D. Candidate peptide sequences were subsequently confirmed by comparison of their mass spectra acquired for synthetic analogs. All six peptides had related sequences, with four being 9 amino acids long, and two being 10 amino acids long (Table I) . The four 9-mers (P1-P4) were identical in the first eight amino acids and differed at the COOH terminus, where the amino acids were valine (P1), methionine (P2), phenylalanine (P3), and cysteine (P4). The two 10-mers, P5 and P6, were identical to P2 and P3, respectively, except for an additional alanine at the COOH terminus. When these sequences were entered into the protein database, they were found to be homologous to a human cyclin B1 sequence derived from HeLa cells. These sequences were also homologous to mouse and rat cyclin B1 (Table I).
Table I.
Cyclin B1 Sequences Derived from the Tumor and Database Bind to HLA-A2.1
| Sequencea | HLA-A2.1 bindingb |
|
|---|---|---|
| Cyclin B1 peptides from the tumor | ||
| P1 | AGYLMELCV | 1.60 |
| P2 | AGYLMELCM | 1.48 |
| P3 | AGYLMELCF | 1.42 |
| P4 | AGYLMELCC | 1.61 |
| P5 | AGYLMELCMA | 1.58 |
| P6 | AGYLMELCFA | 2.08 |
| Cyclin B1 peptides from the database | ||
| Human cyclin B1 (CB9) | AKYLMELTM | 2.28 |
| Human cyclin B1 (CB10) | AKYLMELTML | 2.25 |
| Mouse cyclin B1 | AKYLMELSML | − |
| Rat cyclin B1 | AKYLMELSML | − |
Tumor-derived peptides were sequenced by electrospray ionization tandem mass spectrometry, yielding six peptides (P1–P6) having high homology to human HeLa cyclin B1 (GenBank/EMBL/DDBJ accession number: P14635), as well as mouse (GenBank/EMBL/DDBJ accession number: P24860) and rat cyclin B1 (GenBank/EMBL/DDBJ accession number: P30277).
X-Fold increase = (mean fluorescent intensity of T2 cells loaded with peptide/mean fluorescent intensity of T2 cells with no peptide). An X-Fold increase of >1 indicates that the peptide binds to HLA-A2.1. The positive control consisted of T2 cells loaded with the Flu matrix peptide (GILGFVFTL), which had an X-Fold increase of 2.2.
We synthesized peptides P1-P6 along with the homologous 9 and 10 residue HeLa sequences (CB9 and CB10) from the database and tested them for their ability to bind HLA-A2.1 using the T2 cell line and class I stabilization assay. As leucine and isoleucine have identical masses and are not distinguished by low-energy MS/MS analysis, the peptides were synthesized with leucines at positions 4 and 7 to match the leucines present at these same positions in the HeLa cyclin B1 sequence, as well as in the mouse and rat cyclin B1 sequences. All the peptides bound to HLA-A2.1 with various affinities, as measured by increases in mean fluorescent intensity in anti–HLA-A2.1 staining of peptide-loaded T2 cells. The HeLa-derived cyclin B1 peptides also bound to HLA-A2.1. Affinities of the HeLa-derived cyclin B1 peptides were higher than that of the tumor-derived peptides, except for P6 (Table I).
CD8+ T Cells from an HLA-A*0201 Healthy Donor Can Be Primed to Synthetic Cyclin B1 Peptide P4
As the cyclin B1 peptides were derived from a first dimension HPLC fraction that primed tumor-specific CTLs from an HLA-A2.1+ donor, we sought to determine whether these peptides were indeed responsible for the immunostimulatory activity. We used naive CD8+ T cells from another healthy HLA-A2.1+ donor and autologous DCs loaded with the individual synthetic peptides (P1–P6). No T cell responses were detected against the peptides in the absence of in vitro stimulation in either this donor or another HLA-A2.1+ donor (data not shown). However, after four rounds of stimulation, we detected antigen-specific IFN-γ secretion by CD8+ T cells in response to P4, and not to other peptides (P1, P2, P5, P6) or peptide HIV-POL (ILKEPGSHV) that is known to bind HLA-A2.1 and served here as the negative control (Fig. 1) . We were able to block the T cell response to P4 using the anti–class I antibody W6/32, showing that the P4-specific responses were HLA class I restricted. These in vitro primed T cells that specifically recognized P4-loaded DCs were unable to kill the original tumor from which the peptides were derived (data not shown). This was not unexpected considering that these T cells were primed with high concentrations of peptide (50 μM), and are thus expected to be of low affinity and incapable of recognizing the comparatively lower levels of the same HLA-peptide complexes on the tumor.
Figure 1.

HLA class I–restricted T cell response of a healthy HLA-A2+ donor to cyclin B1 peptides. CD8+ T cells generated in vitro by priming to synthetic cyclin B1 peptides were used in an IFN-γ ELISPOT assay after the fourth restimulation. Number of T cells per well is indicated in parenthesis.
HLA Class I–restricted Memory T Cell Responses against Cyclin B1 Peptides in HLA-A*0201 Breast Cancer Patients
We tested PBMCs from six breast cancer patients who had undergone surgery but had not yet started chemotherapy, for their ability to recognize the cyclin B1 peptides in an IFN-γ ELISPOT assay. Four out of the six HLA-A2+ breast cancer patients tested exhibited secondary responses against one or more of the cyclin B1 peptides (Fig. 2) . Patient A exhibited strong HLA class I–restricted secondary T cell responses to three of the three peptides tested, P4, CB9, and CB10, after only one in vitro stimulation. There was no recognition of the HIV-POL control peptide. Patient B appeared to have a weak secondary response to one of the three peptides tested, P1, and only after two in vitro stimulations. This patient was later found to be HLA-A*0206, suggesting that, if the anti-P1 response is real, P1 may also bind to HLA-A*0206.
Figure 2.
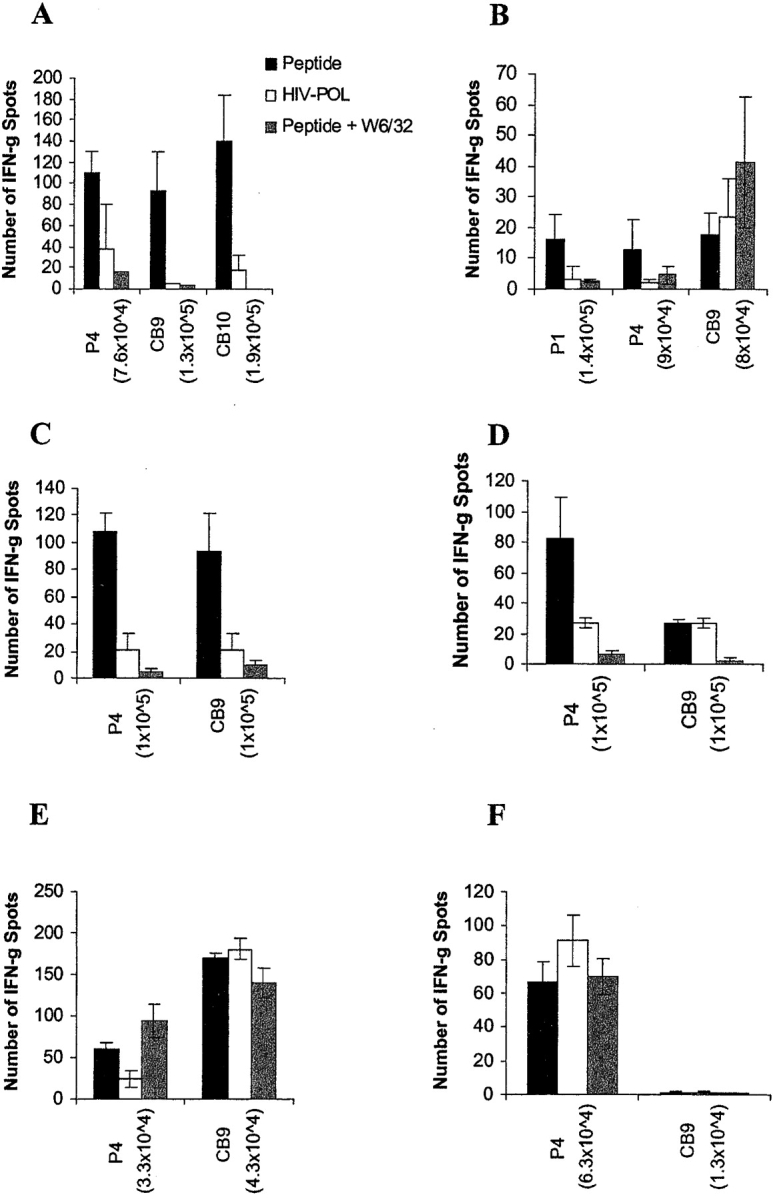
HLA class I–restricted T cell responses to cyclin B1 peptides in HLA-A2+ breast cancer patients. PBMCs were tested for recognition of cyclin B1 peptides after one in vitro stimulation (A, B, E, and F) or no in vitro stimulation (C and D) in an IFN-γ ELISPOT assay. Number of T cells per well is indicated in parenthesis.
Most interestingly, we detected secondary responses in the absence of any in vitro stimulation. Patient C showed peptide-specific HLA class I–restricted T cell responses to two of two peptides tested, P4 and CB9. Patient D showed strong HLA class I–restricted T cell response to P4 and lower responses against both the HIV-POL peptide and CB9. However, only CB9 response was blocked by the anti-MHC class I antibody, confirming antigen specificity. Patients E and F did not respond to either of the two peptides tested, P4 and CB9. We also did not detect responses to any of the peptides in an HLA-A*0201 negative patient (data not shown).
HLA Class I–restricted Memory T Cell Responses to Cyclin B1 Peptides in HLA-A*0201 SCCHN Patients
We also examined PBMCs from five HLA-A*0201 SCCHN patients for their ability to respond to the cyclin B1 peptides in an IFN-γ ELISPOT assay. These patients were chosen for having either stable disease or being in long term remission. We detected HLA class I–restricted T cell responses to one or more of the cyclin B1 peptides in four of the five patients tested (Fig. 3) . Patient A exhibited HLA class I–restricted T cell responses to five of eight peptides, after only one in vitro stimulation. For two of these peptides, P4 and CB9, we also detected peptide-specific T cells in the absence of any in vitro stimulation (data not shown).
Figure 3.

HLA class I–restricted T cell responses to cyclin B1 peptides in HLA-A2.1+ SCCHN patients. PBMCs were tested for recognition of cyclin B1 peptides after one (A) or two in vitro stimulations (B–D) in an IFN-γ ELISPOT assay.
Patient B exhibited HLA class I-restricted T cell responses to three of the five peptides (P3, P4, CB9), and not to the other two (P1, P2). Patient C showed heightened T cell responses to all five peptides tested, but only responses against P2 and CB10 could be blocked with anti-class I antibody. Patient D had the same heightened T cell response that appeared specific for three of the six peptides tested, P4, P5, and CB9. The fifth SCCHN patient failed to show a detectable response to any of the peptides even after three in vitro stimulations (data not shown).
T Cells from a SCCHN Patient Can Lyse the Tumor from Which the Peptides Were Derived
Because T cells from healthy individuals primed to synthetic cyclin B1 peptides in vitro could not kill tumor cells, we wanted to determine whether T cells from patients that we assumed had been primed in vivo could lyse tumor cells. We restimulated PBLs from one SCCHN patient once in vitro with either peptide P4 or peptide CB9 and tested the T cells for their ability to kill the original tumor MS-A2 from which the peptides were derived. We included in the assay the same tumor transduced with the CD80 gene to provide “costimulation” for T cell activation (MS-A2/CD80). As shown in Fig. 4 A, T cells restimulated once with P4 were able to lyse the MS-A2/CD80 tumor and to a lesser extent, MS-A2 tumor, but not the HLA-A2− target MS or K562 that was a control target for LAK activity. Similarly, in Fig. 4 B, T cells restimulated with CB9 peptide were able to lyse the MS-A2+/CD80+ tumor and not the other targets.
Figure 4.

T cells from an HLA-A2.1+ SCCHN patient restimulated to cyclin B1 peptides in vitro are able to kill the original tumor. T cells were restimulated with P4 (A), and CB9 (B) for 5 d and tested in a CTL assay.
Cyclin B1 Protein Is Overexpressed in Epithelial Tumor Cell Lines
To understand the reason why cyclin B1 peptides would elicit T cell responses in cancer patients, we examined by immunohistochemistry the cyclin B1 expression in the original tumor cell line MS-A2 from where they were first isolated and identified (Fig. 5, A and B) . There was intense staining of cyclin B1 protein in the tumor cells, predominantly found in the cytoplasm. Fig. 5, C and D, depict similar intense cytoplasmic staining of cyclin B1 in a human lung adenocarcinoma cell line 201T. No cyclin B1 staining was observed in normal cells, represented by primary cultures of human airway bronchoepithelial cells (Fig. 5 E).
Figure 5.
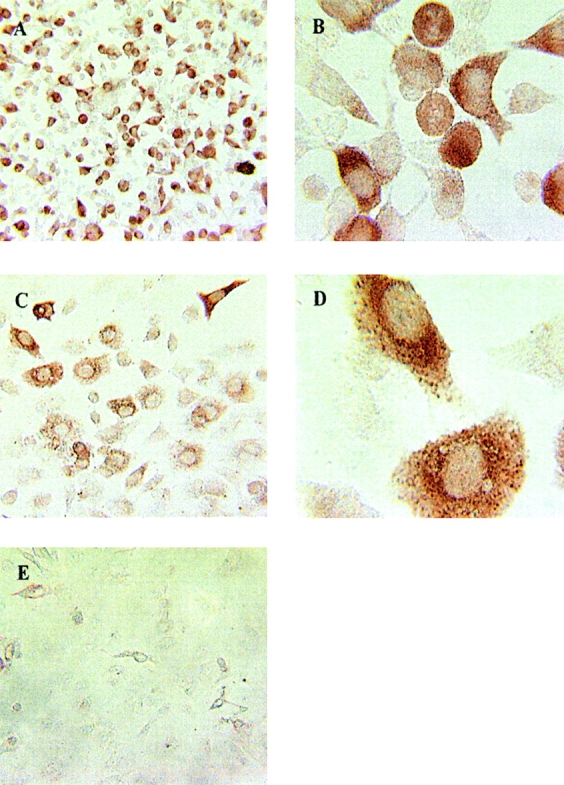
Cyclin B1 protein is overexpressed in epithelial tumor cells. (A) MS-A2 cells, original magnification, ×20. (B) MS-A2 cells, original magnification, ×40. (C) 201T cells, original magnification, ×20. (D) 201T cells, original magnification, ×40. (E) Human airway bronchoepithelial cells, original magnification, ×20.
Cyclin B1 Protein Is Also Overexpressed in SCCHN
To rule out that the intense staining of cyclin B1 observed in the tumor cell lines might be a result of a prolonged in vitro culture, we examined a tumor cell line as well as tumor tissue sections obtained from the SCCHN patients whom we had analyzed for cyclin B1–specific T cell responses. Fig. 6, A and B , shows intense cytoplasmic staining of cyclin B1 in the tumor cell line PCI-13, derived from the tumor of patient A in Fig. 3. Very high expression of cyclin B1 in the cell line correlates with strong cyclin B1–specific T cell responses observed in this patient. Intense cytoplasmic cyclin B1 staining was also observed in the tumor tissue samples (Fig. 6, C and D; E and F) of two other patients who exhibited cyclin B1-specific T cell responses (patients C and D, respectively; Fig. 3). No cyclin B1 staining was detected in the normal mucosa surrounding the tumor. The tumor seen in Fig. 6, G and H, showed weak and diffuse cyclin B1 staining that was not convincingly positive. Patient B from whom the tumor was obtained did have cyclin B1–specific T cell responses (Fig. 3). The same weak staining was seen in the tumor shown in Fig. 6, I and J, derived from a patient who did not exhibit any HLA class I–restricted T cell responses against the cyclin B1 peptides (data not shown).
Figure 6.
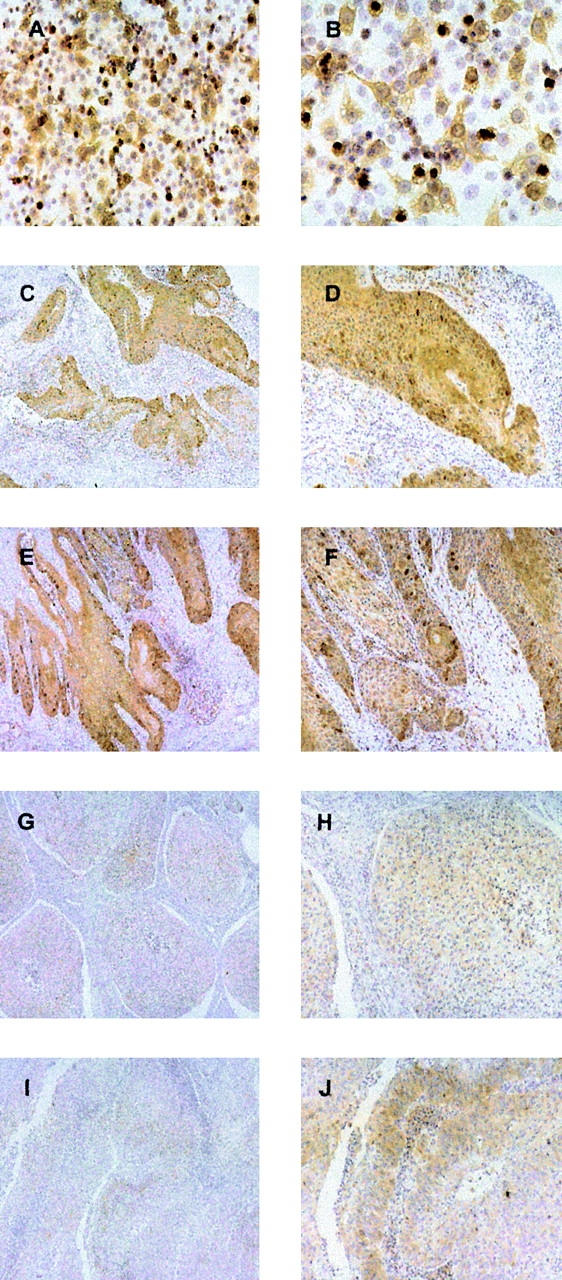
Cyclin B1 protein is overexpressed in SCCHN. A and B, SCCHN cell Line PCI-13. C–J, SCCHN tumor sections. A, C, E, G, and I, original magnification, ×10. B, D, F, H, and J, original magnification, ×20.
Differences in the COOH Terminus of the Cyclin B1 Peptides Are Not the Result of DNA or RNA Mutations
As cyclin B1 peptides P1-P6 that we isolated from MS-A2 tumor class I molecules differed from the HeLa sequence in the second and eighth amino acid and from each other in the COOH-terminal residues, we set out to examine if these differences were a result of mutations in the MS-A2 cells DNA or RNA. We sequenced the area in the genomic DNA corresponding to the region encoding the peptides. As the only human cyclin B1 sequence available in the database was from HeLa cells, we also sequenced the same region of genomic DNA from two normal donors, as well as from the SCCHN cell line PCI-13. The sequences were all identical in the region of interest (Table II) , ruling out mutations at the DNA level as the cause for generation of these differences in the cyclin B1 peptides. We next examined the RNA by cloning and sequencing the corresponding cyclin B1 cDNA from normal lung tissue and MS tumor. Most of the MS sequences were identical, except for a thymine to adenine substitution in clone MS6.1, that would render an amino acid change from Glutamic acid (E) to Aspartic acid (D) (Table II). This change, however, does not contribute to the sequences of P1–P6. The other sequences in the region of interest were identical, ruling out mutations at the RNA level as the cause for the generation of multiple cyclin B1 peptides.
Table II.
Partial Sequence Alignment Analysis of Cyclin B1 DNA and cDNA of the Region Encoding Peptides P1–P6
| DNA | |
| A K Y L M E L T M L | |
| HeLa cyclin B1 cDNA | TGATGTCGAGCAACATACTTTGGCCAAATACCTGATGGAACTAACTATGTTGGAC-TATGACATGGTGCA |
| Normal cyclin B1 DNA (1) | …-… |
| Normal cyclin B1 DNA (2) | …-… |
| MSA2 cyclin B1 DNA | …A…-… |
| PCI13 cyclin B1 DNA | …A… |
| cDNA | |
| A K Y L M E L T M L | |
| Normal cyclin B1 cDNA | TGATGTCGAGCAACATACTTTGGCCAAATACCTGATGGAACTAACTATGTTGGACTATGACATGGTGCA |
| HeLa cyclin B1 cDNA | … |
| MS6.1-cDNA | …T… |
| MS6.2-cDNA | …A… |
| MS6.3-cDNA | …A… |
| MS6.4-cDNA | …A… |
| MS6.5-cDNA | … |
| MS6.6-cDNA | .-…A… |
| MS6.7-cDNA | …N… |
| MS6.9-cDNA | … |
| MS6.10-cDNA | …A… |
Discussion
In this study, we report the identification of cyclin B1 as a shared epithelial tumor antigen. We used our recently described approach of tumor antigen discovery that relies on DCs as APCs to prime naive CD8+ T cells against tumor-derived peptides (27). Although a variety of approaches have been used to identify HLA class I–restricted tumor antigens, most depended on the availability of tumor-specific T cell lines or clones derived from cancer patients. Our approach employed T cells from healthy donors and explored the full repertoire of anti-tumor responses that could be generated in healthy, immunocompetent individuals to tumor antigens presented by professional APCs.
Cyclin B1 is an important molecule involved in the transition from G2 to M phase of the cell cycle (28). It associates with the active form of the cdc2 (cdk1) kinase in the cytoplasm, and translocates into the nucleus where it initiates chromosome condensation, destruction of the nuclear membrane, and assembly of the mitotic spindle. It then rapidly gets ubiquitinated and targeted to the proteasome for degradation (37). Anaphase is then initiated and the cell progresses to complete the cell cycle. Cyclin B1 is expressed only at certain points of the cell cycle, starting with the gradual accumulation of the protein at G1, to its peak at G2 where it acquires the threshold needed to initiate mitosis. Overexpression of cyclin B1, detected by immunochemistry, has been reported in a variety of tumors, i.e., breast, colon, prostate, oral, esophageal, and non-small cell lung cancers (38–43). We detected overexpression of cyclin B1 in a panel of SCCHN that showed intense cyclin B1 staining in the tumor cells and not in the surrounding mucosa. This was also observed in adenocarcinomas of the lung and breast (data not shown). The overexpressed protein was found predominantly in the cytoplasm, which is different from the nuclear localization in normal dividing cells. Cyclin B1 overexpression in tumors correlated with the presence of cyclin B1–specific memory T cell responses in their PBLs, suggesting that T cells in these SCCHN patients had been primed in vivo to cyclin B1 derived from their tumors. We hypothesize that the overexpression of cyclin B1 in the cytoplasm in these tumors and its degradation in the proteasome increases dramatically HLA class I-cyclin B1 peptide complexes on the tumor cell surface making them tumor-specific targets for T cells. It would be interesting to examine in a larger number of patients if cyclin B1 overexpression in SCCHN tumors and the presence of cyclin B1-specific T cell responses might correlate with a better prognosis and increased survival.
As these cyclin B1 peptides were originally isolated from a fraction that supported the growth of peptide-specific CTLs that lysed the original tumor cells (27), we were surprised that the synthetic peptide-primed T cells from a different HLA-A2+ healthy donor were unable to recognize and kill the same tumor (data not shown). We believe that this discrepancy between T cells recognizing peptide-loaded targets versus tumor probably lies in the comparatively lower density of HLA-cyclin B1 peptide complexes on the tumor, being below the threshold necessary for recognition by the low-affinity T cells generated on synthetic peptides in vitro. However, we expanded cyclin B1 peptide-specific T cell lines from a SCCHN patient's T cells that had been primed in vivo and showed that these T cells were capable of lysing the original tumor cells, especially if the costimulatory molecule CD80 was present on the target cell. Having been primed in vivo to physiological concentrations of antigen that we were not able to mimic in vitro, these T cells from a cancer patient were better at recognizing the tumor cells compared with in vitro primed T cells from a healthy donor.
The precise mechanism that is responsible for the small amino acid differences we see between the peptides we isolated from an HPLC fraction and the sequence found from the database has not been elucidated. One possibility we considered was the polymorphism in human cyclin B1 genes, similar to what has been reported for rat cyclin B1 (44). Very little work has been done on human cyclin B1 genes. Only one has been cloned and sequenced from the HeLa cell line and nothing is known about the degree of polymorphism of cyclin B1 genes in that line. We have sequenced from normal and tumor cells the portion of the cyclin B1 gene that contains the region encoding the peptides. We observed differences in several nucleotides but all were outside the region of interest (data not shown). No differences were found in the specific region that encoded the peptides, ruling out changes at the gene level as the cause for the multiple related cyclin B1 peptides. We also did not detect any differences at the mRNA level, suggesting that the changes were most likely occurring at the protein level. A recent study has suggested that up to 30% of proteins in a cell are mistranslated or misfolded giving rise to defective ribosomal products (DRIPs), that are quickly directed to the proteasome for degradation (45). This process may even be exaggerated in transformed cells. Inasmuch as cyclin B1 also uses the ubiquitin-proteasome system for degradation (37), we propose that the cyclin B1 peptides that we identified were derived from overexpressed and mistranslated cyclin B1 protein accumulating in the cytoplasm of tumor cells.
To our knowledge, this is the first report of a human cyclin as a tumor antigen recognized by T cells. Although deregulation of the cell cycle is one of the hallmarks of human cancer, little attention has been given to exploring cyclins as targets of an immune response, with an exception of a brief report regarding the presence of antibody responses against cyclin B1 in hepatocellular carcinoma patients (46). Our work did not specifically set out to test cyclin B1 molecule as a tumor antigen either, until we found that peptides derived from this molecule were stimulators of a T cell response. Pathologists have previously reported overexpression of various cyclins in certain cancers, such as overexpression of cyclin D1 and cyclin E in breast cancer (47), and cyclins A and B1 in melanomas (48), breast cancer (38, 49, 50), and in oral carcinomas (41). Studies in our lab have shown that in some tumors, G1 cyclins are found to be more abundant while in others the G2 cyclins predominate (data not shown). Their aberrant expression may also lead to presentation of immunogenic peptides on the surface of tumors. While here we have presented data only on peptides derived from cyclin B1, we believe that the whole family of human cyclins could be candidate tumor antigens provided their expression in tumor cells differs significantly from their expression in normal cells.
Acknowledgments
The authors wish to thank Joseph M. Pilewski and Joseph D. Latoche for helpful assistance with the immunohistochemical staining of the tumor cell lines; Robert Bast (MD Anderson, Houston, TX) for the collection of PBMCs and tumor digests from breast cancer patients; Elaine Elder (University of Pittsburgh Tissue Bank Core Facility) for the collection of PBMCs from SCCHN patients; Daniel F. Graziano and Jessica C. Kettel for assistance with the ELISPOT, and Nehad Alajez for help with sequencing.
This work was supported by Department of Defense grant DAMD 17-9-1-7057 to H. Kao, National Institutes of Health grant 2R37 A133993 to D.F. Hunt, National Institutes of Health grant 5PO1CA 73743 to O.J. Finn, and National Institutes of Health grant PO1DE 12321 to T.L. Whiteside.
H. Kao's present address is Department of Pathology and Immunology, Washington University School of Medicine, St. Louis, MO 63110.
Footnotes
Abbreviations used in this paper: DC, dendritic cell; ELISPOT, enzyme-linked immunospot; MS, mass spectrometry; RP, reverse phase; SCCHN, squamous cell carcinomas of the head and neck.
References
- 1.Brichard, V., A. Van Pel, T. Wolfel, C. Wolfel, E. De Plaen, B. Lethe, P. Coulie, and T. Boon. 1993. The tyrosinase gene codes for an antigen recognized by autologous cytolytic T lymphocytes on HLA-A2 melanomas. J. Exp. Med. 178:489–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Coulie, P.G., V. Brichard, A. Van Pel, T. Wolfel, J. Schneider, C. Traversari, S. Mattei, E. De Plaen, C. Lurquin, J.P. Szikora, et al. 1994. A new gene coding for a differentiation antigen recognized by autologous cytolytic T lymphocytes on HLA-A2 melanomas. J. Exp. Med. 180:35–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hunt, D.F., H. Michel, T.A. Dickinson, J. Shabanowitz, A.L. Cox, K. Sakaguchi, E. Appella, H.M. Grey, and A. Sette. 1992. Peptides presented to the immune system by the murine class II major histocompatibility complex molecule I-Ad. Science. 256:1817–1820. [DOI] [PubMed] [Google Scholar]
- 4.Hunt, D.F., R.A. Henderson, J. Shabanowitz, K. Sakaguchi, H. Michel, N. Sevilir, A.L. Cox, E. Appella, and V.H. Engelhard. 1992. Characterization of peptides bound to the class I MHC molecule HLA-A2.1 by mass spectrometry. Science. 255:1261–1263. [DOI] [PubMed] [Google Scholar]
- 5.Fisk, B., T.L. Blevins, J.T. Wharton, and C.G. Ioannides. 1995. Identification of an immunodominant peptide of HER-2/neu protooncogene recognized by ovarian tumor-specific cytotoxic T lymphocyte lines. J. Exp. Med. 181:2109–2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blake, J., J.V. Johnston, K.E. Hellstrom, H. Marquardt, and L. Chen. 1996. Use of combinatorial peptide libraries to construct functional mimics of tumor epitopes recognized by MHC class I-restricted cytolytic T lymphocytes. J. Exp. Med. 184:121–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tureci, O., U. Sahin, and M. Pfreundschuh. 1997. Serological analysis of human tumor antigens: molecular definition and implications. Mol. Med. Today. 3:342–349. [DOI] [PubMed] [Google Scholar]
- 8.Henderson, R.A., and O.J. Finn. 1996. Human tumor antigens are ready to fly. Adv. Immunol. 62:217–256. [DOI] [PubMed] [Google Scholar]
- 9.Traversari, C., P. van der Bruggen, I.F. Luescher, C. Lurquin, P. Chomez, A. Van Pel, E. De Plaen, A. Amar-Costesec, and T. Boon. 1992. A nonapeptide encoded by human gene MAGE-1 is recognized on HLA-A1 by cytolytic T lymphocytes directed against tumor antigen MZ2-E. J. Exp. Med. 176:1453–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gaugler, B., B. Van den Eynde, P. van der Bruggen, P. Romero, J.J. Gaforio, E. De Plaen, B. Lethe, F. Brasseur, and T. Boon. 1994. Human gene MAGE-3 codes for an antigen recognized on a melanoma by autologous cytolytic T lymphocytes. J. Exp. Med. 179:921–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boel, P., C. Wildmann, M.L. Sensi, R. Brasseur, J.C. Renauld, P. Coulie, T. Boon, and P. van der Bruggen. 1995. BAGE: a new gene encoding an antigen recognized on human melanomas by cytolytic T lymphocytes. Immunity. 2:167–175. [DOI] [PubMed] [Google Scholar]
- 12.Van den Eynde, B., O. Peeters, O. De Backer, B. Gaugler, S. Lucas, and T. Boon. 1995. A new family of genes coding for an antigen recognized by autologous cytolytic T lymphocytes on a human melanoma. J. Exp. Med. 182:689–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen, Y.T., M.J. Scanlan, U. Sahin, O. Tureci, A.O. Gure, S. Tsang, B. Williamson, E. Stockert, M. Pfreundschuh, and L.J. Old. 1997. A testicular antigen aberrantly expressed in human cancers detected by autologous antibody screening. Proc. Natl. Acad. Sci. USA. 94:1914–1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kawakami, Y., S. Eliyahu, C. Jennings, K. Sakaguchi, X. Kang, S. Southwood, P.F. Robbins, A. Sette, E. Appella, and S.A. Rosenberg. 1995. Recognition of multiple epitopes in the human melanoma antigen gp100 by tumor-infiltrating T lymphocytes associated with in vivo tumor regression. J. Immunol. 154:3961–3968. [PubMed] [Google Scholar]
- 15.Kawakami, Y., S. Eliyahu, C.H. Delgado, P.F. Robbins, K. Sakaguchi, E. Appella, J.R. Yannelli, G.J. Adema, T. Miki, and S.A. Rosenberg. 1994. Identification of a human melanoma antigen recognized by tumor-infiltrating lymphocytes associated with in vivo tumor rejection. Proc. Natl. Acad. Sci. USA. 91:6458–6462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wolfel, T., M. Hauer, J. Schneider, M. Serrano, C. Wolfel, E. Klehmann-Hieb, E. De Plaen, T. Hankeln, K.H. Meyer zum Buschenfelde, and D. Beach. 1995. A p16INK4a-insensitive CDK4 mutant targeted by cytolytic T lymphocytes in a human melanoma. Science. 269:1281–1284. [DOI] [PubMed] [Google Scholar]
- 17.Robbins, P.F., M. El-Gamil, Y.F. Li, Y. Kawakami, D. Loftus, E. Appella, and S.A. Rosenberg. 1996. A mutated beta-catenin gene encodes a melanoma-specific antigen recognized by tumor infiltrating lymphocytes. J. Exp. Med. 183:1185–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang, R.F., X. Wang, A.C. Atwood, S.L. Topalian, and S.A. Rosenberg. 1999. Cloning genes encoding MHC class II-restricted antigens: mutated CDC27 as a tumor antigen. Science. 284:1351–1354. [DOI] [PubMed] [Google Scholar]
- 19.Ikeda, H., B. Lethe, F. Lehmann, N. van Baren, J.F. Baurain, C. de Smet, H. Chambost, M. Vitale, A. Moretta, T. Boon, and P.G. Coulie. 1997. Characterization of an antigen that is recognized on a melanoma showing partial HLA loss by CTL expressing an NK inhibitory receptor. Immunity. 6:199–208. [DOI] [PubMed] [Google Scholar]
- 20.Ropke, M., J. Hald, P. Guldberg, J. Zeuthen, L. Norgaard, L. Fugger, A. Svejgaard, S. Van der Burg, H.W. Nijman, C.J. Melief, and M.H. Claesson. 1996. Spontaneous human squamous cell carcinomas are killed by a human cytotoxic T lymphocyte clone recognizing a wild-type p53-derived peptide. Proc. Natl. Acad. Sci. USA. 93:14704–14707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ressing, M.E., A. Sette, R.M. Brandt, J. Ruppert, P.A. Wentworth, M. Hartman, C. Oseroff, H.M. Grey, C.J. Melief, and W.M. Kast. 1995. Human CTL epitopes encoded by human papillomavirus type 16 E6 and E7 identified through in vivo and in vitro immunogenicity studies of HLA- A*0201-binding peptides. J. Immunol. 154:5934–5943. [PubMed] [Google Scholar]
- 22.Lennette, E.T., G. Winberg, M. Yadav, G. Enblad, and G. Klein. 1995. Antibodies to LMP2A/2B in EBV-carrying malignancies. Eur J Cancer. 31A(no. 11):1875–1878. [DOI] [PubMed] [Google Scholar]
- 23.Shievely, J., and J. Beatty. 1985. CEA related antigens: molecular, biological and clinical significance. CRC Crit. Rev. Oncol. Hematol. 2:355–399. [DOI] [PubMed] [Google Scholar]
- 24.Wang, M.C., L.D. Papsider, M. Kuriyama, L.A. Valenzuela, G.P. Murphy, and T.M. Chu. 1981. Prostate antigen: a new potential marker for prostatic cancer. Prostate. 2:89–96. [DOI] [PubMed] [Google Scholar]
- 25.Disis, M.L., and M.A. Cheever. 1998. HER-2/neu oncogenic protein: issues in vaccine development. Crit. Rev. Immunol 18:37–45. [DOI] [PubMed] [Google Scholar]
- 26.Finn, O.J., K.R. Jerome, R.A. Henderson, G. Pecher, N. Domenech, J. Magarian-Blander, and S.M. Barratt-Boyes. 1995. MUC-1 epithelial tumor mucin-based immunity and cancer vaccines. Immunol. Rev. 145:61–89. [DOI] [PubMed] [Google Scholar]
- 27.Kao, H., A.A. Amoscato, P. Ciborowski, and O.J. Finn. 2001. A new strategy for tumor antigen discovery based on in vitro priming of naive T cells with dendritic cells. Clin. Cancer Res. 7(Suppl):773s–780s. [PubMed] [Google Scholar]
- 28.King, R.W., P.K. Jackson, and M.W. Kirschner. 1994. Mitosis in transition. Cell. 79:563–571. [DOI] [PubMed] [Google Scholar]
- 29.Hiltbold, E.M., M.D. Alter, P. Ciborowski, and O.J. Finn. 1999. Presentation of MUC1 tumor antigen by class I MHC and CTL function correlate with the glycosylation state of the protein taken up by dendritic cells. Cell. Immunol. 194:143–149. [DOI] [PubMed] [Google Scholar]
- 30.Yasumura, S., H. Hirabayashi, D.R. Schwartz, J.F. Toso, J.T. Johnson, R.B. Herberman, and T.L. Whiteside. 1993. Human cytotoxic T-cell lines with restricted specificity for squamous cell carcinoma of the head and neck. Cancer Res. 53:1461–1468. [PubMed] [Google Scholar]
- 31.Martin, S.E., J. Shabanowitz, D.F. Hunt, and J.A. Marto. 2000. Subfemtomole MS and MS/MS peptide sequence analysis using nano-HPLC micro-ESI fourier transform ion cyclotron resonance mass spectrometry. Anal. Chem. 72:4266–4274. [DOI] [PubMed] [Google Scholar]
- 32.Shabanowitz, J., R.E. Settlage, J.A. Marto, R.E. Christian, F.W. White, P.S. Russo, S.E. Martin, and D.F. Hunt. 2000. Sequencing the primordial soup. Mass Spectrometry in Biology and Medicine. A.L. Burlingame, S.A. Carr, and M.A. Baldwin, editors. Humana Press, Totowa, NJ. 163–177.
- 33.Eng, J.K., A.L. McCormack, and J.R. Yates. 1994. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass. Spectrom. 5:976–989. [DOI] [PubMed] [Google Scholar]
- 34.Clauser, K.R., P. Baker, and A.L. Burlingame. 1999. Role of accurate mass measurement (+/− 10 ppm) in protein identification strategies employing MS or MS/MS and database searching. Anal. Chem. 71:2871–2882. [DOI] [PubMed] [Google Scholar]
- 35.Zeh, H.J., III, G.H. Leder, M.T. Lotze, R.D. Salter, M. Tector, G. Stuber, S. Modrow, and W.J. Storkus. 1994. Flow-cytometric determination of peptide-class I complex formation. Identification of p53 peptides that bind to HLA-A2. Hum. Immunol. 39:79–86. [DOI] [PubMed] [Google Scholar]
- 36.Herr, W., J. Schneider, A.W. Lohse, K.H. Meyer zum Buschenfelde, and T. Wolfel. 1996. Detection and quantification of blood-derived CD8+ T lymphocytes secreting tumor necrosis factor alpha in response to HLA-A2.1-binding melanoma and viral peptide antigens. J. Immunol. Methods. 191:131–142. [DOI] [PubMed] [Google Scholar]
- 37.Murray, A. 1995. Cyclin ubiquitination: the destructive end of mitosis. Cell. 81:149–152. [DOI] [PubMed] [Google Scholar]
- 38.Kawamoto, H., H. Koizumi, and T. Uchikoshi. 1997. Expression of the G2-M checkpoint regulators cyclin B1 and cdc2 in nonmalignant and malignant human breast lesions: immunocytochemical and quantitative image analyses. Am. J. Pathol. 150:15–23. [PMC free article] [PubMed] [Google Scholar]
- 39.Wang, A., N. Yoshimi, N. Ino, T. Tanaka, and H. Mori. 1997. Overexpression of cyclin B1 in human colorectal cancers. J. Cancer Res. Clin. Oncol. 123:124–127. [DOI] [PubMed] [Google Scholar]
- 40.Mashal, R.D., S. Lester, C. Corless, J.P. Richie, R. Chandra, K.J. Propert, and A. Dutta. 1996. Expression of cell cycle-regulated proteins in prostate cancer. Cancer Res. 56:4159–4163. [PubMed] [Google Scholar]
- 41.Kushner, J., G. Bradley, B. Young, and R.C. Jordan. 1999. Aberrant expression of cyclin A and cyclin B1 proteins in oral carcinoma. J. Oral Pathol. Med. 28:77–81. [DOI] [PubMed] [Google Scholar]
- 42.Murakami, H., M. Furihata, Y. Ohtsuki, and S. Ogoshi. 1999. Determination of the prognostic significance of cyclin B1 overexpression in patients with esophageal squamous cell carcinoma. Virchows Arch. 434:153–158. [DOI] [PubMed] [Google Scholar]
- 43.Soria, J.C., S.J. Jang, F.R. Khuri, K. Hassan, D. Liu, W.K. Hong, and L. Mao. 2000. Overexpression of cyclin B1 in early-stage non-small cell lung cancer and its clinical implication. Cancer Res. 60:4000–4004. [PubMed] [Google Scholar]
- 44.Petit, J., M. Riviere, J. Szpirer, and C. Szpirer. 1999. High level of single-nucleotide polymorphism in the rat cyclin B1 gene. Mamm. Genome. 10:635–637. [DOI] [PubMed] [Google Scholar]
- 45.Schubert, U., L.C. Anton, J. Gibbs, C.C. Norbury, J.W. Yewdell, and J.R. Bennink. 2000. Rapid degradation of a large fraction of newly synthesized proteins by proteasomes. Nature. 404:770–774. [DOI] [PubMed] [Google Scholar]
- 46.Covini, G., E.K. Chan, M. Nishioka, S.A. Morshed, S.I. Reed, and E.M. Tan. 1997. Immune response to cyclin B1 in hepatocellular carcinoma. Hepatology. 25:75–80. [DOI] [PubMed] [Google Scholar]
- 47.Steeg, P.S., and Q. Zhou. 1998. Cyclins and breast cancer. Breast Cancer Res. Treat. 52:17–28. [DOI] [PubMed] [Google Scholar]
- 48.Tran, T.A., J.S. Ross, J.A. Carlson, and M.C. Mihm. 1998. Mitotic cyclins and cyclin-dependent kinases in melanocytic lesions. Hum. Pathol. 29:1085–1090. [DOI] [PubMed] [Google Scholar]
- 49.Keyomarsi, K., and A.B. Pardee. 1993. Redundant cyclin overexpression and gene amplification in breast cancer cells. Proc. Natl. Acad. Sci. USA. 90:1112–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dutta, A., R. Chandra, L. Leiter, and S. Lester. 1995. Cyclins as markers of tumor proliferation: immunocytochemical studies in breast cancer. Proc. Natl. Acad. Sci. 92:5386–5390. [DOI] [PMC free article] [PubMed] [Google Scholar]