

Abstract
To understand the physiological function of glutaredoxin, a thiotransferase catalyzing the reduction of mixed disulfides of protein and glutathione (protein-SSG), we generated a line of knockout mice deficient in the cytosolic glutaredoxin 1 (Grx1). To our surprise, mice deficient in Grx1 were not more susceptible to acute oxidative insults in models of heart and lung injury induced by ischemia/reperfusion and hyperoxia, respectively; suggesting that changes in S-glutathionylation status of cytosolic proteins are not the major cause of such tissue injury. On the other hand, mouse embryonic fibroblasts (MEFs) isolated from Grx1-deficient mice displayed an increased vulnerability to diquat and paraquat, but they were not more susceptible to cell death induced by hydrogen peroxide (H2O2) and diamide. A deficiency in Grx1 also sensitized MEFs to protein S-glutathionylation in response to H2O2 treatment and retarded deglatuthionylation of the S-glutathionylated proteins, especially evident for an unspecified protein of approximately 44 kDa. Additional experiments showed that MEFs lacking Grx1 were more tolerant to apoptosis induced by tumor necrosis factor α plus actinomycin D. These findings suggest that different oxidants may damage the cells via distinct mechanisms in which Grx1-dependent de-glutathionylation may or may not be protective, and Grx1 may exert its function on specific target proteins.
Keywords: Reaction oxygen species, protein glutathionylation, thiol oxidation, cell death, gene targeting
Introduction
In mammals, reaction oxygen species (ROS) are constantly generated in cells by leakage of electrons from the mitochondrial electron transport chain or by cytosolic flavoproteins, enzymatic reactions catalyzed by various oxidases, as well as autooxidation of a number of oxygen-containing reaction intermediates generated in enzymatic reactions [1]. ROS at low concentrations may act as secondary messengers in redox signaling pathways [2 – 4]; however, at higher concentrations they may disrupt normal cell functions through oxidative modification of cellular constituents such as proteins and lipids. The cells counterbalance the production of ROS with a complex system of nonenzymatic and enzymatic antioxidants. The latter consists of three isoforms of superoxide dismutase functioning in converting superoxide anion radical to hydrogen peroxide, which can then be decomposed by catalase and/or by various forms of glutathione peroxidase and peroxiredoxin [5, 6].
However, depending on the subcellular sites and extent of ROS generation, as well as the distribution and concentrations of antioxidants in the cells, some of the ROS produced may escape the cellular antioxidant defense, leading to oxidative damage of cells. Oxidant-induced cell damage and tissue injury are evident in mammals under certain pathogenic conditions in which the rate of ROS production is enhanced such as exposure to radiation, xenobiotics, and hyperoxia; tissue ischemia followed by reperfusion; and inflammation while the phagocytic cells are activated [7]. One of the major cellular targets of oxidation is protein. ROS can oxidize both aliphatic and aromatic amino acid residues of proteins [8], leading to irreversible structural changes. Additionally, the cysteine residues of proteins, particularly following ionization to cysteine thiolate anions, are vulnerable to oxidation to form sulfenic acids (−SOH). The cysteine sulfenic acids in proteins are very unstable and can have several fates; they can react readily with vicinal protein thiols or non-protein thiols, especially glutathione (GSH), to form intra- and interprotein disulfides and mixed disulfides of proteins with GSH (protein-SSG), respectively. Alternatively, in the absence of thiols in close vicinity, they can be further oxidized to cysteine sulfinic (RSO2H) and sulfonic (RSO3H) acids. The chemical changes that result from thiol oxidation can affect the catalytic and/or structural function of a protein.
Remarkably, cells are equipped with enzyme systems to specifically reduce most of these forms of oxidized thiols. Cysteine sulfinic acid is generally thought to represent an irreversible oxidative product of protein thiols that that can progress to the sulfonic acid oxidation state. However, recent data have documented an exception, showing that the sulfinic acid moiety in 2-Cys peroxiredoxins in cells under oxidative stress can be reduced by two ATP-dependent reductases, sulfiredoxin and sestrin [9 – 12].
The other forms of oxidized protein thiols can be reduced by two members of the thiol-disulfide oxidoreductase superfamily, thioredoxin (Trx) and glutaredoxin (Grx), that contain conserved Cys-X-X-Cys motifs and serve as hydrogen donors for ribonucleotide reductase [13]. Although both Trx1 and Grx1 exhibit activity in regeneration of oxidatively damaged proteins, further studies have shown that they have different substrate preferences [14, 15]. Trx has been reported to preferentially reduce protein sulfenic acids as well as intra- and inter-protein disulfides. Two isoforms of thioredoxin have been found in mammals. Trx1 is a 104 amino-acid protein and is mainly present in the cytosol [16]. The mature protein of Trx2 is approximately 100 amino-acids long and is located in the mitochondria [17]. Since any protein bearing an accessible thiol for activity is susceptible to oxidation, and thiol oxidation often alters the structure and function of many proteins, Trx is believed to regulate a variety of cellular functions through reduction of the target proteins. Indeed, both Trx1 and Trx2 have been shown to function in modulation of cellular redox signaling and in defense against oxidant-mediated injury in vitro and in vivo [18].
In contrast, Grx is believed to play a unique role in the repair of oxidized protein thiols, specifically catalyzing the reduction of protein-SSG [3, 15]. Since GSH is the most abundant non-protein thiol in cells (at concentrations from 0.5 to 20 mM) [19], the majority of protein mixed disulfides formed inside cells under oxidative stress is protein-SSG, and to a lesser extent intra- and inter-protein disulfides. For the latter two species of protein disulfides, formation of intra-protein disulfides is believed to be more favorable than that of inter-protein disulfides in cells under oxidative stress, because the chance of having two thiol groups residing on two proteins in close vicinity is relatively low. The protein-SSG mixed disulfides are reduced efficiently by Grx via a monothiol mechanism, involving a nucleophilic double displacement reaction in which GSH serves to recycle the enzyme [20]. Furthermore, previous tissue and cell culture studies have indicated that glutathione-dependent catalysis by Grx accounts for most of the deglutathionylation activity [21, 22]. Therefore, Grx is believed to play a pivotal role in defense against oxidative stress and in redox regulation of cellular function [3].
Two glutaredoxin (Glrx) genes have been characterized in mammals. The Glrx1 gene encodes the cytosolic enzyme Grx1 [23]. The second Glrx (Glrx2) gene encodes two proteins as a result of alteration of RNA splicing. One of the Grx2 isoforms is expressed with a N-terminal leader sequence that directs it to the mitochondria, and both have C-terminal sequences that suggest nuclear localization [24, 25]. As expected, cells overexpressing Grx1 are more resistant to doxorubicin-induced toxicity compared to control cells [26]. In addition, treatment of cultured cerebellar granule neurons with recombinant Grx protein of E. coli prevents cell death induced by dopamine, and the protection appears to be mediated through activation of the Ras/PI3K/Ref-1/Akt/NF-κB pathway [27, 28]. This protective mechanism was further elaborated in another study in which Grx1 overexpression was shown to protect H9c2 cells against toxicity of hydrogen peroxide (H2O2) [29]. Apparently, Grx1 maintains the level of phosphorylated Akt (activated Akt), a general mediator of cellular survival signals, by dissociating the interaction between Akt and protein phosphatase 2A. Interestingly, Grx1 also affects other cellular signaling pathways. Lee and colleagues showed that Grx1 protects cells against oxidant injury from glucose starvation, probably through binding to the C-terminus of apoptosis signal-regulating kinase 1 (ASK1) [30, 31]. In addition, a number of studies with a variety of cells in culture have documented a role of Grx1 in regulation of redox signal transduction [3]. Despite these in vitro studies, the function of Grx1 in vivo has not yet been characterized. In this report, we describe the generation of a line of knockout mice deficient in Grx1, along with the tests of potential physiological consequences of the enzyme deficiency.
Materials and Methods
Targeted disruption of the mouse glutaredoxin 1 (Glrx1) gene
Custom PCR (polymerase chain reaction) screening of a bacterial artificial chromosome (BAC) library carrying strain 129SV mouse genomic DNA was performed by Incyte Genomics (St. Louis, MO) using two pairs of primers derived from the sequence of a mouse cDNA coding for Grx1 (NCBI accession #AA239905). Three BAC clones (GS Control Number 20424, 20428, and 20429) were identified from the screening. Multiple DNA fragments from BAC clone 20424 were then subcloned into plasmid pKS (Stratagene, La Jolla, CA) and DNA sequence determined. The mouse Glrx1 gene contains three exons and the protein coding sequence is located in the first two exons (Fig. 1A). Exon 3 only codes for the 3’ untranslated sequence of the mRNA. The structure of the mouse Glrx1 gene is virtually identical to that of the human gene, except intron 1 of the mouse gene is unusually long − 7.2 kb, compared to 1.0 kb in humans [32].
Fig. 1.

Targeted disruption of the mouse Glrx1 gene. (A) Genomic structure and partial restriction map of the mouse Glrx1 locus (top), the targeting vector (middle), and the targeted locus (bottom) are shown. The opened and black boxes represent the protein coding regions and noncoding regions in the exons. The number of the exon is indicated under each exon. The shaded box on top of the restriction map of the Glrx1 locus represents the 3' external sequence used for probing the DNA blot filters. B, BamHI; E, EcoRI; H, HindIII; N, NotI; P, PstI; neo, neomycin resistance cassette; TK, herpes thymidine kinase gene under the control of a mouse promoter of the phosphoglycerate kinase-1 (PGK-1) gene [33]. Arrows show the directions of transcription of the neo and TK genes. The sizes of the PstI restriction fragments from the wild-type and targeted loci hybridized with the probe are shown on the top and bottom of the figure, respectively. (B) DNA blot analysis of wild-type, heterozygous Glrx1 knockout, and homozygous Glrx1 knockout mice. DNA isolated from mouse tails was digested with PstI and probed with the 3' external probe shown in (A). +/+, +/−, and −/− represent wild-type, heterozygous Glrx1 knockout, and homozygous Glrx1 knockout mice, respectively. The 4.3 kb hybridization band is derived from the wild-type allele, and 2.9 kb hybridization band the mutated allele.
To construct the targeting vector, the 3.5-kb HindIII DNA fragment containing the 5’ flanking sequence of the mouse Glrx1 gene was first inserted into the HindIII site of plasmid pKS with the direction of transcription of the mouse genomic DNA fragment opposite to that of the ampicillin resistance gene (coding for β-lactamase). The 3.5-kb HindIII DNA fragment was subsequently released from the pKS vector by digestion with enzymes KpnI and XbaI (which are located in the multiple cloning sites of pKS vector) and cloned into the corresponding sites in plasmid pPNT [33], resulting in plasmid pPNT-5’Glrx1. The plasmid pPNT-5’Glrx1 was digested with KpnI and SalI, blunt-ended by treatment with mung bean nuclease, and then religated. These procedures removed the XhoI site, that was derived from the multiple cloning sites of pKS, in plasmid pPNT-5’Grx1 to generate plasmid pPNT-5’Glrx1(−X). For inserting the 3’ portion of the mouse Glrx1 gene into the targeting vector, the 1.9-kb BamHI DNA fragment containing exon 3 of the mouse Glrx1 gene was initially inserted into the BamHI site of plasmid pKS with transcription direction opposite to that of the ampicillin resistance gene. This BamHI DNA fragment was then removed from the pKS plasmid by digestion with enzymes XhoI and NotI (which are located in the multiple cloning sites of pKS vector) and cloned into the corresponding sites in plasmid pPNT-5’Glrx1(−X). The resulting targeting vector, in which exons 1 and 2 were replaced by a neomycin resistance cassette, was linearized with NotI digestion and then transfected into R1 embryonic stem (ES) cells [34]. Fourteen clones (2.8%) were identified from a total of 503 clones screened to contain the targeted Grx1 allele. Chimeric mice were then generated by microinjecting clones #118 and #256 into blastocysts isolated from strain C57BL/6 mice [35]. All chimeric mice transmitted the targeted Glrx1 allele into offspring. Heterozygous knockout mice were interbred to generate homozygous knockout mice.
Preparation of tissue samples for gene expression studies
Tissues from wild-type (Glrx1+/+), heterozygous Glrx1 knockout (Glrx1+/−), and homozygous Glrx1 knockout (Glrx1−/−) mice were homogenized in guanidinium isothiocynante solution, and total RNA was isolated according to the method described Chirgwin et al. [36]. Thirty micrograms of total RNA were denatured with glyoxyal and subjected to blot analysis according to the procedures described by Thomas [37]. For protein analysis, the tissues were homogenized in 1.5 to 2 ml of lysis buffer (50 mM potassium phosphate buffer, pH 7.8, 0.5% Triton X-100, and 3% glycerol) containing protease inhibitor cocktail (P-8340, Sigma, St. Louis, MO) and 1 mM phenylmethylsulfonyl fluoride with a Polytron homogenizer, followed by sonication. The homogenates were clarified by centrifugation at 20,000 × g for 15 min and stored at −70 °C. Protein concentrations of tissue homogenates were determined by the use of a bicinchoninic acid protein assay kit (Pierce, Rockford, IL). Thirty micrograms of tissue protein were separated on a SDS-polyacrylamide gel for protein blot analysis. The protein blot membrane was reacted with polyclonal antibodies against the human Grx1 protein generated in rabbits (kindly provided by Dr. Marjorie Lou of University of Nebraska at Lincoln, Lincoln, NE) and then with polyclonal antibodies against copper-zinc superoxide dismutase (Santa Cruz, SantaCruz, CA).
Preparation of tissue samples and assay for activity of Grx
Frozen tissue samples were thawed on ice and 100 mg of them were weighed and transferred to individual 15 ml conical tubes prechilled on ice. 1.5 ml of complete homogenization buffer [0.1 M K-monobasic/Na-dibasic pH7.5, supplemented with protease inhibitors aprotinin and leupeptin (20 ng/ml)] was added to each tube and then homogenized twice for 30 seconds on ice using a Polytron homogenizer. The homogenate obtained were transferred to 2 ml centrifuge tubes and centrifuged at 100 × g for 10 min at 4 °C (to remove the cell debris). The supernatant (free of cell debris) was transferred into fresh 1.5 ml tubes and centrifuged at 10,000 × g for 30 min at 4 °C. The resulting supernatant was used for assays of protein content and Grx activity.
Bovine serum albumin-SSG[35S] mixed disulfide ([35S]BSA-SSG) was used as substrate in a radiolabel assay for determining the activity of Grx (both Grx1 and Grx2). [35S]BSA-SSG was prepared as described previously with the following modifications [38]. S-carboxymethyl BSA was initially reacted with N-succinimidylpyridyl bis (3,3’-dithio-propionate) and the reaction was then quenched with glycine. The modified BSA was subsequently separated from small molecules by dialysis against 100 mM sodium phosphate pH 7.0 overnight with two changes of buffer, following by reaction with 4 mM [35S]GSH for 1 hour at room temperature. The resulting [35S]BSA-SSG product was separated from [35S]GSH by G-25 chromatography, and it typically has ≥ 0.9 GS-equivalent/mol of BSA.
The activity of Grxs in mouse tissues was measured by monitoring time-dependent release of radioactivity from [35S]BSA-SSG. This assay of GSH-dependent de-glutathionylation of protein-SSG is highly selective for Grx [22]. Also, the radiolabel assay avoids the multiple flaws associated with the GSSG reductase-coupled spectrophotometric analysis of tissue homogenates, including interference by light scattering and non-specific NADPH oxidase activity. Aliquots of homogenates and assay buffer (0.1 M Na/K phosphate buffer, pH 7.4, containing 0.5 mM GSH and 0.2 mM NADPH) were pre-warmed to 30°C, mixed, and then an aliquot of [35S]BSA-SSG (final concentration of 0.1 mM) was added to initiate the reaction (total volume 0.5 ml). Aliquots of the reaction mixtures were precipitated with ice-cold trichloroacetic acid (final concentration of 10%) at 12 sec, 1, 2, and 3 min of incubation. After centrifugation, the supernatants were analyzed for 35S by scintillation counting with ≥ 97% efficiency (cpm). The total rates of de-glutathionylation (slopes of [35S]GSH-equivalents released versus time) were corrected for non-enzymatic de-glutathionylation by subtracting the rate of [35S]GSH-equivalents released by GSH in the absence of tissue homogenates. Enzymatic rates are expressed as nanomoles of product/min/mg of protein.
Mouse model of regional ischemia/reperfusion-induced heart injury
Mice at 10 to 12 weeks of age were anesthetized with tribromoethanol (275 mg/kg, i.p.). An endotracheal tube (PE 90) was inserted 5 to 8 mm from the larynx, and the mice were ventilated with room air (a tidal volume of 0.5 ml) using a rodent respirator (Columbus Instruments International, Columbus, OH) set at 110–120 beats/min. The heart was exposed through a left thoracotomy incision and the left anterior descending coronary artery (LAD) was ligated as described previously [39]. After 60 min of LAD ligation, the ligature was released to allow reperfusion for 4 h. The mouse was again anesthetized with tribromoethanol and thoracostomy performed. The heart was then perfused as Langendorff preparations for 5 min. The left coronary artery was re-occluded, and 1% Evans blue was infused into the aorta and coronary arteries to determine the area at risk. The heart was then cut transversely into 5 sections, with one section made at the site of the ligature, and stained with triphenyltetrazolium chloride (TTC) for measurement of the infarct size as described previously [39]. The area at risk (unstained by Evans blue) was expressed as a percentage of the left ventricle (LV). The infarct size (area unstained by TTC) was expressed both as a percentage of the area at risk and as a percentage of LV [39].
Assessment of hyperoxia-induced lung injury in mice
The Glrx1+/+ and Glrx1−/− mice at 10 weeks of age were used in exposure to >99% oxygen in polystyrene chambers. The CO2 concentration was maintained at less than 5% by providing approximately 12 complete gas exchanges per hr. During the exposure, food and water ad libitum were provided, and the mice were kept under a 12-h on, 12-h off light cycle at all times. The mice were sacrificed at 72 hrs of exposure by overdose of pentobarbital and the lungs isolated. The left lobe was gently blotted dry with a piece of paper towel and the total weight (wet weight) measured. The lungs were then dried at 80 °C under vacuum overnight and weighted again for measuring the dry weight.
Preparation of mouse embryonic fibroblasts (MEFs)
The homozygous Glrx1 knockout mice in a 129SV and C57BL/6 hybrid background and genetically matched wild-type mice were used for preparation of MEFs. Briefly, mouse embryos were dissected out from the pregnant dams between 12.5 to 14.5 days post-coitus and internal organs removed. The embryos were then chopped into small pieces and stirred in 50 ml of Hanks’ Balanced Salt Solution (without calcium and magnesium) containing 0.05% trypsin and 0.53 mM EDTA (25300-054, Invitrogen, Carlsbad, CA) in an Erlenmeyer flask at 37 °C for 1 hr. Ten milligrams of DNase were added at the end of incubation to decrease the viscosity of the cell suspension due to lysis of cells. The cell suspension was then passed through two layers of cheese cloth to remove the undigested and large chunks of tissues. The cells were then collected by centrifugation and cultured in Dulbecco’s modified Eagle medium (Catalog #11995-065, Invitrogen, Carlsbad, CA) containing 10% fetal bovine serum, penicillin, and streptomycin. MEFs between passages 2 to 4 were used for the study.
Determination of cytotoxicity in MEFs by MTT reduction assay
24-well culture plates, each well seeded with 2 × 105 MEFs prepared from either Glrx1+/+ or Glrx1−/− mice, were used for cytotoxicity studies. For studying H2O2-induced cell death, the overnight cultures of MEFs were washed once with Dulbecco’s phosphate-buffered saline (D-PBS, Base Catalogue No. 14040, Invitrogen, Carlsbad, CA) supplemented with glucose (1 g/liter) and then treated with the same solution containing various concentrations of H2O2 (1 ml/well) at 37 °C with 5% CO2 for one hour. The D-PBS was removed at the end of treatment and cells were further incubated in 1 ml of culture medium containing 0.5 mg 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT) for 3 hours. The MTT-medium was subsequently removed and replaced with 0.5 ml of isopropyl alcohol containing 0.01 N HCl to dissolve the formazan crystals that were formed from reduction of MTT in the cells [40]. 0.2 ml of the dissolved formazan was then transferred into a 96-well plate and the absorbance at 570 nm measured using the SPECTRAmax® Microplate Spectrophotometer (Molecular Devices, Sunnyvale, CA). The net absorbance from the wells of MEFs treated with D-PBS without H2O2 was taken as 100% of MTT reduction. Cell viability was calculated by dividing the absorbance of H2O2-treated MEFs with that of the same MEFs treated with D-PBS without H2O2.
The same procedures were also used for quantifying cell viability following treatment with diamide [azodicarboxylic acid bis (dimethylamide)], diquat (1,1′-ethylene-2,2′-bipyridyldiylium dibromide), paraquat (1,1′-dimethyl-4,4′-bipyridinium dichloride), and tumor necrosis factor α (TNFα) plus actinomycin D, except the cells were treated in culture medium instead of D-PBS, and the treatment lasted 24 hrs.
Detection of protein S-glutathionylation in MEFs by protein blot analysis
For determining the effect of Grx1 deficiency on protein S-glutathionylation, 35-mm culture dishes, each seeded with 5 × 105 Glrx1+/+ or Glrx1−/− MEFs, were treated with 2.5 ml of D-PBS containing glucose (1 g/liter) and various concentrations of H2O2 for one hour as described above. At the end of treatment, the attached cells were scraped into the D-PBS in which they were treated (including the floating cells as a result of detachment of cell monolayers due to treatment), and then precipitated by centrifugation. The cell pellet was re-suspended in 0.3 ml of lysis buffer (50 mM potassium phosphate buffer, pH 7.8, containing 0.5% Triton X-100, 3% glycerol) containing protease inhibitor cocktail (P-8340, Sigma, St. Louis, MO), 1 mM phenylmethylsulfonyl fluoride, and 28.5 mM N-ethylmaleimide to prevent S-glutathionylation of proteins during sample manipulation. The cells were then lysed by freezing-and-thawing once, followed by sonication. The cell lysate was clarified by centrifugation and protein concentration determined by the use of a bicinchoninic acid protein assay kit (Pierce, Rockford, IL). Total cellular proteins were then separated on a non-reducing SDS-polyacrylamide gel and transferred to a piece of nitrocellulose membrane. The protein blot membrane was reacted with a monoclonal antibody against GSH (ViroGen, Watertown, MA), followed by the standard procedures of chemiluminescence to visualize the antibody-interactive proteins.
Statistical analysis
The data were first analyzed by one-way analysis of variance (ANOVA) followed by Newman-Keuls multiple comparison. For myocardial ischemia/reperfusion study, difference between two groups was established by Student’s t-test. Differences with a p value <0.05 were considered statistically significant.
Results
Generation and characterization of knockout mice deficient in Grx1
We have previously generated a line of Glrx1 knockout mice in which exon 2 and parts of introns 1 and 2 of the mouse Glrx1 gene were replaced by a neomycin resistance cassette. Unexpectedly, a species of Glrx1 mRNA smaller than the wild-type Glrx1 mRNA, presumably resulting from fusion of exons 1 and 3 of the gene, is expressed in all tissues of homozygous knockout mice examined (Ho et al. unpublished data). Therefore we repeated gene targeting of ES cells using a new construct in which both exons 1 and 2 of the mouse Glrx1 gene were deleted (Fig. 1A). Chimeric mice generated from the targeted ES cells successfully transmitted the mutated Glrx1 allele to their offspring and homozygous knockout mice were generated by interbreeding two heterozygous knockout mice (Fig. 1B).
An expression study was then performed to demonstrate that deletion of exons 1 and 2 inactivates the mouse Glrx1 gene. As shown in Fig. 2A, an approximately 50% decrease of Glrx1 mRNA was found in tissues of Glrx1+/− mice in comparison to that of Glrx1+/+ mice, and no Glrx1 mRNA could be detected in tissues of Glrx1−/− mice. To determine whether expression of the Glrx2 gene is altered in tissues of Glrx1 knockout mice, the same RNA blot membrane was re-hybridized with a mouse cDNA coding for Grx2. When standardized against the mRNA level of glyceraldehydes 3-phosphate dehydrogenase (Gapd), the levels of Glrx2 mRNA in brain, kidney, liver, and lungs of mice with three different Glrx1 genotypes were equivalent (Fig. 2A). (Please note that less RNA from brain and kidney of a Glrx1−/− mouse than that of the corresponding tissues of Glrx1+/− and Glrx1+/− mice was loaded on the gel, as revealed by the levels of Gapd mRNA.) However, the level of Glrx2 mRNA in heart of a Glrx1−/− mouse is slightly decreased relative to those of Glrx1+/+ and Glrx1+/− mice.
Fig. 2.
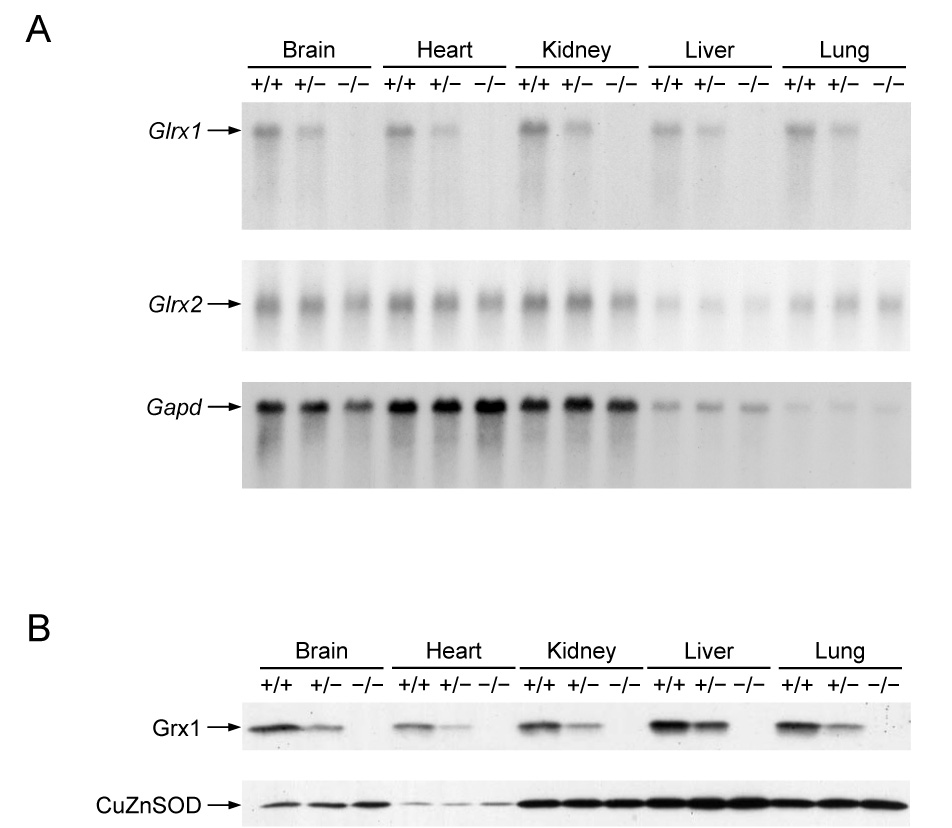
Expression analysis of Glrx1 knockout mice. (A) RNA blot analysis of Glrx1 mRNA expression in tissues of wild-type and Glrx1 knockout mice. The RNA blot membrane was first hybridized with a mouse Glrx1 cDNA, and then re-hybridized with a mouse cDNA for glutaredoxin 2 (Glrx2), and then a rat cDNA for glyceraldehydes 3-phosphate dehydrogenase (Gapd) (B) Protein blot analysis of Glrx1 in tissues of wild-type and Glrx1 knockout mice. The protein blot membrane was initially reacted with rabbit anti-human Grx1 antibodies and then rereacted with rabbit anti-human copper-zinc superoxide dismutase (CuZnSOD) antibodies. In (A) and (B), +/+, +/−, and −/− represent wild-type, heterozygous Glrx1 knockout, and homozygous Glrx1 knockout mice, respectively.
Abolishment of Grx1 expression in tissues of Glrx1−/− mice was also confirmed by protein blot analysis (Fig. 2B), where cytosolic CuZnSOD was used as the loading control because its specific content was unchanged in tissues of the knockout mice (see below). In addition, the specific Grx activity in tissue homogenates of Glrx1−/− mice was also determined. In all cases there was a lack of detectable deglutathionylase activity in homogenates of the five tissues from the homozygous knockout mice. These data also suggest that the contribution of the mitochondrial and nuclear Grx2 to total Grx activity in these mouse tissues is negligible, and that no compensatory deglutathionylase activity is developed in the knockout mice.
To determine whether a deficiency in Grx1 affects expression of other antioxidant enzymes in heart and lungs, the activities of catalase and glutathione peroxidase were determined by activity assays and the levels of copper-zinc and manganese superoxide dismutases measured by protein blot studies. No changes in the activities and protein levels of these enzymes could be found in these two organs of Grx1-deficient mice compared to those of wild-type mice (data not shown). Further attempts to determine the level of the Grx2 protein in tissues of Glrx1−/− mice were not successful, because the three antibodies against the human Grx2 protein available (two of them purchased from LabFrontier, Seoul, Korea; the other provided by Dr. Marjorie Lou of University of Nebraska at Lincoln, Lincoln, NE) either do not react or react weakly with the mouse Grx2 protein (data not shown).
The deficiency in Grx1 seems to have very limited effect on mouse development and physiology in general, since the homozygous knockout mice are healthy and fertile upon observation until one year of age.
Mice deficient in Grx1 do not display greater myocardial injury after an ischemia/reperfusion insult
ROS have been postulated to participate in the pathogenesis of ischemia/reperfusion (I/R)-induced heart injury [7]. This hypothesis is supported by the findings that knockout mice deficient in copper-zinc superoxide dismutase (CuZnSOD) or cellular glutathione peroxidase (Gpx1) are more sensitive to cardiac I/R injury, and transgenic mice overexpressing either of these enzymes exhibit a resistant phenotype [41 – 45]. Analogously, overexpression of Grx1 in H9c2 cardiomyocyte cells in vitro protects them from H2O2-induced apoptosis [29], however an in vivo effect of genetic manipulation of Grx1 has not been reported. The newly generated Glrx1 knockout mice allowed us to investigate whether a deficiency in Grx1 renders mice more susceptible to regional I/R injury in vivo. Toward this end, Glrx1+/+ and Glrx1−/− mice were subjected to 60 min of LAD ligation, followed by 4 h of reperfusion. As shown in Fig. 3, the areas at risk, expressed as the percentage of the left ventricle (LV), in hearts of Glrx1+/+ and Glrx1−/− mice were comparable (46 ± 6% vs. 43 ± 5%, respectively). The infarct sizes of hearts of Glrx1+/+ and Glrx1−/− mice, standardized against either LV or area at risk, were equivalent (p>0.05). These results indicate that a deficiency in Grx1 does not affect the infarct size in mouse hearts in an in vivo I/R model.
Fig. 3.
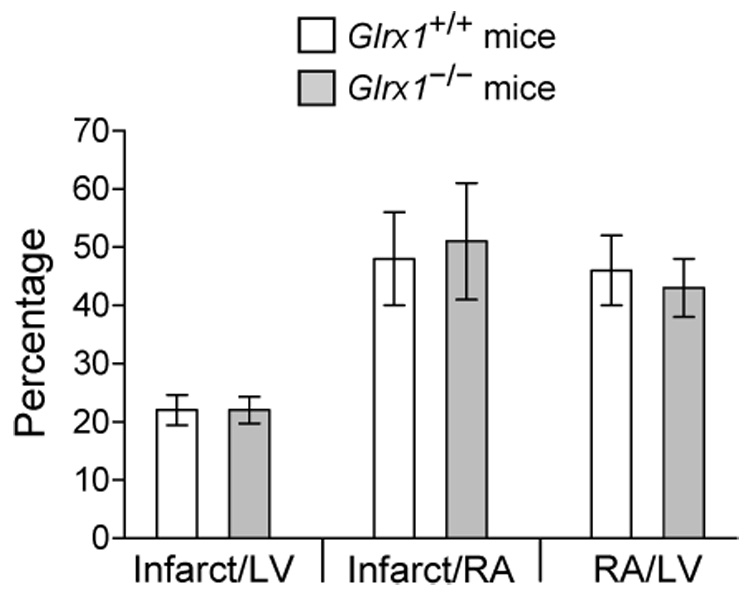
Lack of effect of Grx1 deficiency on myocardial infarction in mice. Animals were subjected to 60 min of LAD coronary artery ligation followed by 4 h of reperfusion. The infarct size was standardized against LV and area at risk (AR). Each value represent mean ± SD of 5 to 6 hearts.
Deficiency in Grx1 does not increase hyperoxia-induced lung injury in mice
Exposure of animals to hyperoxia is known to cause an increased production of ROS in the lungs, leading to destruction of alveolar epithelium, inflammation, edema, and eventually death of animals [46]. Previous studies have shown that overexpression of manganese superoxide dismutase, extracellular superoxide dismutase (ECSOD), or 1-Cys peroxiredoxin (Prdx6) in the lungs of transgenic mice greatly attenuates lung damage resulting from exposure to hyperoxia [47 – 49]. Conversely, knockout mice deficient in ECSOD or Prdx6 are particularly vulnerable to the same model of lung injury [50, 51]. These data have demonstrated the role of oxidative stress in hyperoxia-induced lung damage as well as the protective function of different antioxidant enzymes. However, the molecular and cellular mechanisms by which ROS cause lung injury are not understood. The available Glrx1 knockout allowed us to investigate whether a decreased capacity in Grx1-mediated de-glutathionylation of proteins would sensitize mice to hyperoxia-induced lung injury. As shown in Fig. 4, exposure to >99% oxygen caused lung edema in both Glrx1+/+ and Glrx1−/− mice as evident in the increased lung wet-to-dry ratios. Although the wet-to-dry ratio of Grx1-deficient lungs (7.2 ± 1.1) is higher than that wild-type lungs (6.2 ± 1.1), the difference is not statistically significant.
Fig. 4.
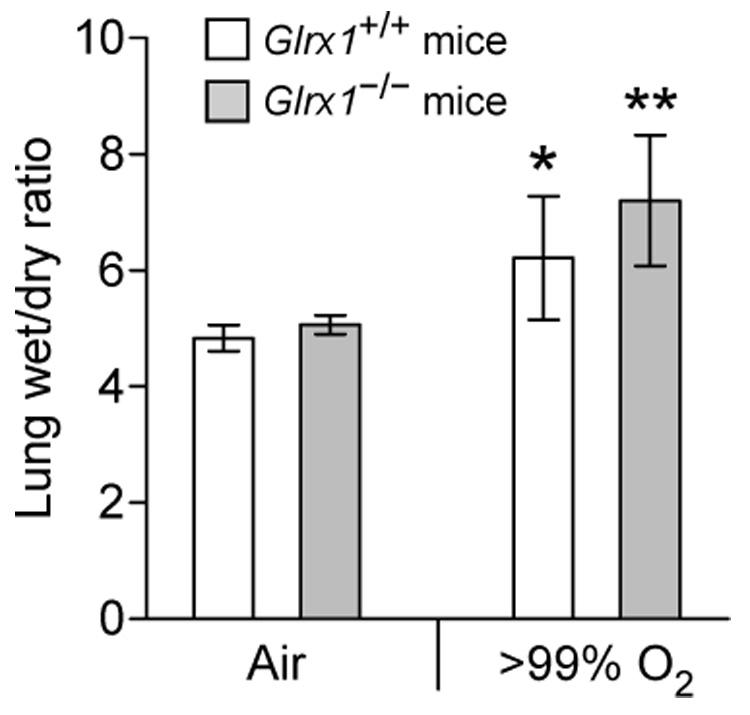
Lung edema (wet/dry ratio) in Glrx1+/+ and Glrx1−/− mice following exposure to >99% oxygen for 72 hours. *, p<0.05 vs. Glrx1+/+ mice exposed to air. **, p<0.01 vs. Glrx1−/− mice exposed to air. N ≥ 4 for each group of mice. Each value represent mean ± SD.
Grx1-deficient MEFs are more susceptible to cell damage induced by diquat and paraquat but not H2O2 and diamide
The function of Grx1 in cell defense against oxidative stress was also studied in fibroblasts prepared from embryos of Glrx1+/+ and Glrx1−/− mice. Fig. 5 shows the dose dependent decreases in viability of mouse embryonic fibroblasts (MEFs), as determined by the declines in the capabilities of cells to reduce MTT, following treatment with different oxidants. The time points of treatment were determined in separate experiments (data not shown) and were one hour for treatment with H2O2 and 24 hours for diamide, diquat, and paraquat. As show in Fig. 5A, a deficiency in Grx1 does not render MEFs more susceptible to loss of cell viability due to toxicity of H2O2. Likewise, treatment of MEFs with diamide, a thiol oxidizing agent, is not more potent in decreasing viability of Grx1-deficient MEFs compared to its effect on wild-type MEFs (Fig. 5B). On the other hand, MEFs lacking Grx1 are more vulnerable to cell damage induced by either diquat or paraquat, two chemicals that generate superoxide anion radical through the redox cycling mechanism (Fig. 5C and 5D). The increased susceptibility of Grx1-deficient MEFs to diquat and paraquat can also be concluded from the severity of morphological changes of the cells following treatment (Fig. 6).
Fig. 5.
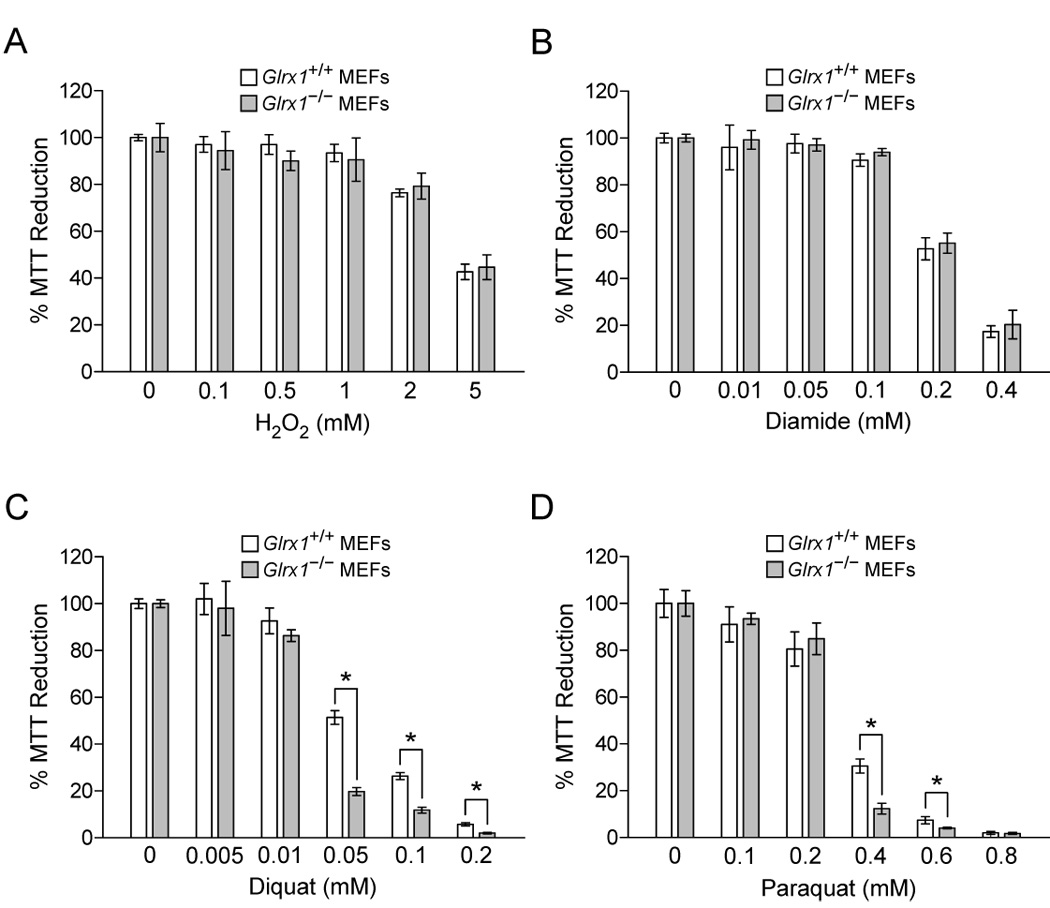
The susceptibility of MEFs to oxidant-induced cell damage. MEFs isolated from Glrx1+/+ and Glrx1−/− mice were treated with different concentrations of H2O2 in D-PBS for 1 hr (A), or with different concentrations of diamide (B), diquat (C), and paraquat (D) in culture medium for 24 hrs. Cell viability was determined by the MTT assay. Results are shown as the percent absorbance of oxidant-treated MEFs compared to that of the same MEFs cultured in D-PBS or medium without oxidants. The data from Glrx1+/+ and Glrx1−/− MEFs with the same treatment were compared. Data are representatives from at least three independent experiments. Each bar represents mean ± SD, n = 4 measurements. *, p<0.001, Glrx1+/+ MEFs vs. Glrx1−/− MEFs with the same treatment.
Fig. 6.
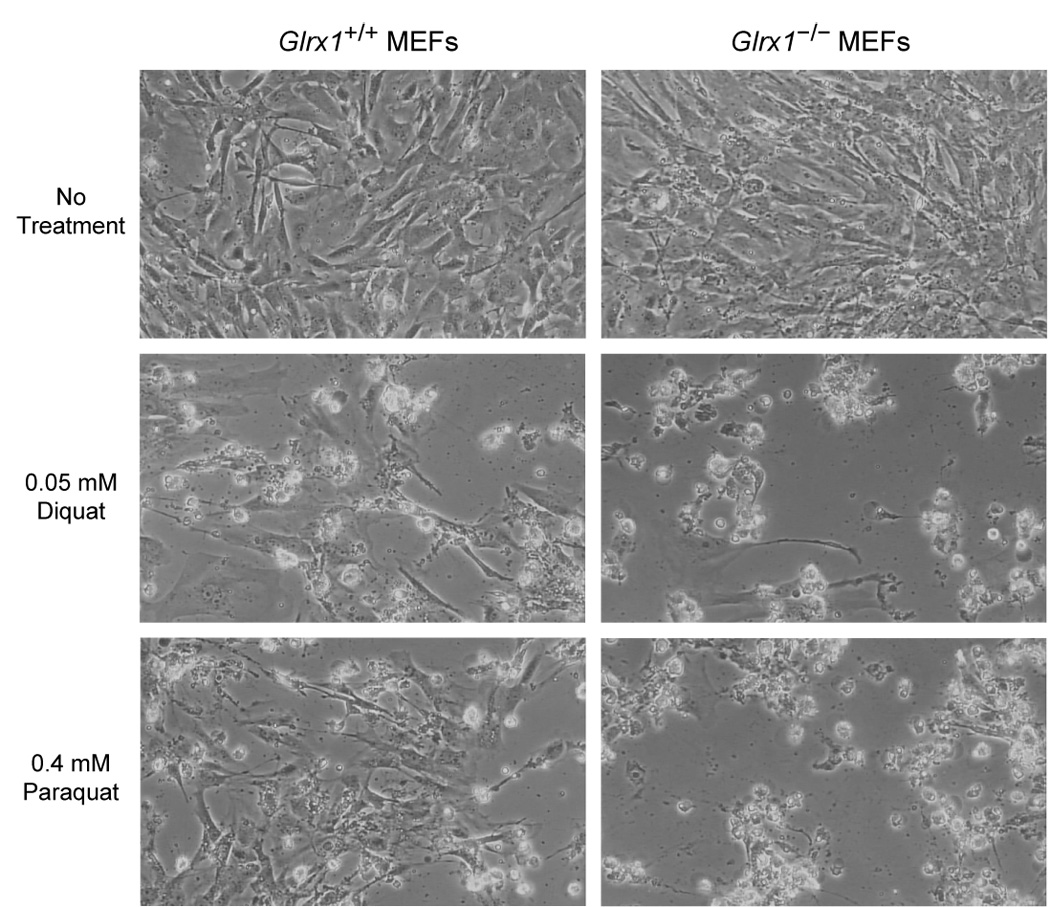
Morphology of MEFs treated with diquat and paraquat. Glrx1+/+ and Glrx1−/− MEFs were cultured in medium in the absence (as control) or presence of 0.05 mM diquat or 0.4 mM paraquat at 37 °C for 24 hrs. The genotypes of the MEFs and treatments are shown on top and left of the figure, respectively. A more extensive detachment of cell monolayer and cell death (shown as rounded cells) were found in Glrx1−/− MEFs treated with diquat or paraquat compared to Glrx1+/+ MEFs.
Grx1-deficient MEFs are more resistant to apoptosis induced by tumor necrosis factor a plus actinomycin D
The involvement of ROS in cell apoptosis has been implicated in numerous studies [52]. We therefore determined whether a deficiency in Grx1 affects the execution of the cell apoptotic pathway. As shown in Fig. 7, apoptosis induced with tumor necrosis factor a plus actinomycin D causes a 36 % decrease in viability of wild-type MEFs compared to controls, and the extent of cell death is attenuated in MEFs deficient in Grx1.
Fig. 7.
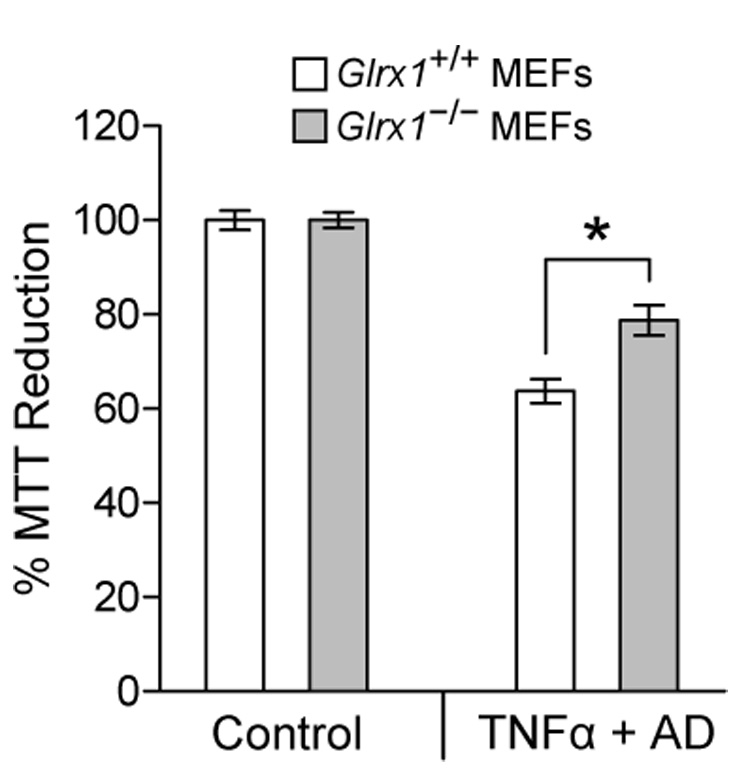
Grx1-deficient MEFs are more tolerant to apoptosis induced by tumor necrosis factor α (TNFα) plus actinomycin D. Glrx1+/+ and Glrx1−/− MEFs were cultured medium in the absence (as control) or presence of 10 ng/ml tumor necrosis factor α (TNFα) plus 50 ng/ml actinomycin D (AD) at 37 °C for 24 hrs. Cell viability was determined by the MTT assay as described in Fig. 5. Data are representatives from two independent experiments. Each bar represents mean ± SD, n = 4 measurements. *, p<0.001, Glrx1+/+ MEFs vs. Glrx1−/− MEFs.
Grx1 deficiency affects formation and reduction of protein-SSG mixed disulfides in MEFs in response to oxidative stress
Since Grx1 catalyzes de-glutathionylation of protein-SSG mixed disulfides, we sought to investigate whether a deficiency in Grx1 affects the extent of S-glutathionylated proteins in MEFs in response to treatment with H2O2. As shown in Fig. 8A, two proteins with apparent molecular masses of 44 kDa and 34 kDa become more prominently S-glutathionylated in Grx1-deficient MEFs compared with those of wild-type MEFs following treatment with H2O2 at concentrations of 0.5, 1, and 2 mM. The levels of S-glutathionylation of other higher molecular-weight proteins were markedly increased in both Glrx1+/+ and Glrx1−/− MEFs after being treated with 5 mM H2O2 for one hour, but the protein-SSG levels in the Glrx1−/− cells were clearly higher than those in the Glrx1+/+ cells. These observations indicate that cells deficient in Grx1 are more susceptible to H2O2-induced accumulation of S-glutathionylatied proteins. We then examined the time course of de-glutathionylation of the protein-SSG mixed disulfides that are formed due to H2O2 treatment. In this experiment, MEFs were treated with 5 mM of H2O2 for one hour and then cultured in regular medium in the absence of H2O2 for different periods of time before harvest for measurement of S-glutathionylated proteins. As shown in Fig. 8B, the glutathionyl moiety of most of the S-glutathionylated proteins, with the exception of a few high molecular-weight proteins, are removed in wild-type MEFs within one hour after termination of H2O2 treatment, consistent with rapid Grx1-catalyzed de-glutathionylation which occurs within minutes for most cellular proteins [22]. For Grx1-deficient MEFs also, the majority of protein-SSG mixed disulfides were de-glutathionylated at one hour, consistent with a typical non-enzymatic time course. There was a notable exception, however, namely de-glutathionylation of a 44-kDa protein was particularly retarded in the absence of Grx1 so that it remained extensively S-glutathionylated in Glrx1−/− cells even at three hours following removal of H2O2.
Fig. 8.

Formation and decay of protein-SSG mixed disulfides in Glrx1+/+ and Glrx1−/− MEFs treated with 5 mM H2O2. (A) S-glutathionylation of proteins in Glrx1+/+ and Glrx1−/− MEFs in response to different concentrations of H2O2. The cells were treated with H2O2 in D-PBS for 1 hr and total proteins were separated on a 12% SDS-gel without the use of reducing agent (β-mercaptoethanol) for blot analysis using an anti-GSH antibody. The 44-kDa and 34-kDa glutathionylated proteins are indicated by arrows on right of the figure. (B) Reversal of protein S-glutathionylation in Glrx1+/+ and Glrx1−/− MEFs. The cells were treated with 5 mM H2O2 in DPBS for 1 hr and then cultured in regular medium for different periods of time before harvesting for blot analysis as described in (A). The proteins were separated on a 10% non-reducing SDS-gel to allow better separation of the high-molecular-weight proteins. The 44-kDa glutathionylated protein is indicated by an arrow on right of the figure.
Discussion
In eukaryotes, a precise control of oxidation/reduction of protein thiols is essential for normal cell physiology. For example, formation of intra- and inter-protein disulfides is required for maturation, secretion, and function of a large number of secretory proteins, and is also critical for the function of some of the intracellular proteins. Oxidative protein folding in the endoplasmic reticulum (ER) of cells can be catalyzed by protein disulfide isomerase which is maintained in its oxidized state by Ero1 (ER oxidoreductin 1), as well as by members belonging to the family of Quiescin-sulfhydryl oxidase (QSOX) [53, 54]. In addition, Erv1p, a member of the QSOX family that is located in the mitochondrial intermembrane space, is involved in mitochondrial biogenesis and assembly of cytosolic iron-sulfur cluster-containing proteins [55, 56], suggesting the role of disulfide bond formation in these critical cellular processes. These understandings illustrate the importance of thiol oxidation in normal protein function and cell physiology. On the other hand, the cytosolic compartment of cells is highly reduced and therefore most of the cysteine residues of intracellular proteins are in the reduced state. For these proteins, thiol oxidation as a result of oxidative stress may represent an undesirable event. Fortunately, certain forms of oxidized protein thiols can be reduced by members of the thioredoxin fold family, Trx and Grx. This study set out to further our understanding of the in vivo functions of Grx1 using a line of knockout mice deficient in this enzyme. In contrast to mice deficient in Trx1 or Trx2, which die in utero [57, 58], mice lacking Grx1 develop normally and are healthy upon observation to one year of age, indicating the distinct functions of Trxs and Grx1 in mouse development.
Due to the high intracellular concentration of GSH, S-glutathionylation of proteins occurs in cells under normal culturing condition and is further enhanced in cells under oxidative stress (Fig. 7A). S-glutathionylation can inactivate the catalytic function of many proteins such as transcription factors NF-κB and NF-1 [59, 60], protein tyrosine phosphatase 1B (PTP-1B) [61, 62], protein kinase C-α [63], etc. However, the activities of a number of other proteins, such as HIV-1 protease, glutathione S-transferase, and Ras, are enhanced by S-glutathionylation [64 – 66]. In certain cases, the altered protein function due to S-glutathionylation has been shown to play a role in regulation of cell physiology. For example, S-glutathionylation-mediated inactivation of PTP-1B in cells treated with epidermal growth factor may be critical for sustaining/enhancing the receptor tyrosine kinase-mediated signaling pathway [67]. On the other hand, activation of Ras due to S-glutathionylation in vascular smooth muscle cells in response to angiotensin II results in stimulation of protein synthesis, potentially leading to cell hypertrophy [66]. Therefore, S-glutathionylation of proteins may represent a novel mechanism by which redox modulates cell signaling and physiology [3]. However, extensive S-glutathionylation of proteins in cells following oxidative stress may represent the underlying mechanism for oxidant-induced tissue injury. In this report, we tested whether a deficiency in Grx1, and therefore a decreased capacity in de-glutathionylation, would sensitize mice to injury in heart and lungs induced by ischemia/reperfusion and hyperoxia, respectively, two models in which the role of ROS in causing tissue damage has been established. Our results show that mice lacking Grx1 are not more vulnerable to these two models of oxidant-mediated tissue injury, suggesting that generalized S-glutathionylation of cytosolic proteins, which is reversible by Grx1, is not a major cause of tissue damage. Unfortunately, attempts to measure the levels of S-glutathionylated proteins in heart and lung samples of Glrx1+/+ and Glrx1−/− mice by protein blot analysis using the monoclonal anti-GSH antibody were not successful. This is because the extensive signals resulting from the interaction between the secondary anti-mouse IgG antibodies (conjugated to horseradish peroxidase) and the endogenous mouse IgG in tissue samples greatly mask the signals generated from other proteins that interact with the anti-GSH antibody.
However, these studies do not exclude the physiological role of Grx1 in other models of oxidant-mediated tissue response and injury. For example, Cys-179 of the inhibitory κB kinase β (IKK-β) subunit of IκB kinase is a target of oxidation, resulting in formation of IKK-β-SSG mixed disulfide [68]. This greatly abrogates the kinase activity of IKK-β, thereby decreasing the phosphorylation of the inhibitory κB (IκB) and the subsequent activation of the transcription factor NF-κB. The function of Grx1 in preventing oxidation-mediated inactivation of IKK-β has been demonstrated by experiments using cultured cells in which expression of Grx1 is altered by gene silencing using interference RNA. This conclusion is further supported by the observations that primary tracheal epithelial cells isolated from Glrx1−/− mice exhibit attenuated binding activity of NF-κB, nuclear translocation of the RelA subunit of NF-κB, and activation of expression of chemokines in response to treatment with lipopolysaccharide compared to the same cells isolated from Glrx1+/+ mice [68].
A second example is illustrated by the results that MEFs deficient in Grx1 are more tolerant to apoptosis induced by TNFα plus AD (Fig. 7). The molecular mechanism underlying this observation was suggested by a recent report of Pan and Beck [69]. In their studies, down-regulation of Grx1 expression by interference RNA desensitizes human umbilical vein endothelial cells (HUVECs) to cell death induced by TNF-α plus cycloheximide. This is because treatment of HUVECs with these agents enhances S-glutathionylation of procaspase 3 which inhibits its cleavage and activation during the apoptotic response. In this model of programmed cell death, Grx1 actually functions in promoting apoptosis by de-glutathionylating the procaspase 3-SSH mixed disulfide, thereby facilitating formation of the active caspase 3. These insights indicate that the consequence of protein glutathionylation and therefore the role of Grx1 in reversing this process may vary in different models of cell injury and response involving oxidative stress. Thus, this newly generated line of knockout mice should prove a valuable tool in addressing the functional role of Grx1 in a variety of disease models in which cellular redox is perturbed.
The effect of Grx1 deficiency on the response of MEFs to oxidants was also studied. Lack of Grx1 did not exacerbate the toxic effects of H2O2 or surprisingly, diamide, an agent that more selectively promotes protein-SSG formation, on the viability of cells. On the other hand, MEFs deficient in Grx1 were more susceptible to injury induced by diquat or paraquat, suggesting that different oxidants may damage the cells via distinct mechanisms, some of which may not be attenuated by Grx1-dependent de-glutathionylation. Furthermore, in the model of H2O2-induced cell injury, the degree of protein S-glutathionylation seems not to correlate with the extent of cell death, as more extensive S-glutathionylation of proteins in Grx1-deficient MEFs is not associated with an increase in cell death (Fig. 5, Fig. 6, and Fig. 8). This is also true for MEFs treated with diquat and paraquat. Both agents cause more cell death in Grx1-deficient MEFs than wild-type MEFs, yet no increases of protein S-glutathionylation can be found in either MEFs at 12 hrs and 24 hrs post-treatment (data not shown). Although some of these observations seem to contradict the hypothesis that oxidation of protein thiols, particularly formation of protein-SSG mixed disulfides, contributes to oxidant-induced cell damage, there are several mitigating considerations. Firstly, the time frame of commitment to cell death is likely much shorter than the time for progression to cell death. Therefore it is important to examine early events associated with changes in S-glutathionylation status and relative rates of de-glutathionylation. Secondly, examination of gross changes in protein-SSG status may not be reflective of specific molecular events associated with commitment to cell death. Since the sensitivity of the monoclonal antibody against GSH has not yet been characterized and the separation on an SDS-gel is relatively limited, S-glutathionylation of certain proteins, that play a critical role in cellular injury response to oxidants, may not be detected due to the limited sensitivity of the antibody or may be masked by the proteins with a high degree of S-glutathionylation (Fig. 8). Thirdly, the use of cell death as an end point may not be appropriate for correlating the extent of cell injury to the level of protein S-glutathionylation. Nevertheless, overexpression of Grx1 has been shown to prevent cells from oxidant-mediated damage in response to dopamine, H2O2, and high concentrations of glucose [26 – 31]. These results support the hypothesis that oxidant-induced cell injury is partly mediated through S-glutathionylation of proteins. However, direct measures of protein-SSG status of specific proteins in these studies were not performed, so it remains to be determined whether an increased expression of Grx1 affects the levels of protein S-glutathionylation in each case.
Finally, MEFs deficient in Grx1 are more vulnerable to S-glutathionylation of proteins induced by a high concentration of H2O2 than wild-type MEFs (Fig. 8A), indicating that Grx1 is an important cellular mechanism of de-glutathionylation, as shown in several previous studies [21, 22, 66, 68 – 71]. For example, de-glutathionylation of actin in NIH3T3 fibroblast cells, and its polymerization and migration to the cell periphery, in response to EGF-stimulation was shown to be abolished in cells in which Grx1 was knocked down by interference RNA [71]. Consistent with this observation, we speculate that the approximately 44-kDa protein that is so slowly de-glutathionylated in the Glrx1−/− cells is actin (Fig. 8B). The current and other earlier studies suggest that Grx1 may preferentially de-glutathionylate certain cellular proteins including actin, a known cellular target for oxidant-induced S-glutathionylation [70, 71]. Further studies to identify the protein targets of Grx1-mediated de-glutathionylation should advance our understanding in redox regulation of cellular function and oxidant-induced cell damage.
In summary, a line of knockout mice deficient in Grx1 was generated in this study for testing the role of this protein in oxidant-mediated cell and tissue injury. Our studies showed that a deficiency in Grx1 does not affect the susceptibility of mice to acute injury of heart and lungs induced by ischemia/reperfusion and hyperoxia, respectively. This observation may preclude a role for Grx1 in protection of the heart and lung cells. However, at present we can not exclude the possibility that alternative or compensatory regulatory mechanisms may have developed in these mice which are deprived of Grx1 at the embryonic stage. On the other hand, we observed that Grx1 deficiency selectively sensitizes the MEFs to oxidant-induced cell death and that Grx1 may exert its function on specific protein targets. These results indicate the complexity of the mechanisms by which each oxidant causes cell injury. Identification of the cellular protein targets of S-glutathionylation that are formed due to alteration of cellular redox status and following oxidative stress using this line of knockout mice should provide new insights to our understanding in cell and tissue physiology in both health and disease.
Acknowledgements
We thank Dr. Richard Mulligan of Harvard University and Dr. Andras Nagy of Mount Sinai Hospital at Toronto for the gifts of plasmid pPNT and R1 embryonic stem cells, respectively; Syntex Inc, Palo Alto, CA for the gift of ganciclovir, and Ms. Ping T. Ho for sequencing the entire mouse Glrx1 gene.
Abbreviations
- ROS
reactive oxygen species
- GSH
glutathione
- protein-SSG
protein-GSH mixed disulfides
- Grx1
glutaredoxin 1
- Trx
thioredoxin
- MEFs
mouse embryonic fibroblasts
- BAC
bacterial artificial chromosomes
- TTC
triphenyltetrazolium chloride
- D-PBS
Dulbecco’s phosphate-buffered saline
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide
- diamide
azodicarboxylic acid bis (dimethylamide)
- diquat
1,1′-ethylene-2,2′-bipyridyldiylium dibromide
- paraquat
1,1′-dimethyl-4,4′-bipyridinium dichloride
- TNFα
tumor necrosis factor α
- AD
actinomycin D
- LAD
left anterior descending coronary artery
Footnotes
The work was supported by NIH grant HL63317 and American Heart Association Greater Midwest Affiliate grant 0455876Z to Y.-S. H., by NIH grant HL87271, a grant from the Department of Veterans Affairs Merit Review and a grant-in-aid from American Heart Association Southeast Affiliate to B.H.L.C., and by NIH grants AG024413 and AG15885, and a Merit Review grant from the Department of Veterans Affairs to J. J. M. The use of equipment in the Imagine and Cytometry Facility Core and Cell Culture Facility Core was supported by a Center grant P30 ES06639.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Freeman B, Crapo JD. Biology of disease: free radicals and tissue injury. Lab. Invest. 1982;47:412–426. [PubMed] [Google Scholar]
- 2.Finkel T. Redox-dependent signal transduction. FEBS Lett. 2000;476:52–54. doi: 10.1016/s0014-5793(00)01669-0. [DOI] [PubMed] [Google Scholar]
- 3.Shelton MD, Chock PB, Mieyal JJ. Glutaredoxin: role in reversible protein S-glutathionylation and regulation of redox signal transduction and protein translocation. Antioxid. Redox Signal. 2005;7:348–366. doi: 10.1089/ars.2005.7.348. [DOI] [PubMed] [Google Scholar]
- 4.Rhee SG. Cell signaling: H2O2, a necessary evil for cell signaling. Science. 2006;312:1882–1883. doi: 10.1126/science.1130481. [DOI] [PubMed] [Google Scholar]
- 5.Forman HJ, Fisher AB. Antioxidant defense. In: Gilbert DL, editor. Oxygen and Living Processes: An Interdisciplinary Approach. New York: Springer-Verlag; 1982. pp. 235–249. [Google Scholar]
- 6.Rhee SG, Chae HZ, Kim K. Peroxiredoxins: a historical overview and speculative preview of novel mechanisms and emerging concepts in cell signaling. Free. Radic. Biol. Med. 2005;38:1543–1552. doi: 10.1016/j.freeradbiomed.2005.02.026. [DOI] [PubMed] [Google Scholar]
- 7.Cross CE, Halliwell B, Borish ET, Pryor WA, Ames BN, Saul RL, McCord JM, Harman D. Oxygen radicals and human disease. Annals Int. Med. 1987;107:526–545. doi: 10.7326/0003-4819-107-4-526. [DOI] [PubMed] [Google Scholar]
- 8.Stadtman ER. Oxidation of free amino acids and amino acid residues in proteins by radiolysis and by metal-catalyzed reactions. Annu. Rev. Biochem. 1993;62:797–821. doi: 10.1146/annurev.bi.62.070193.004053. [DOI] [PubMed] [Google Scholar]
- 9.Biteau B, Labarre J, Toledano MB. ATP-dependent reduction of cysteine-sulphinic acid by S. cerevisiae sulphiredoxin. Nature. 2003;425:980–984. doi: 10.1038/nature02075. [DOI] [PubMed] [Google Scholar]
- 10.Budanov AV, Sablina AA, Feinstein E, Koonin EV, Chumakov PM. Regeneration of peroxiredoxins by p53-regulated sestrins, homologs of bacterial AhpD. Science. 2004;304:596–600. doi: 10.1126/science.1095569. [DOI] [PubMed] [Google Scholar]
- 11.Chang TS, Jeong W, Woo HA, Lee SM, Park S, Rhee SG. Characterization of mammalian sulfiredoxin and its reactivation of hyperoxidized peroxiredoxin through reduction of cysteine sulfinic acid in the active site to cysteine. J. Biol. Chem. 2004;279:50994–51001. doi: 10.1074/jbc.M409482200. [DOI] [PubMed] [Google Scholar]
- 12.Woo HA, Jeong W, Chang TS, Park KJ, Park SJ, Yang JS, Rhee SG. Reduction of cysteine sulfinic acid by sulfiredoxin is specific to 2-cys peroxiredoxins. J. Biol. Chem. 2005;280:3125–3128. doi: 10.1074/jbc.C400496200. [DOI] [PubMed] [Google Scholar]
- 13.Holmgren A. Thioredoxin and glutaredoxin systems. J. Biol. Chem. 1989;264:13963–13966. [PubMed] [Google Scholar]
- 14.Yoshitake S, Nanri H, Fernando MR, Minakami S. Possible differences in the regenerative roles played by thioltransferase and thioredoxin for oxidatively damaged proteins. J. Biochem. 1994;116:42–46. doi: 10.1093/oxfordjournals.jbchem.a124500. [DOI] [PubMed] [Google Scholar]
- 15.Gravina SA, Mieyal JJ. Thioltransferase is a specific glutathionyl mixed disulfide oxidoreductase. Biochemistry. 1993;32:3368–3376. doi: 10.1021/bi00064a021. [DOI] [PubMed] [Google Scholar]
- 16.Wollman EE, d’Auriol L, Rimsky L, Shaw A, Jacquot J-P, Wingfield P, Graber P, Dessarps F, Robin P, Galibert F, Bertoglio J, Fradelizi D. Cloning and expression of a cDNA for human thioredoxin. J. Biol. Chem. 1998;263:15506–15512. [PubMed] [Google Scholar]
- 17.Spyrou G, Enmark E, Miranda-Vizuete A, Gustafsson J. Cloning and expression of a novel mammalian thioredoxin. J. Biol. Chem. 1997;272:2936–2941. doi: 10.1074/jbc.272.5.2936. [DOI] [PubMed] [Google Scholar]
- 18.Lillig CH, Holmgren A. Thioredoxin and related molecules – from biology to health and disease. Antioxi. Redox Signal. 2007;9:25–57. doi: 10.1089/ars.2007.9.25. [DOI] [PubMed] [Google Scholar]
- 19.Meister A, Anderson ME. glutathione. Annu. Rev. Biochem. 1983;52:711–760. doi: 10.1146/annurev.bi.52.070183.003431. [DOI] [PubMed] [Google Scholar]
- 20.Yang Y, Jao S, Nanduri S, Starke DW, Mieyal JJ, Qin J. Reactivity of the human thioltransferase (glutaredoxin) C7S, C25S, C78S, C82S mutant and NMR solution structure of its glutathionyl mixed disulfide intermediate reflect catalytic specificity. Biochemistry. 1998;37:17145–17156. doi: 10.1021/bi9806504. [DOI] [PubMed] [Google Scholar]
- 21.Jung CH, Thomas JA. S-glutathiolated hepatocyte proteins and insulin disulfides as substrates for reduction by glutaredoxin, thioredoxin, protein disulfide isomerase, and glutathione. Arch Biochem. Biophys. 1996;335:61–72. doi: 10.1006/abbi.1996.0482. [DOI] [PubMed] [Google Scholar]
- 22.Chrestensen CA, Starke DW, Mieyal JJ. Acute cadmium exposure inactivates thioltransferase (Glutaredoxin), inhibits intracellular reduction of protein-glutathionyl-mixed disulfides, and initiates apoptosis. J. Biol. Chem. 2000;275:26556–26565. doi: 10.1074/jbc.M004097200. [DOI] [PubMed] [Google Scholar]
- 23.Wells WW, Yang Y, Deits TL, Gan ZR. Thioltransferases. Adv. Enzymol. Relat. Areas Mol. Biol. 1993;66:149–201. doi: 10.1002/9780470123126.ch4. [DOI] [PubMed] [Google Scholar]
- 24.Lundberg M, Johansson C, Chandra J, Enoksson M, Jacobsson G, Ljung J, Johansson M, Holmgren A. Cloning and expression of a novel human glutaredoxin (Grx2) mitochondrial and nuclear isoforms. J. Biol. Chem. 2001;276:26269–26275. doi: 10.1074/jbc.M011605200. [DOI] [PubMed] [Google Scholar]
- 25.Gladyshev VN, Liu A, Novoselov SV, Krysan K, Sun QA, Kryukov VM, Kryukov GV, Lou MF. Identification of a new mammalian glutaredoxin (thiotransferase), grx2. J. Biol. Chem. 2001;276:30374–30380. doi: 10.1074/jbc.M100020200. [DOI] [PubMed] [Google Scholar]
- 26.Meyer EB, Wells WW. Thiotransferase increases resistance of MCF-7 cells to adriamycin. Free Radic. Biol. Med. 1999;26:770–776. doi: 10.1016/s0891-5849(98)00247-0. [DOI] [PubMed] [Google Scholar]
- 27.Daily D, Vlamis-Gardikas A, Offen D, Mittelman L, Melamed E, Holmgren A, Barzilai A. Glutaredoxin protects cerebellar granule neurons from dopamine-induced apoptosis by activating NF-κB via Ref-1. J. Biol. Chem. 2001;276:1335–1344. doi: 10.1074/jbc.M008121200. [DOI] [PubMed] [Google Scholar]
- 28.Daily D, Vlamis-Gardikas A, Offen D, Mittelman L, Melamed E, Holmgren A, Barzilai A. Glutaredoxin protects cerebellar granule neurons from dopamine-induced apoptosis by duel activation of the Ras-phosphoinositide 3-kinase and Jun N-terminal kinase pathways. J. Biol. Chem. 2001;276:21618–21626. doi: 10.1074/jbc.M101400200. [DOI] [PubMed] [Google Scholar]
- 29.Murata H, Ihara Y, Nakamura H, Yodoi J, Sumikawa K, Kondo T. Glutaredoxin exerts an antiapoptotic effect by regulating the redox state of Akt. J. Biol. Chem. 2003;278:50266–50233. doi: 10.1074/jbc.M310171200. [DOI] [PubMed] [Google Scholar]
- 30.Song JJ, Rhee JG, Suntharalingam M, Walsh SA, Spitz DR, Lee YJ. Role of glutaredoxin in metabolic oxidative stress. Glutaredoxin as a sensor of oxidative stress mediated by H2O2. J. Biol. Chem. 2002;277:46566–46577. doi: 10.1074/jbc.M206826200. [DOI] [PubMed] [Google Scholar]
- 31.Song JJ, Lee YJ. Differential role of glutaredoxin and thioredoxin in metabolic oxidative stress-induced activation of apoptosis signal-regulating kinase 1. Biochemical J. 2003;373:845–853. doi: 10.1042/BJ20030275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Park JB, Levine M. The human glutaredoxin gene: determination of its organization, transcription start point, and promoter analysis. Gene. 1997;197:189–193. doi: 10.1016/s0378-1119(97)00262-x. [DOI] [PubMed] [Google Scholar]
- 33.Tybulewicz VCJ, Crawford CE, Jackson PK, Bronson RT, Mulligan RC. Neonatal lethality and lymphopenia in mice with a homozygous disruption of the c-abl proto-oncogene. Cell. 1991;65:1153–1163. doi: 10.1016/0092-8674(91)90011-m. [DOI] [PubMed] [Google Scholar]
- 34.Nagy A, Rossant J, Nagy R, Abramow-Newerly W, Roder JC. Derivation of completely cell culture-derived mice from early-passage embryonic stem cells. Proc. Natl. Acad. Sci. USA. 1993;90:8424–8428. doi: 10.1073/pnas.90.18.8424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bradley A. Production and analysis of chimaeric mice. In: Roberston EJ, editor. Tetratocarcinomas and Embryonic Stem Cells: A Practical Approach. Oxford: IRL Press at Oxford University Press; 1987. pp. 113–151. [Google Scholar]
- 36.Chirgwin JM, Przybyla AE, MacDonald RJ, Rutter WJ. Isolation of biologically active ribonucleic acid from sources enriched in ribonuclease. Biochemistry. 1979;18:5294–5299. doi: 10.1021/bi00591a005. [DOI] [PubMed] [Google Scholar]
- 37.Thomas PS. Hybridization of denatured RNA and small DNA fragments transferred to nitrocellulose. Proc. Natl. Acad. Sci. USA. 1980;77:5201–5205. doi: 10.1073/pnas.77.9.5201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Srinivasan U, Mieyal PA, Mieyal JJ. pH profiles indicative of rate-limiting nucleophilic displacement in thioltransferase catalysis. Biochemistry. 1997;36:3199–3206. doi: 10.1021/bi962017t. [DOI] [PubMed] [Google Scholar]
- 39.Chen Z, Siu B, Ho Y-S, Vincent R, Hamdy RC, Chua BHL. Overexpression of MnSOD protects against myocardial ischemia/reperfusion injury in transgenic mice. J. Mol. Cell. Cardiol. 1998;30:2281–2289. doi: 10.1006/jmcc.1998.0789. [DOI] [PubMed] [Google Scholar]
- 40.Berridge MV, Tan AS. Characterization of the cellular reduction of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT): subcellular localization, substrate dependence, and involvement of mitochondrial electron transport in MTT reduction. Arch. Biochem. Biophys. 1993;303:474–482. doi: 10.1006/abbi.1993.1311. [DOI] [PubMed] [Google Scholar]
- 41.Yoshida T, Maulik N, Engelman RM, Ho Y-S, Das DK. Targeted disruption of the mouse Sod1 gene makes the hearts vulnerable to ischemic reperfusion injury. Circ. Res. 2000;86:264–269. doi: 10.1161/01.res.86.3.264. [DOI] [PubMed] [Google Scholar]
- 42.Yoshida T, Maulik N, Engelman RM, Ho Y-S, Magnenat J-L, Rousou JA, Flack JE, 3rd, Deatron D, Das DK. Glutathione peroxidase knockout mice are susceptible to myocardial ischemia reperfusion injury. Circulation. 1996;96 Suppl II:II-216–II-220. [PubMed] [Google Scholar]
- 43.Wang P, Chen H, Qin H, Sankarapandi S, Becher MW, Wong PC, Zweier JL. Overexpression of human copper, zinc-superoxide dismutase (SOD1) prevents postischemic injury. Proc. Natl. Acad. Sci. USA. 1998;95:4556–4560. doi: 10.1073/pnas.95.8.4556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen Z, Oberley TD, Ho Y-S, Chua CC, Siu B, Hamdy RC, Epstein CJ, Chua BHL. Overexpression of CuZnSOD in coronary vascular cells attenuates myocardial ischemia/reperfusion injury. Free Radic. Biol. Med. 2000;29:589–596. doi: 10.1016/s0891-5849(00)00363-4. [DOI] [PubMed] [Google Scholar]
- 45.Yoshida T, Watanabe M, Engleman DT, Engleman RM, Schley J, Maulik N, Ho Y-S, Oberley TD, Das DK. Transgenic mice overexpressing glutathione peroxidase are resistant to myocardial ischemia reperfusion injury. J. Mol. Cell. Cardiol. 1996;28:1759–1767. doi: 10.1006/jmcc.1996.0165. [DOI] [PubMed] [Google Scholar]
- 46.Crapo JD, Barry BE, Foscue HA, Shelburne J. Structural and biochemical changes in rat lungs occurring during exposure to lethal and adaptive doses of oxygen. Am. Rev. Respir. Dis. 1980;122:123–143. doi: 10.1164/arrd.1980.122.1.123. [DOI] [PubMed] [Google Scholar]
- 47.Wispe JR, Warner BB, Clark JC, Dey CR, Neuman J, Glasser SW, Crapo JD, Chang L-Y, Whitsett JA. Human Mn-superoxide dismutase in pulmonary epithelial cells of transgenic mice confers protection from oxygen injury. J. Biol. Chem. 1992;267:23937–23941. [PubMed] [Google Scholar]
- 48.Folz RJ, Abushamaa AM, Suliman HB. Extracellular superoxide dismutase in the airways of transgenic mice reduces inflammation and attenuates lung toxicity following hyperoxia. J. Clin. Invest. 1999;103:1055–1066. doi: 10.1172/JCI3816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang Y, Phelan Shelley A, Manevich Y, Feinstein SI, Fisher AB. Am. J. Respir. Cell Mol. Biol. 2006;34:481–486. doi: 10.1165/rcmb.2005-0333OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Carlsson LM, Jonsson J, Edlund T, Marklund SL. Mice lacking extracellular superoxide dismutase are more sensitive to hyperoxia. Proc. Natl. Acad. Sci. USA. 1995;92:6264–6268. doi: 10.1073/pnas.92.14.6264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang Y, Feinstein SI, Manevich Y, Ho Y-S, Fisher AB. Lung injury and mortality with hyperoxia are increased in peroxiredoxin 6 gene-targeted mice. Free Radic. Biol. Med. 2004;37:1736–1743. doi: 10.1016/j.freeradbiomed.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 52.Le Bras M, Clement MV, Pervaiz S, Brenner C. Reactive oxygen species and the mitochondrial signaling pathway of cell death. Histol. Histopathol. 2005;20:205–219. doi: 10.14670/HH-20.205. [DOI] [PubMed] [Google Scholar]
- 53.Tu BP, Weissman JS. Oxidative protein folding in eukaryotes: mechanisms and consequences. J. Cell Biol. 2004;164:341–346. doi: 10.1083/jcb.200311055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Thorpe C, Hoober KL, Raje S, Glynn NM, Burnside J, Turi GK, Coppock DL. Sulfhydryl oxidases: emerging catalysts of protein disulfide bond formation in eukaryotes. Arch. Biochem. Biophys. 2002;405:1–12. doi: 10.1016/s0003-9861(02)00337-5. [DOI] [PubMed] [Google Scholar]
- 55.Lisowsky T. ERV1 is involved in the cell-division cycle and the maintenance of mitochondrial genomes in Saccharomyces cerevisiae. Curr. Genet. 1994;26:15–20. doi: 10.1007/BF00326299. [DOI] [PubMed] [Google Scholar]
- 56.Lange H, Lisowsky T, Gerber J, Muhlenhoff U, Kispal G, Lill R. An essential function of the mitochondrial sulfhydryl oxidase Erv1p/ALR in the maturation of cytosolic Fe/S proteins. EMBO Rep. 2001;2:715–720. doi: 10.1093/embo-reports/kve161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Matsui M, Oshima M, Oshima H, Takaku K, Maruyama T, Yodoi J, Taketo MM. Early embryonic lethality caused by targeted disruption of the mouse thioredoxin gene. Devel. Biol. 1996;178:179–185. doi: 10.1006/dbio.1996.0208. [DOI] [PubMed] [Google Scholar]
- 58.Nonn L, Williams RR, Erickson RP, Powis G. The absence of mitochondrial thioredoxin 2 causes massive apoptosis, exencephaly, and early embryonic lethality in homozygous mice. Mol. Cell. Biol. 2003;23:916–922. doi: 10.1128/MCB.23.3.916-922.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pineda-Molina E, Klatt P, Vazquez J, Marina A, de Lacoba MG, Perez-Sala D, Lamas S. Glutathionylation of the p50 subunit of NF-kappaB: a mechanism for redox-induced inhibition of DNA binding. Biochemistry. 2001;40:14134–14142. doi: 10.1021/bi011459o. [DOI] [PubMed] [Google Scholar]
- 60.Bandyopadhyay S, Starke DW, Mieyal JJ, Gronostajski RM. Thioltransferase (glutaredoxin) reactivates the DNA-binding activity of oxidation-inactivated nuclear factor I. J. Biol. Chem. 1998;273:392–397. doi: 10.1074/jbc.273.1.392. [DOI] [PubMed] [Google Scholar]
- 61.Barrett WC, DeGnore JP, Konig S, Fales HM, Keng Y, Zhong-Yin Z, Yim MB, Chock PB. Regulation of PTP1B via glutathionylation of the active site cysteine 215. Biochemistry. 1999;38:6699–6705. doi: 10.1021/bi990240v. [DOI] [PubMed] [Google Scholar]
- 62.Barrett WC, DeGnore JP, Keng Y, Zhang Z, Yim MB, Chock PB. Roles of superoxide radical anion in signal transduction mediated by reversible regulation of protein-tyrosine phosphatase 1B. J. Biol. Chem. 1999;274:34543–34546. doi: 10.1074/jbc.274.49.34543. [DOI] [PubMed] [Google Scholar]
- 63.Ward NE, Stewart JR, Ioannides CG, O’Brian CA. Oxidant-induced S-glutathiolation inactivates protein kinase C-α (PKC-α): a potential mechanism of PKC isozyme regulation. Biochemistry. 2000;39:10319–10329. doi: 10.1021/bi000781g. [DOI] [PubMed] [Google Scholar]
- 64.Davis DA, Newcomb FM, Starke DW, Ott DE, Mieyal JJ, Yarchoan R. Thioltransferase (glutaredoxin) is detected within HIV-1 and can regulate the activity of glutathionylated HIV-1 protease in vitro. J. Biol. Chem. 1997;272:25935–25940. doi: 10.1074/jbc.272.41.25935. [DOI] [PubMed] [Google Scholar]
- 65.Dafre AL, Sies H, Akerboom T. Protein S-thiolation and regulation of microsomal glutathione transferase activity by the glutathione redox couple. Arch. Biochem. Biophys. 1996;332:288–294. doi: 10.1006/abbi.1996.0344. [DOI] [PubMed] [Google Scholar]
- 66.Adachi T, Pimentel DR, Heibeck T, Hou X, Lee YL, Jiang B, Ido Y, Cohen RA. S-glutathiolation of Ras mediates redox-sensitive signaling by angiotensin II in vascular smooth muscle cells. J. Biol. Chem. 2004;279:29857–29862. doi: 10.1074/jbc.M313320200. [DOI] [PubMed] [Google Scholar]
- 67.Lee SR, Kwon KS, Kim SR, Rhee SG. Reversible inactivation of protein-tyrosine phosphatase 1B in A431 cells stimulated with epidermal growth factor. J. Biol. Chem. 1998;273:15366–15372. doi: 10.1074/jbc.273.25.15366. [DOI] [PubMed] [Google Scholar]
- 68.Reynaert NL, van der Vliet A, Guala AS, McGovern T, Hristova M, Pantano C, Heintz NH, Heim J, Ho Y-S, Matthews DE, Wouters EFM, Janseen-Heininger YMW. Dynamic redox control of NF-κB through glutaredoxin-regulated S-glutathionylation of inhibitory κB kinase β. Proc. Natl. Acad. Sci. USA. 2006;103:10386–10391. doi: 10.1073/pnas.0603290103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pan S, Berk BC. Glutathiolation regulates tumor necrosis factor-alpha-induced caspase-3 cleavage and apoptosis: key role for glutaredoxin in the death pathway. Circ. Res. 2007;100:213–219. doi: 10.1161/01.RES.0000256089.30318.20. [DOI] [PubMed] [Google Scholar]
- 70.Wang J, Boja ES, Tan W, Tekle E, Fales HM, English S, Mieyal JJ, Chock PB. Reversible glutathionylation regulates actin polymerization in A431 cells. J. Biol. Chem. 201;276:47763–47766. doi: 10.1074/jbc.C100415200. [DOI] [PubMed] [Google Scholar]
- 71.Wang J, Tekle E, Outrahim H, Mieyal JJ, Stadtman ER, Chock PB. Stable and controllable RNA interference: Investigating the physiological function of glutathionylated actin. Proc. Natl. Acad. Sci. USA. 2003;100:5103–5106. doi: 10.1073/pnas.0931345100. [DOI] [PMC free article] [PubMed] [Google Scholar]