

Abstract
We previously showed that a slowly replicating, minimally pathogenic clone of simian immunodeficiency virus (SIV), SIVmneCl8, evolves increased ability to replicate in T cells with the onset of AIDS in pig-tailed macaques. Moreover, molecular clones derived from late stages of infection (SIVmne170 and SIVmne027) replicate to high levels in vivo compared to SIVmneC 8. Here, we investigated the role of rt mutations in SIVmne variant replication. We demonstrate selection for rt alleles that enhance viral infectivity and replication capacity in CD4+ T cells. Moreover, the ability of SIVmne to be induced from resting CD4+ T cells by anti-CD3/CD28 stimulation is more strongly influenced by the variant rt alleles than nef alleles. Taken together, our data underscore the importance of RT determinants for pathogenicity of SIV and for the capacity to replicate in CD4+ T cell populations.
Keywords: SIV, reverse transcriptase, variant selection, CD4, T cells, infectivity
INTRODUCTION
During the course of human immunodeficiency virus type 1 (HIV-1) infections, viral variants with distinct characteristics emerge that may be important for persistence and disease progression. Typically, these variants have greater ability to infect and replicate in CD4+ T cells compared to the viruses present at the early stages of infection, suggesting that mutations evolve to enhance replicative capacity (reviewed in Kimata, 2006). The level of HIV-1 replication in the host heralds the onset of AIDS, which underscores the potential importance of viral replicative capacity in driving disease progression (Connor et al., 1993; Embretson et al., 1993; Ho et al., 1989; Mellors et al., 1996; Mellors et al., 1997; Pantaleo et al., 1993; Schnittmen et al., 1990; Zhang et al., 1999). However, defining the exact evolutionary changes in the virus that influence HIV-1 replication is difficult because of the absence of an animal model to evaluate HIV-1 replication, persistence, and disease. Similar evolution of viral variants has been observed with simian immunodeficiency virus (SIV), making it an important model for identifying and examining mutations that enhance the replicative capacity and pathogenesis of emerging variant viruses (Kimata, 2006; Whetter et al., 1999).
We and others have previously shown that infection of pig-tailed macaques (Macaca nemestrina) with a variant, SIVmneCl8, results in the emergence of viral variants that demonstrate increased replicative capacity in CD4+ T cells, cytopathicity, and pathogenicity (Kimata et al., 1999; Overbaugh et al., 1991; Rudensey et al., 1995). Importantly, phenotypic differences of the parental virus and variants are mainly revealed by infection of primary cells rather than cell lines, which efficiently propagate all the viruses. Like early stage HIV-1 variants, SIVmneCl8 is macrophage-tropic, slowly replicating in T cells and minimally cytopathic. By contrast, we have shown that late-stage molecular variants with increased pathogenicity, SIVmne027 and SIVmne170, demonstrate increased replicative capacity in phytohemagglutinin (PHA)-stimulated macaque peripheral blood mononuclear cells (PBMCs) compared to the early virus, SIVmneCl8 (reviewed in Kimata, 2006). Surprisingly, the ability to replicate in dendritic cell (DC)-T cell co-cultures was significantly different between these late variant viruses (Kimata et al., 1998; Kimata et al., 2004; Kimata and Overbaugh, 1997). SIVmne027 replicates to high levels in the DC-T cell co-cultures, whereas replication of SIVmne170 is limited, similar to SIVmneCl8 (Kimata et al., 2004). Interestingly, a pol determinant was found to contribute to the enhanced infectivity of SIVmne027, but not SIVmne170 (Patel et al., 2002). Further characterization of the pol determinant of SIVmne027 showed that it contributes not only to increased infectivity, but also greater virus replication in DC-T cell co-cultures (Kimata et al., 2004). Here, we further examine the contributions of mutations in pol, focusing on rt variants and the effects of rt alleles on viral infectivity and replication in DC-T cell co-cultures and in CD4+ T cells activated through CD3/CD28 engagement.
RESULTS
Pol contributes to the replicative capacity of a highly pathogenic SIVmne
Initially, to determine the contribution of pol to the increased replication capacity of a variant virus that emerged late in infection, we infected DC-T cell co-cultures with chimeric viruses containing the pol region of SIVmne027 (containing five amino acid differences in the rt sequence compared to SIVmneCl8: I73M, K412E, K434E, K466R and A486T) in the SIVmneCl8 backbone (chimera 8/027pol, see Fig. 1) and compared the level of replication with that of the parental viruses SIVmneCl8 and SIVmne027. DC-T cell co-cultures were used because HIV and SIV replication in CD4+ T cells is more efficient the presence of DCs (Cameron et al., 1992; Gummuluru et al., 2002; Kimata et al., 2004; Messmer et al., 2000; Pope et al., 1994).
Figure 1. Schematic of Viruses.
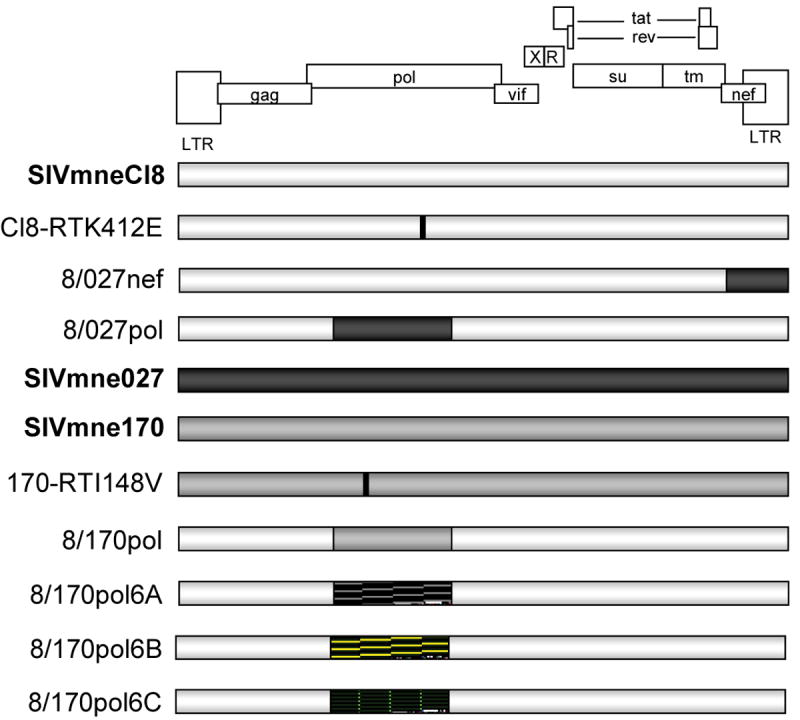
Parental viruses are shown in bold. Chimeric viruses are shown with the corresponding regions of the parental viruses shaded accordingly. Point mutations are indicated with a black horizontal line. Hatched areas (
 ) indicate regions cloned from viruses isolated 6 weeks post-infection from SIVmne170-infected macaques; these regions were cloned into the SIVmneCl8 background.
) indicate regions cloned from viruses isolated 6 weeks post-infection from SIVmne170-infected macaques; these regions were cloned into the SIVmneCl8 background.
A summary of the characteristics of the parental viruses SIVmneCl8 (early-variant virus) and SIVmne170 and SIVmne027 (late-variant viruses) is shown in Table 1 with accompanying references. The molecular cloning and initial in vitro phenotypic characterizations of SIVmneCl8, SIVmne170 and SIVmne027 have been described in the indicated references. Each virus was obtained by lambda-phage cloning of provirus (Kimata et al., 1998; Kimata and Overbaugh, 1997). Although cloned from a pig-tailed macaque with lymphoma, SIVmneCl8 is phenotypically similar to variants found early after infection, whereas SIVmne170 and SIVmne027 are representative of variants found at later stages of infection following inoculation with SIVmneCl8 (Kimata et al., 1998; Kimata and Overbaugh, 1997; Rudensey et al., 1995). SIVmne170 is derived from PBMCs and SIVmne027 was cloned from lymph node tissue. The in vivo pathogenicities of these viruses were previously reported (Kimata et al., 1999).
Table 1.
Summary of in vivo and in vitro replication profiles of SIV variants.
| Virus Type | Name | Plasma Viral Load# | In vitro Replication Capacity | References | |||||
|---|---|---|---|---|---|---|---|---|---|
| Peak | Set-point | PHA-Stimulated Lymphoblasts | Macrophages | Dendritic Cells | Resting CD4+ T Cells | DC-T Cell Co-cultures | |||
| Early | SIVmneCl8 | 1.4 × 107 | 2.4 × 103 | Low | Moderate | ND* | ND | Low | Kimata et al., 2004; Kimata and Overbaugh, 1997; Patel et al., 2002; Rudensey et al., 1995 |
| Late (blood) | SIVmne170 | 5.3 × 107 | 6.0 × 106 | High | Low | ND | ND | Low | Kimata et al., 1999; Kimata et al., 2004; Kimata and Overbaugh, 1997; Patel et al., 2002; Rudensey et al., 1995 |
| Late (lymph node) | SIVmne027 | 4.2 × 108 | 8.3 × 106 | High | Low | ND | Low | High | Kimata et al., 1998; Kimata et al., 1999; Patel et al., 2002 |
RNA copies per mL. Peak viral load for SIVmne170 is delayed by 2-3 weeks.
ND, not detected
As shown in Fig. 2A, the chimeric virus 8/027pol replicated to a 60-fold higher level than the early parental virus, SIVmneCl8. The replication level was similar to a control chimeric virus, 8/027nef, which contains the nef allele from SIVmne027 in the background of SIVmneCl8 and has increased infectivity relative to SIVmneCl8 (Patel et al., 2002). Neither determinant alone was sufficient to completely confer a replication profile similar to that of SIVmne027.
Figure 2. Viral Replication in T cells.
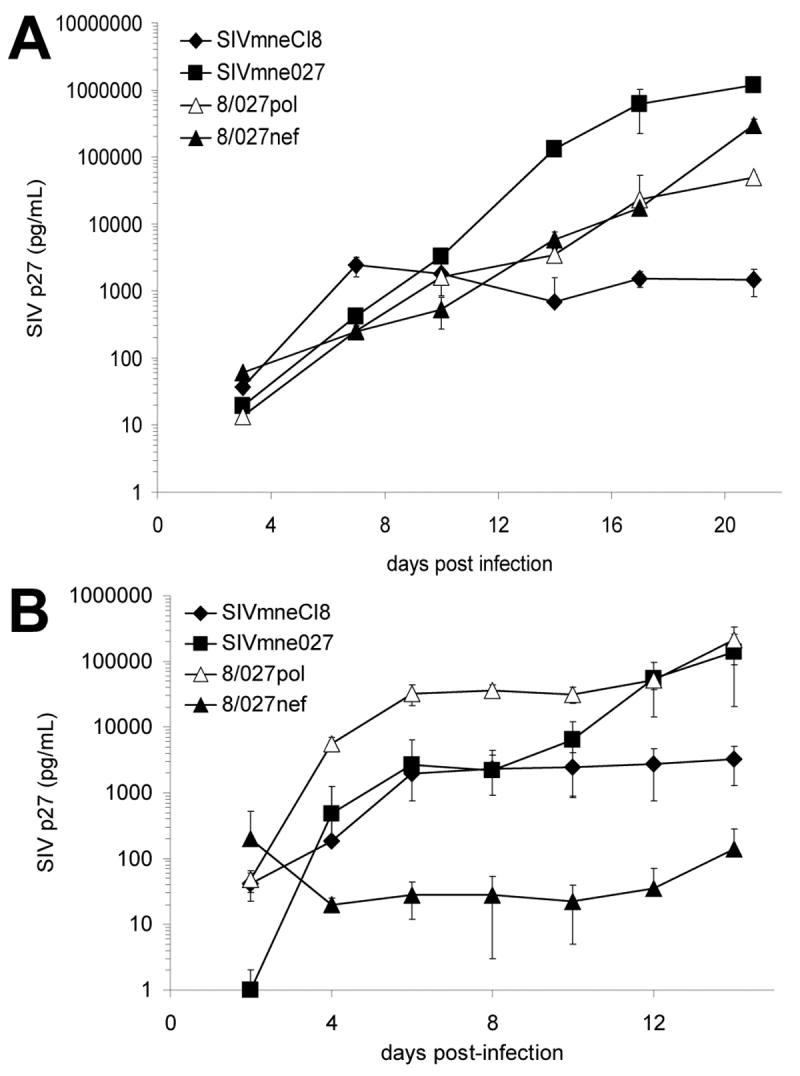
(A) Replication of SIV variants in pig-tailed macaque dendritic cell-T cell co-cultures. Monocyte-derived dendritic cells were co-cultured with autologous T cells in triplicate and infected with chimeric and parental viruses as described (Kimata et al., 2004). Viral replication was monitored by measuring SIV p27 levels in supernatants of the co-cultures by ELISA. (B) Replication of SIV variants in pig-tailed macaque CD4+ T cells costimulated with anti-CD3/anti-CD28 antibodies. T cells were infected with virus in triplicate, washed, and incubated in plates coated with anti-CD3 and anti-CD28 for 24 hours. Cultures were allowed to continue for 2 weeks. Supernatants were harvested and media replaced every 3-4 days. In both A and B, average p27 levels ± SD are shown. Data are representative of six and two independent experiments in A and B, respectively.
The ability of the chimeric viruses to infect resting CD4+ T cells and replicate after subsequent activation was then measured and compared. As shown in Fig. 2B, replication of 8/027pol was high in response to anti-CD3/anti-CD28 costimulation, which mimics normal T cell stimulation and activation. Replication was similar to SIVmne027 and nearly 100-fold greater than the parental virus, SIVmneCl8. By contrast, the 8/027nef chimera failed to replicate efficiently and produced 10-100 fold lower levels of p27 antigen than SIVmneCl8. Together, these data suggest that the SIVmne027 pol region is an important determinant for increased viral replication in T cells. Additionally, because the pol determinant mainly consists of sequences from the rt gene, the data suggest that differences in the rt gene may account for the enhanced replication capacity of SIVmne027.
A connection domain mutation enhances replication of SIVmneCl8
To identify specific amino acid residues of the variant-specific rt alleles that could account for the minimal infectivity of SIVmneCl8, we compared the proviral DNA sequences of the SIVmne027 and SIVmneCl8 rt alleles with those of other SIV clones. We identified two unique amino acid mutations, a lysine at position 412 within the connection domain and an alanine at 486, in the SIVmneCl8 rt gene (Fig 3.). Since both of these amino acids occur only in the minimally pathogenic virus, SIVmneCl8, and not in the more pathogenic viruses SIVmne027 and SIVmne170 (both of which contain the consensus glutamate and threonine at positions 412 and 486, respectively) we examined the effects of these mutations on infectivity and viral replication. Site-directed mutagenesis was used to introduce either a single amino acid mutation in RT at position 412 that changes the lysine to the consensus glutamic acid (mutant Cl8-RTK412E) or position 486 that changes the alanine to threonine in SIVmneCl8 (Fig. 3). Infectivity of the two mutant viruses was tested in sMAGI cells and only the Cl8-RTK412E mutant demonstrated an increase in infectivity relative to the parental virus, SIVmneCl8, (data not shown) and therefore, was further characterized. Interestingly, the Cl8-RTK412E mutant demonstrated a 12-fold increase in infectivity compared to the parental virus SIVmneCl8 in the sMAGI assay. This increase was similar to that of the 8/027pol chimera (Fig. 4A). Additionally, the Cl8-RTK412E mutant showed a 10-fold enhanced replication in CD4+ T cells in response to anti-CD3/anti-CD28 costimulation relative to the parental SIVmneCl8 virus (Fig. 4B). This was similar to the 8/027pol chimeric virus. Together with the data in Fig. 2 these data indicate a role for mutations in the connection domain of RT in influencing viral infectivity and replication in CD4+ T cells.
Figure 3. Predicted Amino Acid Sequences of RT Variants.

Variant reverse transcriptase alleles cloned from PBMC DNA of SIVmne170-infected pig-tailed macaques at 6 weeks post-infection were sequenced and the amino acid sequences were determined. Sequences labeled 393-6 were derived from macaque F94393 and those labeled with 233-6 were from macaque J94233. The infection of these animals with SIVmne170 was previously reported (Kimata et al., 1999). Recombinant viruses, 8/170pol6A, 8/170pol6B, and 8/170pol6C, that were used for infectivity studies in Fig. 5A include the variant rt alleles from 393-6.13, 233-6.22, and 233-6.28, respectively, in the background of SIVmneCl8; restriction enzyme sites for the recombinants lie outside of the region presented in this figure. Recombinant viruses were constructed as described in the materials and methods (Patel et al., 2002). The connection domain of RT is highlighted in gray.
Figure 4. Infectivity and Replication of Cl8-RTK412E.
(A) The infectivity of RT mutant, Cl8-RTK412E, was compared to the parental virus, SIVmneCl8, and chimeric virus 8/027pol using the sMAGI infectivity assay as described (Patel et al., 2002). Infections were carried out in triplicate. The average numbers of infected cells per ng p27 ± SD are shown. One representative of four independent experiments is shown. (B) Replication of the mutant Cl8-RTK412E in pig-tailed macaque CD4+ T cells after costimulation with anti-CD3/anti-CD28 monoclonal antibodies. The average p27 levels ± SD are shown and the data are representative of independent experiments using cells from two different pig-tailed macaque donors.
Primary infection and RT infectivity
SIVmne170, like SIVmne027, is also a late stage clone, but replication in DC-T cell co-cultures and infection of resting PBMCs or T cells is significantly impaired compared to SIVmne027 (Table 1) (Kimata et al., 1998; Kimata et al., 2004; Patel et al., 2002). Furthermore, when SIVmne170 was inoculated into pig-tailed macaques, peak plasma viral loads were delayed by 2-3 weeks and nearly 10-fold lower than viral loads in SIVmne027-infected animals, although set-point viral loads were comparable for both viruses (Kimata et al., 1999). These data suggest that SIVmne170 may undergo early selection for mutations with increased replicative capacity. To identify residues in the SIVmne170 rt gene that may confer its low infectivity, variant rt alleles were cloned and sequenced from PBMC specimens taken at 6 weeks post-infection of two pig-tailed macaques infected with the molecular clone SIVmne170 (Kimata et al., 1999). Variant and parental rt allelic sequences were compared. A single point mutation (I148V) was found to be common to all of the variant alleles (Fig. 3). Mutant chimeric viruses containing variant rt alleles in the background of SIVmneCl8 were generated using previously described methods (Patel et al., 2002). The infectivity of three representative clones (8/170pol6A containing the following rt changes from the parental virus SIVmne170: I148V and Q474P; 8/170pol6B containing the following rt changes from the parental virus SIVmne170: I148V, R173G, P419S and G489R; and 8/170pol6C containing the following rt changes from the parental virus SIVmne170: V108I, I148V, R173K, G211S, F227L, I257M and T288A) was compared to the early variant SIVmneCl8, the late variant SIVmne027 (both of which contain valine at position 148) as well as a chimeric virus containing the pol region of SIVmne170 in the backbone of SIVmneCl8 (Fig. 5A). Constructing the chimeric viruses in the SIVmneCl8 background enabled us to limit the effects on infectivity to mutations contained only in the pol region and ‘normalized’ effects of other regions of the viral genome. All of the pol chimeric viruses were similar to the chimeric virus 8/027pol and showed 2 to 3-fold increases in infectivity compared to either SIVmneCl8 or 8/170pol, suggesting that the valine at 148 enhances viral infectivity. Interestingly, earlier biochemical studies of RTs containing either isoleucine or valine at position 148 have demonstrated that I148-containing RT (found in SIVmne170) has lower dNTP binding affinity compared to RT dNTP binding with a corresponding valine (Diamond et al., 2003). This decrease in dNTP binding resulted in an increase in replication fidelity in the SIVmne170 variant (Diamond et al., 2001).
Figure 5. Infectivity and Replication of SIV mutants.

(A) RT clones recovered from SIVmne170-infected macaques were sequenced and compared (see Fig. 3). RT coding sequences of three representative isolates were placed into the SIVmneCl8 background and the influence of the mutations on infectivity of the chimeric viruses (8/170pol6A, 8/170pol6B, and 8/170pol6C) were examined using the sMAGI infectivity assay. The average numbers of infected cells/ng p27 ± SD are shown. One representative of two independent experiments is shown. (B) Infectivity of a SIVmne170 mutant containing a valine at position 148 of RT (170-RTI148V) was compared to the infectivity of the early virus, SIVmneCl8, late variant SIVmne027, and the isogenic parental virus, SIVmne170 using the sMAGI infectivity assay. The average numbers of infected cells per ng p27 ± SD are shown. One representative of three independent experiments is shown. (C) Replication of the mutant, 170-RTI148V, in pig-tailed macaque DC-T cell co-cultures. (D) Replication of the mutant 170-RTI148V in pig-tailed macaque CD4+ T cells after costimulation with anti-CD3/anti-CD28 monoclonal antibodies. In C and D, the average p27 levels ± SD are shown and the data are representative of two independent experiments using cells from two different pig-tailed macaque donors.
Since each of the tested chimeric viruses contained its own unique set of mutations in addition to the I148V mutations, we were unable to conclude that the I148V mutation in these chimeras was solely responsible for the increased infectivity. Therefore, to confirm the importance of the I148V RT mutation in viral infection and replication, we introduced a valine at position 148 of RT within SIVmne170 (170-RTI148V), and compared the infectivity and replication capacity of the mutant with SIVmneCl8, SIVmne027, and parental virus, SIVmne170. In the sMAGI assay, infectivity of 170-RTI148V was similar to SIVmne027 and 3 to 4-fold more infectious than SIVmne170 (Fig. 5B). In DC-T cell co-cultures, 170-RTI148V replicated to a high level, similar to the variant SIVmne027, while the replication level of the parental virus, SIVmne170 was less than that of SIVmneCl8 (Fig. 5C). Furthermore, when resting CD4+ T cells were infected and then activated with anti-CD3/anti-CD28 costimulation, 170-RTI148V replicated to significantly higher levels than the parental virus, SIVmne170 (Fig. 5D). Together, these data confirm that functionality of RT in SIV is a significant determinant of viral infectivity, which impacts infection and replication in CD4+ T cells.
DISCUSSION
The capacity for HIV-1 and SIV to infect and spread through the CD4+ T cell population is central to AIDS pathogenesis (Haase, 2005). Both plasma viral load and CD4+ T cell depletion are linked to the extent to which the infecting virus is able to spread through the T cell population. Recent studies suggest that the target cell population consists mainly of memory CD4+ T cells that have resting phenotypes (Mattapallil et al., 2005; Veazey et al., 2000). Also, we and others have demonstrated that plasma viral load is a reflection of the replicative capacity of the infecting SIV variant in primary CD4+ T cells, but not macrophages or cell lines (Kimata et al., 1999). Similar observations have been reported for HIV-1 infection (Campbell et al., 2003). Furthermore, RT mutations that modulate viral replicative capacity of HIV-1 infection influence plasma viral load, even in the absence of or prior to antiretroviral therapy (Barbour et al., 2004; Daar et al., 2005). Additionally, Nef has been shown to have a significant role in increasing replication and spread of SIV through the CD4+ T cell population (Mohri et al., 2001; Roeth and Collins, 2006). Here, we present data indicating that RT mutations in SIV also modulate viral infection and replicative capacity in CD4+ T cells and influence the contribution of Nef to viral replication.
We found that the pol region of a late stage variant SIVmne027 was able to enhance viral replication in the background of SIVmneCl8. When the chimeric virus 8/027pol was compared with 8/027nef, it was observed that the pol determinant played a more significant role in infectivity and replication in resting CD4+ T cells than did nef. Thus, while both determinants enable replication in DC-T cell co-cultures, pol is the more important determinant for replication in resting CD4+ T cells in response to costimulation via CD3/CD28. Because an earlier study did not identify a functional defect in the enzymatic activity of the SIVmneCl8 RT (Diamond et al., 2003), we focused our further analysis on the unique SIVmneCl8 mutation within the connection domain of RT. An introduction of a single mutation in the connection domain, K412E, of the SIVmneCl8 RT was able to enhance infectivity of SIVmneCl8 to levels comparable to the chimeric virus 8/027pol. Mutations in the HIV-1 RT connection domain are known to affect polymerization, RNase H activity, dimerization of the large (p66) and small (p51) RT subunits and inhibit RT activity (Argyris et al., 1999; Cen et al., 2004; Divita et al., 1994; Julias et al., 2003; Nikolenko et al., 2007), therefore it is possible that the K412E mutation in the connection domain of SIVmneCl8 also affects dimerization of the two RT subunits, flexibility of the protein or binding affinity for an unknown cellular co-factor necessary for efficient reverse transcription.
Next, we demonstrated that an RT mutation dramatically enhances replicative capacity of another SIV variant, SIVmne170, in DC-T cell co-cultures and in resting CD4+ T cells following costimulation. Previous studies found that while SIVmne170 could replicate efficiently in cell lines and PHA-stimulated PBMCs, it fails to replicate in DC-T cell co-cultures and resting PBMC cultures (Kimata et al., 1998; Kimata et al., 1999; Kimata and Overbaugh, 1997). Here, when we infected resting CD4+ T cells, this virus did not replicate in response to costimulation. This phenotype occurred despite the fact that SIVmne170 encodes a functional Nef protein that enhances infectivity (Patel et al., 2002). Interestingly, unlike the variant SIVmne027, which replicates efficiently in DC-T cell co-cultures and in resting CD4+ T cells in response to costimulation, SIVmne170 does not encode an rt allelic determinant that increases viral infectivity (Patel et al., 2002). These data suggest that the rt gene modulates replication in CD4+ T cells.
We defined a single mutation from isoleucine to valine at position 148 (I148V) of RT in SIVmne170 that was able to enhance both infectivity and replication levels of SIVmne170 to levels comparable to SIVmne027 in both DC-T cell co-cultures and in resting CD4+ T cells in response to activation signals. We also found that valine 148 is rapidly selected during primary infection of SIVmne170-infected macaques. Interestingly, it was demonstrated earlier that the SIVmne170 RT has lower dNTP binding affinity than other SIV RTs due to the isoleucine at 148, and conversion to the consensus valine increases dNTP binding affinity (Diamond et al., 2003). That the valine at 148 is critical for high dNTP binding affinity, infectivity and replication suggests its importance for infection of T cells, particularly resting CD4+ T cells which have lower levels of dNTPs compared to activated T cells (Diamond et al., 2004). Moreover, as we observed with the SIVmneCl8/SIVmne027 chimeric viruses, the functionality of RT determined whether Nef asserted a pronounced negative effect on SIVmne170 replication in CD4+ T cells or if the virus could overcome the effect and replicate. These results suggest that RT mutations that increase dNTP binding affinity enable SIV, and by analogy HIV-1, to exploit and replicate in cellular environments that otherwise would be unfavorable for viral replication. Interestingly, competitive replication assays using SIVmne variants also demonstrate that a key determinant of viral replication fitness is located in the 5’ half of the viral genome (Voronin et al., 2005), which when considered with our results implicates the pol domain or, more specifically, the rt sequence of the viral genome as an important replication determinant.
A number of functions have been ascribed to Nef that may have a role in HIV-1 or SIV replication, including downregulation of CD4, major histocompatibility complex I, CD3, and CD28, and modulation of viral infectivity and T cell activation (reviewed in Roeth and Collins, 2006). In earlier studies, we and others showed that the parental virus SIVmneCl8 encodes a nef allele with minimal activity. As this virus evolves in vivo, it acquires mutations in nef that primarily increase viral infectivity and enable CD4 downregulation (Heidecker et al., 1998; Patel et al., 2002). The SIVmne170 and SIVmne027 nef alleles are representative of variants that have acquired these activities. Whether the mutations in these nef variants also affect CD3/CD28 downregulation, thereby interfering with T cell activation is unknown, but should be further investigated as a potential explanation for the inhibitory effect these alleles have on viral replication in resting CD4+ T cells after activation via CD3/CD28. Interestingly, in DC-T cell co-cultures, we found that the inhibitory effect of Nef in context of low functioning RTs is not observed, suggesting that activation signals derived from DC-T cell interactions and which increase viral replication, may occur through molecules other than CD3 and CD28, bypassing the negative effects of Nef. Alternatively, DC infection may induce cytokines and chemokines that render T cells permissive for viral infection.
In conclusion, our data identify SIV RT as a significant infectivity and replication determinant. The data provide further evidence that the pathogenicity of SIV may be linked to its capacity to spread in the CD4+ T cell population. Whether mutations in RT allow infection of specific T cell populations based on the activation status of the T cells needs to be explored further. This is significant in light of the discovery that the primary targets during early stages of HIV infection are memory CD4+ T cells with a ‘resting’ phenotype (Haase, 2005; Li et al., 2005; Zhang et al., 1999). While it may be argued that the SIVmneCl8 RT variant studied here resulted from happenstance and is unlikely to predominate at any time during infection of the host, the SIVmneCl8 genotype has been shown to predominate in primary infection following intrarectal inoculation of an uncloned population of SIVmne variants (Polacino et al., 1999). Additionally, SIVmneCl8 replicates as well as late variants, SIVmne170 and SIVmne027, in cell lines and to a higher level in monocyte-derived macrophages (Kimata et al., 2004). These data suggest that SIVmneCl8 is not inherently defective or unfit. Further, it is important to again note that post-acute viral burden in the host has been correlated with replicative capacity of RT variants (Campbell et al., 2003). Thus, RT variants of different functional capacity may influence viral replication and disease.
MATERIALS AND METHODS
Cell lines
The hybrid T cell-B cell line, CEM×174 (Russell et al., 1985), was cultured in RPMI supplemented with 10% heat-inactivated fetal bovine serum (FBS), 100 U/mL penicillin, 100 μg/mL streptomycin and 2 mM L-glutamine. The indicator cell line, sMAGI (Chackerian et al., 1995), was cultured in RPMI medium supplemented with 0.2 mg/mL G418 and 50 U/mL Hygromycin B (sMAGI medium). 293T cells (Lebkowski et al., 1985) were cultured in DMEM supplemented with 10% heat-inactivated FBS, 100 U/mL penicillin, 100 μg/mL streptomycin and 2 mM L-glutamine (DMEM medium). All cultures were incubated in 37°C/5% CO2 incubators.
Viruses
Chimeric viruses that contain either the pol region (8/027pol) or nef gene (8/027nef) of SIVmne027 in the background of SIVmneCl8 or pol region of SIVmne170 in the background of SIVmneCl8 (8/170pol) were previously described (Kimata et al., 2004; Kimata and Overbaugh, 1997). Recombinant viruses 8/170pol6A, 8/170pol6B and 8/170pol6C were constructed by excising a NsiI (position 1886)-HpaI (position 3868) pol fragment from SIVmneCl8 and replacing it with the homologous PCR-amplified variant pol sequences from animals infected with SIVmne170.
Stocks of SIVmneCl8, SIVmne027, SIVmne170, and chimeric viruses were generated by transfection of 293T cells with cloned proviral sequences using the FuGENE 6 transfection kit (Invitrogen) according to the manufacturer’s instructions. The cultures were incubated for 48 hours post-transfection, at which time supernatant was harvested and passed through 0.22 μm filters. Viral supernatants were titered on sMAGI cells and either frozen at -80°C until needed or were then mixed with CEM×174 cells to amplify the viral stocks. Supernatants from CEM×174 cells were collected on days 7, 10 and 12 days post-infection, passed through 0.22 μm filters, titered on sMAGI cells and the highest titer viral supernatants were stored at -80°C until use.
PCR Cloning of pol Fragments
Pig-tailed macaque genomic DNA specimens from PBMCs isolated at 6 weeks after inoculation with the molecular clone SIVmne170 were kindly provided by Julie Overbaugh (Fred Hutchinson Cancer Research Center) (Kimata et al., 1999). The virus stock of SIVmne170 used for infection was produced by transfecting CEM×174 cells and passaging 7 days. The proviral load in each DNA specimen was determined by quantitative real-time PCR essentially as described (Williams and Overbaugh, 2004). A volume of DNA sample estimated to contain one SIV genome was used to amplify and clone individual pol sequences by nested PCR. Pol sequences were amplified using TaqPlus long polymerase mixture (Stratagene, La Jolla, CA) to minimize mutations due to PCR. The first round PCR directly amplified pol from the genomic DNA sample. Two μl of the first round PCR were used for amplification in the second round. Round-one primers Pr-RT1 (5’-GACACTCTGCAAGGCAATGCAGAGC-3’, 1731-1756) and Pr-RT4 (5’-ATACCTTTGTGTGCTGGCACCCATGC-3’, 3916-3941) amplified a 2.2 kb fragment. Round-two primers Pr-RT2 (5’-CAAGAAGACAGGGCTGCTGGAAATG-3’, 1758-1783) and Pr-RT3 (5’-ATTTGGTTAACTAGCCTGCTCTCTG-3’, 3854-3880) amplified a 2.1 kb fragment. Reactions were performed using the following mixtures in 50 μl: 1× TaqPlus long buffer, 800 nM dNTPs, 200 ng of each primer, and 1.5 U TaqPlus long polymerase. For both rounds 1 and 2, amplification was initiated with denaturation at 94°C for 5 minutes followed by 35 cycles of amplification (denaturation at 94°C for 30 sec, annealing at 56°C for 30 sec and a final extension at 72°C for 2 min). Amplified products were cloned into a TOPO cloning vector (Invitrogen) and the sequences were determined.
Site-Directed Mutagenesis
Specific mutations were introduced into rt gene sequences by QuikChange mutagenesis according to the manufacturer’s protocol (Stratagene, La Jolla, CA). Subclones of SIVmneCl8 or SIVmne170 that contained gag-pol sequences inserted into pBluescript KS+ were used for mutagenesis. To introduce a mutation in the SIVmneCl8 rt gene that would change lysine at amino acid position 412 to glutamate, primers 5’-GGTAACCTGGATACCG G AATGGGATTTTATCTCAAC-3’ and 5’-GTTGAGATAAAATCCCATT C CGGTATCCAGGTTACC-3’ were used. To introduce a mutation in the SIVmneCl8 rt gene that would change alanine at position 486 to threonine, primers 5’-CATTTCTCATGGCATTGACAGACTCAGGGCCAAAG-3’ and 5’-CTTTGGCCCTGAGTCTGTCAATGCCATGAGAAATG-3’ were used for mutagenesis. To introduce a mutation in the SIVmne170 rt gene that would change the isoleucine at position 148 to valine, primers 5’-AAACGATACATTTATAAG G TTCTGCCTCAGGGGTGG-3’ And 5’-CCACCCCTGAGGCAGAA C CTTATAAATGTATCGTTT-3’ were used for mutagenesis. Mutations were confirmed by automated DNA sequencing and NsiI-HpaI fragments were cloned into the background of the respective isogenic parental provirus, either SIVmneCl8 or SIVmne170.
sMAGI Assay
Infection of sMAGI indicator cells was performed as described (Chackerian et al., 1995). Infectivity was determined by averaging the number of blue (infected) cells in triplicate cultures, subtracting the average number of blue cells in the uninfected control cultures and dividing by the volume of input viral supernatant. sMAGI assays were performed with different viral stocks for each experiment.
Dendritic Cell/CD4+ T Cell Infection
Peripheral blood mononuclear cells (PBMCs) were purified from pig-tailed macaque blood by Ficoll centrifugation as described previously (Kimata et al., 2004). To generate monocyte-derived DCs, CD14+ monocytes were isolated using Miltenyi anti-CD14 microbeads and miniMACS system according to the manufacturer’s protocol (Miltenyi Biotec, Auburn, CA). Monocytes were cultured in RPMI medium supplemented with 1000 U/mL granulocyte macrophage colony-stimulating factor (GM-CSF) and 1000 U/mL interleukin-4 (IL-4) for one week to induce maturation to dendritic cells as described (Yu Kimata et al., 2002). Dendritic cells were then co-cultured with autologous CD4+ T cells. Co-cultures were incubated with equal amounts of infectious virus (MOI=0.01), as determined by sMAGI assay, for 3 hours and then cells were washed to remove excess virus and reseeded in RPMI medium. Cultures were continued for 2-3 weeks with supernatant samples collected and media replaced every 3-4 days post-infection. Supernatant SIV p27 levels were measured by ELISA (Beckman Coulter, Fullerton, CA). Cells used in duplicate experiments were from different pig-tailed macaque blood samples.
Resting CD4+ T Cell Infection and Activation
PBMCs were purified from pig-tailed macaque blood by Ficoll centrifugation as described previously (Kimata et al., 2004). CD4+ T cells were purified by negative selection using a non-human primate CD4+ T cell isolation kit (Miltenyi Biotec Inc.) according to the manufacturer’s instructions. CD4+ T cells were 90% pure by flow cytometry analysis. After isolation, CD4+ T cells (5 × 105) were immediately incubated with equal amounts of infectious virus (MOI=0.1), as determined by sMAGI assay, for 3 hours in 2 mL RPMI. Cells were washed twice with RPMI to remove unbound virus, resuspended in 2 mL RPMI and incubated for 24 hrs. T cells were then transferred to RPMI media + 50 U/mL of interleukin-2 (IL-2) and 24-well plates coated with 1 μg/well of both anti-CD3 and anti-CD28 antibodies. Cultures were incubated for two weeks, supernatant samples were taken and media replaced (RPMI + IL-2) every two days. Supernatant SIV p27 levels were measured by ELISA (Beckman Coulter, Fullerton, CA). Cells used in duplicate experiments were from different pigtailed macaque blood samples.
Nucleotide Sequence Accession Numbers
Variant pol sequences cloned from SIVmne170-infected macaques were deposited into GenBank (accession numbers EF028382-EF028389).
Acknowledgments
We thank Andy Rice and Dorothy Lewis for insightful discussions, Julie Overbaugh for providing specimens from infected pig-tailed macaques and Maria Viskovska for technical assistance. Pig-tailed macaque blood was obtained from the National Primate Research Center at the University of Washington, supported by NIH grant RR00166. This work was supported by NIAID training grant T32-AI07471 (to T.B.) and NIH grants AI47725 (to J.T.K.) and P30AI036211 (Baylor–UT-Houston CFAR).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Argyris EG, Vanderkooi JM, Venkateswaran PS, Kay BK, Paterson Y. The Connection Domain Is Implicated in Metalloporphyrin Binding and Inhibition of HIV Reverse Transcriptase. Journal of Biological Chemistry. 1999;274:1549–1556. doi: 10.1074/jbc.274.3.1549. [DOI] [PubMed] [Google Scholar]
- Barbour JD, Hecht FM, Wrin T, Segal MR, Ramstead CA, Liegler TJ, Busch MP, Petropoulous CJ, Hellmann NS, Kahn JO, Grant RM. Higher CD4+ T Cell Counts Associated with Low Viral pol Replication Capacity among Treatment-Naive Adults in Early HIV-1 Infection. Journal of Infectious Diseases. 2004;190:251–256. doi: 10.1086/422036. [DOI] [PubMed] [Google Scholar]
- Cameron PU, Freudenthal PS, Barker JM, Gezelter S, Inaba K, Steinman RM. Dendritic Cells Exposed to Human Immunodeficiency Virus Type-1 Transmit a Vigorous Cytopathic Infection to CD4$ˆ+$ T Cells. Science. 1992;257:383–387. doi: 10.1126/science.1352913. [DOI] [PubMed] [Google Scholar]
- Campbell TB, Schneider K, Wrin T, Petropoulos CJ, Connick E. Relationship between In Vitro Human Immunodeficiency Virus Type 1 Replication Rate and Virus Load in Plasma. J Virol. 2003;77:12105–12112. doi: 10.1128/JVI.77.22.12105-12112.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cen S, Niu M, Kleiman L. The connection domain in reverse transcriptase facilitates the in vivo annealing of tRNALys3 to HIV-1 genomic RNA. Retrovirology. 2004;1:33–40. doi: 10.1186/1742-4690-1-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chackerian B, Haigwood NL, Overbaugh J. Characterization of a CD4-Expression Macaque Cell Line That Can Detect Virus after a Single Replication Cycle and Can Be Infected by Diverse Simian Immunodeficiency Virus Isolates. Virology. 1995;213:386–394. doi: 10.1006/viro.1995.0011. [DOI] [PubMed] [Google Scholar]
- Connor RI, Mohri H, Cao Y, Ho DD. Increased viral burden and cytopathicity correlate temporally with CD4+ T-lymphocyte decline and clinical progression in human immunodeficiency virus type 1-infected individuals. J Virol. 1993;67:1772–1777. doi: 10.1128/jvi.67.4.1772-1777.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daar ES, Kesler KL, Wrin T, Petropoulos CJ, Bates M, Lail A, Hellmann NS, Gomperts E, Donfield S Hemophilia Growth and Development Study. HIV-1 pol replication capacity predicts disease progression. AIDS. 2005;19:871–877. doi: 10.1097/01.aids.0000171400.15619.e1. [DOI] [PubMed] [Google Scholar]
- Diamond TL, Kimata JT, Kim B. Identification of a Simian Immunodeficiency Virus Reverse Transcriptase Variant with Enhanced Replicational Fidelity in the Late Stage of Viral Infection. J Biol Chem. 2001;276:23624–23631. doi: 10.1074/jbc.M102496200. [DOI] [PubMed] [Google Scholar]
- Diamond TL, Roshal M, Jamburuthugoda VK, Reynolds HM, Merriam AR, Lee KY, Balakrishnan M, Bambara RA, Planelles V, Dewhurst S, Kim B. Macrophage Tropism of HIV-1 Depends on Efficient Cellular dNTP Utilization by Reverse Transcriptase. Journal of Biological Chemistry. 2004;279:51545–51553. doi: 10.1074/jbc.M408573200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond TL, Souroullas G, Weiss KK, Lee KY, Bambara RA, Dewhurst S, Kim B. Mechanistic Understanding of an Altered Fidelity Simian Immunodeficiency Virus Reverse Transcriptase Mutation, V148I, Identified in a Pig-tailed Macaque. J Biol Chem. 2003;278:29913–29924. doi: 10.1074/jbc.M211754200. [DOI] [PubMed] [Google Scholar]
- Divita G, Restle T, Goody RS, Chermann JC, Baillon JG. Inhibition of human immunodeficiency virus type 1 reverse transcriptase dimerization using synthetic peptides derived from the connection domain. Journal of Biological Chemistry. 1994;269:13080–13083. [PubMed] [Google Scholar]
- Embretson J, Zupancic M, Ribas JL, Burke A, Racz P, Tenner-Racz K, Haase AT. Massive covert infection of helper T lymphocytes and macrophages by HIV during the incubation period of AIDS. Nature. 1993;362:359–362. doi: 10.1038/362359a0. [DOI] [PubMed] [Google Scholar]
- Gummuluru S, KewalRamani VN, Emerman M. Dendritic Cell-Mediated Viral Transfer to T Cells Is Required for Human Immunodeficiency Virus Type 1 Persistence in the Face of Rapid Cell Turnover. J Virol. 2002;76:10692–10701. doi: 10.1128/JVI.76.21.10692-10701.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haase AT. Perils At Mucosal Front Lines For HIV And SIV And Their Hosts. Nat Rev Immunol. 2005;5:783–792. doi: 10.1038/nri1706. [DOI] [PubMed] [Google Scholar]
- Heidecker G, Munoz H, Lloyd P, Hodge D, Ruscetti FW, Morton WR, Hu S-L, Benveniste RE. Macaques Infected with Cloned Simian Immunodeficiency Virus Show Recurring nef Gene Alterations. Virology. 1998;249:260–274. doi: 10.1006/viro.1998.9325. [DOI] [PubMed] [Google Scholar]
- Ho DD, Moudgil T, Alam M. Quantitation of human immunodeficiency virus type 1 in the blood of infected persons. N Engl J Med. 1989;321:1621–1625. doi: 10.1056/NEJM198912143212401. [DOI] [PubMed] [Google Scholar]
- Julias JG, McWilliams MJ, Sarafianos SG, Alvord WG, Arnold E, Hughes SH. Mutation of Amino Acids in the Connection Domain of Human Immunodeficiency Virus Type 1 Reverse Transcriptase That Contact the Template-Primer Affects RNase H Activity. J Virol. 2003;77:8548–8554. doi: 10.1128/JVI.77.15.8548-8554.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimata JT. HIV-1 Fitness and Disease Progression: Insights from the SIV-Macaque Model. Current HIV Research. 2006;4:65–77. doi: 10.2174/157016206775197628. [DOI] [PubMed] [Google Scholar]
- Kimata JT, Kuller L, Anderson DB, Dailey P, Overbaugh J. Emerging cytopathic and antigenic simian immunodeficiency virus variants influence AIDS progression. Nature Medicine. 1999;5:535–541. doi: 10.1038/8414. [DOI] [PubMed] [Google Scholar]
- Kimata JT, Mozaffarian A, Overbaugh J. A Lymph Node-Derived Cytopathic Simian Immunodeficiency Virus Mne Variant Replicates in Nonstimulated Peripheral Blood Mononuclear Cells. J Virol. 1998;72:245–256. doi: 10.1128/jvi.72.1.245-256.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimata JT, Overbaugh J. The cytopathicity of a simian immunodeficiency virus Mne variant is determined by mutations in Gag and Env. J Virol. 1997;71:7629–7639. doi: 10.1128/jvi.71.10.7629-7639.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimata JT, Wilson JM, Patel PG. The increased replicative capacity of a late-stage simian immunodeficiency virus mne variant is evident in macrophage-or dendritic cell-T-cell cocultures. Virology. 2004;327:307–317. doi: 10.1016/j.virol.2004.07.010. [DOI] [PubMed] [Google Scholar]
- Lebkowski JS, Clancy S, Calos MP. Simian virus 40 replication in adenovirus-transformed human cells antagonizes gene expression. Nature. 1985;317:169–171. doi: 10.1038/317169a0. [DOI] [PubMed] [Google Scholar]
- Li Q, Duan L, Estes JD, Ma ZM, Rourke T, Wang Y, Reilly C, Carlis J, Miller CJ, Haase AT. Peak SIV replication in resting memory CD4+ T cells depletes gut lamina propria CD4+ T cells. Nature. 2005;434:1148–1152. doi: 10.1038/nature03513. [DOI] [PubMed] [Google Scholar]
- Mattapallil JJ, Douek DC, Hill B, Nishimura Y, Martin M, Roederer M. Massive infection and loss of memory CD4+ T cells in multiple tissues during acute SIV infection. Nature. 2005;434:1093–1097. doi: 10.1038/nature03501. [DOI] [PubMed] [Google Scholar]
- Mellors JW, Charles RR, Gupta P, White RM, Todd JA, Kingsley LA. Prognosis in HIV-1 Infection Predicted by the Quantity of Virus in Plasma. Science. 1996;272:1167–1170. doi: 10.1126/science.272.5265.1167. [DOI] [PubMed] [Google Scholar]
- Mellors JW, Munoz A, Giorgi JV, Margolick JB, Tassoni CJ, Gupta P, Kingsley LA, Todd JA, Saah AJ, Detels R, Phair JP, Rinaldo CR., Jr Plasma Viral Load and CD4+ Lymphocytes as Prognostic Markers of HIV-1 Infection. Ann Intern Med. 1997;126:946–954. doi: 10.7326/0003-4819-126-12-199706150-00003. [DOI] [PubMed] [Google Scholar]
- Messmer D, Ignatius R, Santisteban C, Steinman RM, Pope M. The Decreased Replicative Capacity of Simian Immunodeficiency Virus SIVmac239Delta nef Is Manifest in Cultures of Immature Dendritic Cells and T Cells. J Virol. 2000;74:2406–2413. doi: 10.1128/jvi.74.5.2406-2413.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohri H, Perelson AS, Tung K, Ribeiro RM, Ramratnam B, Markowitz M, Kost R, Hurley R, Weinberger L, Cesar D, Hellerstein MK, Ho DD. Increased Turnover of T Lymphocytes in HIV-1 Infection and Its Reduction by Antiretroviral Therapy. J Exp Med. 2001;194:1277–1288. doi: 10.1084/jem.194.9.1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolenko GN, Delviks-Frankenberry KA, Palmer S, Maldarelli F, Fivash MJ, Coffin JM, Pathak VK. Mutations in the connection domain of HIV-1 reverse transcriptase increase 3’-azido-3’-deoxythymidine resistance. Proc Natl Acad Sci. 2007;104:317–322. doi: 10.1073/pnas.0609642104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overbaugh J, Rudensey LM, Papenhausen MD, Benveniste RE, Morton WR. Variation in simian immunodeficiency virus env is confined to V1 and V4 during progression to simian AIDS. J Virol. 1991;65:7025–7031. doi: 10.1128/jvi.65.12.7025-7031.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantaleo G, Graziosi C, Demarest JF, Butini L, Montroni M, Fox CH, Orenstein JM, Kotler DP, Fauci AS. HIV infection is active and progressive in lymphoid tissue during the clinically latent stage of disease. Nature. 1993;362:355–358. doi: 10.1038/362355a0. [DOI] [PubMed] [Google Scholar]
- Patel PG, Yu Kimata MT, Biggins JE, Wilson JM, Kimata JT. Highly Pathogenic Simian Immunodeficiency Virus mne Variants That Emerge during the Course of Infection Evolve Enhanced Infectivity and the Ability to Downregulate CD4 but Not Class I Major Histocompatibility Complex Antigens. J Virol. 2002;76:6425–6434. doi: 10.1128/JVI.76.13.6425-6434.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polacino PS, Stallard V, Montefiori DC, Brown CR, Richardson BA, Morton WR, Benveniste RE, Hu S-L. Protection of Macaques against Intrarectal Infection by a Combination Immunization Regimen with Recombinant Simian Immunodeficiency Virus SIVmne gp160 Vaccines. J Virol. 1999;73:3134–3146. doi: 10.1128/jvi.73.4.3134-3146.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pope M, Betjes MGH, Romani N, Hirmand H, Cameron PU, Hoffman L, Gezelter S, Schuler G, Steinman RM. Conjugates of Dendritic Cells and Memory T Lymphocytes from Skin Facilitate Productive Infection with HIV-1. Cell. 1994;78:389–398. doi: 10.1016/0092-8674(94)90418-9. [DOI] [PubMed] [Google Scholar]
- Roeth JF, Collins KL. Human Immunodeficiency Virus Type 1 Nef: Adapting to Intracellular Trafficking Pathways. Microbiol Mol Biol Rev. 2006;70:548–563. doi: 10.1128/MMBR.00042-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudensey LM, Kimata JT, Benveniste RE, Overbaugh J. Progression to AIDS in Macaques Is Associated with Changes in the Replication, Tropism, and Cytopathic Properties of the Simian Immunodeficiency Virus Variant Population. Virology. 1995;207:528–542. doi: 10.1006/viro.1995.1113. [DOI] [PubMed] [Google Scholar]
- Russell DS, David NH, Peter C. Genes regulating HLA class I antigen expression in T-B lymphoblast hybrids. Immunogenetics. 1985;21:235–246. doi: 10.1007/BF00375376. [DOI] [PubMed] [Google Scholar]
- Schnittmen SM, Greenhouse JJ, Psallidopoulos MC, Baseler M, Salzman NP, Fauci AS, Lane HC. Increasing Viral Burden in CD4+ T Cells from Patients with Human Immunodeficiency Virus (HIV) Infection Reflects Rapidly Progressive Immunosuppression and Clinical Disease. Ann Intern Med. 1990;113:438–443. doi: 10.7326/0003-4819-113-6-438. [DOI] [PubMed] [Google Scholar]
- Veazey RS, Tham IC, Mansfield KG, DeMaria M, Forand AE, Shvetz DE, Chalifoux LV, Sehgal PK, Lackner AA. Identifying the Target Cell in Primary Simian Immunodeficiency Virus (SIV) Infection: Highly Activated Memory CD4+ T Cells Are Rapidly Eliminated in Early SIV Infection In Vivo. J Virol. 2000;74:57–64. doi: 10.1128/jvi.74.1.57-64.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voronin Y, Overbaugh J, Emerman M. Simian Immunodeficiency Virus Variants That Differ in Pathogenicity Differ in Fitness under Rapid Cell Turnover Conditions. J Virol. 2005;79:15091–15098. doi: 10.1128/JVI.79.24.15091-15098.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whetter LE, Ojukwu IC, Novembre FJ, Dewhurst S. Pathogenesis of simian immunodeficiency virus infection. J Gen Virol. 1999;80:1557–1568. doi: 10.1099/0022-1317-80-7-1557. [DOI] [PubMed] [Google Scholar]
- Williams D, Overbaugh J. A real-time PCR-based method to independently sample single simian immunodeficiency virus genomes from macaques with a range of viral loads. Journal of Medical Primatology. 2004;33:227–235. doi: 10.1111/j.1600-0684.2004.00073.x. [DOI] [PubMed] [Google Scholar]
- Yu Kimata MT, Cella M, Biggins JE, Rorex C, White R, Hicks S, Wilson JM, Patel PG, Allan JS, Colonna M, Kimata JT. Capture and Transfer of Simian Immunnodeficiency Virus by Macaque Dendritic Cells Is Enhanced by DC-SIGN. J Virol. 2002;76:11827–11836. doi: 10.1128/JVI.76.23.11827-11836.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z-Q, Schuler T, Zupancic M, Wietgrefe S, Staskus KA, Reimann KA, Reinhart TA, Rogan M, Cavert W, Miller CJ, Veazey RS, Notermans D, Little S, Danner SA, Richman DD, Havlir D, Wong J, Jordan HL, Schacker TW, Racz P, Tenner-Racz K, Letvin NL, Wolinsky SM, Haase AT. Sexual Transmission and Propagation of SIV and HIV in Resting and Activated CD4+ T Cells. Science. 1999;286:1353–1357. doi: 10.1126/science.286.5443.1353. [DOI] [PubMed] [Google Scholar]