

Abstract
Lipopolysaccharide (LPS) stimulates immune responses by interacting with the membrane receptor CD14 to induce the generation of cytokines such as tumor necrosis factor (TNF)-α, interleukin (IL)-1, and IL-6. The mechanism by which the LPS signal is transduced from the extracellular environment to the nuclear compartment is not well defined. Recently, an increasing amount of evidence suggests that protein tyrosine kinases especially the Src-family kinases Hck, Fgr, and Lyn, play important roles in LPS signaling. To directly address the physiological function of Hck, Fgr and Lyn in LPS signaling, a genetic approach has been used to generate null mutations of all three kinases in a single mouse strain. hck −/− fgr −/− lyn −/− mice are moderately healthy and fertile; macrophages cultured from these mice express normal levels of CD14 and no other Src-family kinases were detected. Although the total protein phosphotyrosine level is greatly reduced in macrophages derived from hck −/− fgr −/− lyn −/− mice, functional analyses indicate that both elicited peritoneal (PEMs) and bone marrow–derived macrophages (BMDMs) from triple mutant mice have no major defects in LPS-induced activation. Nitrite production and cytokine secretion (IL-1, IL-6, and TNF-α) are normal or even enhanced in hck −/− fgr −/− lyn −/− macrophages after LPS stimulation. The development of tumor cell cytotoxicity is normal in triple mutant BMDMs and only partially impaired in PEMs after LPS stimulation. Furthermore, the activation of the ERK1/2 and JNK kinases, as well as the transcription factor NF-κB, are the same in normal and mutant macrophages after LPS stimulation. The current study provides direct evidence that three Src-family kinases Hck, Fgr, and Lyn are not obligatory for LPS-initiated signal transduction.
Lipopolysaccharide (LPS)1, an outer membrane component of Gram negative bacteria, is a potent activator of monocytes and macrophages. LPS triggers the abundant secretion of many cytokines from macrophages including IL-1 (1), IL-6 (2), and TNF-α (3), which together contributes to the pathophysiology of septic shock. The major cell surface receptor for LPS on macrophages is CD14, a 55-kD glycosyl-phosphatidylinositol–linked membrane protein (4). High-affinity binding of LPS to CD14 is greatly enhanced by the presence of the serum protein LPS-binding protein (5). The crucial role played by CD14 in initiating LPS responses has been demonstrated in animal models. Transgenic mice overexpressing CD14 are hypersensitive to LPS (6), while CD14-deficient mice show no responses to lowdose LPS stimulation (7). At higher concentrations of LPS, macrophages may be activated by a CD14-independent pathway since anti-CD14 mAbs do not block all biological responses (8, 9).
Comparatively less is known about intracellular signaling events after binding of LPS to CD14 (10). There is evidence to suggest that G proteins (11), phospholipase C (12), protein kinase A, and protein kinase C (13) are involved in LPS responsiveness. Strong evidence indicates that protein tyrosine kinases (PTKs) play critical roles in LPS signaling. Within minutes of LPS stimulation, numerous proteins become tyrosine phosphorylated, in particular MAP kinases p42 (ERK2), p44 (ERK1), and p38, the mammalian homologue of the yeast MAPK-like kinase HOG1 (14–17). Synchronous with tyrosine phosphorylation these MAPKs also become enzymatically activated. The c-Jun kinase (JNK) also becomes enzymatically activated within minutes after LPS treatment of macrophages (15). Direct stimulation of the ceramide-activated protein kinase by LPS has also been suggested as a major signaling mechanism (18). Pretreatment of cells with herbimycin A, a general protein tyrosine kinase inhibitor, blocks phosphorylation of the MAPKs and inhibits LPS-induced biological responses (16, 17). More specific tyrosine kinase inhibitors of the tyrphostin family also block LPS-induced TNF-α production and MAPK phosphorylation, and prevent septic shock in mice (19).
Three members of the Src-family of protein tyrosine kinases, Hck, Fgr, and Lyn are strong candidates for the primary signal transducers of LPS responses (20–25). All three of these kinases are rapidly activated after LPS treatment. A portion of intracellular Lyn directly co-associates with CD14 (21). LPS-induced association of Lyn with PI 3-kinase has also been observed (23). Moreover, expression in macrophages of a constitutively active mutant of Hck augments TNF-α production while antisense oligonucleotides to Hck inhibit LPS-induced responses (26). Chronic exposure (24– 48 h) of macrophages to LPS induces increased synthesis of Hck and Lyn, which correlates with the ability of LPS to prime macrophages for respiratory burst (22). All of these observations suggest that protein tyrosine kinases, especially Hck, Fgr, and Lyn play critical roles in LPS-initiated signaling pathways.
To directly test the role of the Src-family kinases, Hck, Fgr, and Lyn in LPS signal transduction, we used macrophages from triple knockout mice that were generated by inter-crossing hck −/−, fgr −/− , and lyn −/− animals. Since these three kinases are the primary Src-family kinases expressed in macrophages (27), cells from hck −/− fgr −/− lyn −/− triple mutant mice have little, if any, Src-family kinase activity. Resting bone marrow macrophages and inflammatory thioglycolate-elicited peritoneal macrophages were used in this study. Surprisingly, LPSinduced cytokine secretion, nitrite production, and tumoricidal activity were all essentially normal in triple knockout macrophages. Although overall tyrosine phosphorylation was dramatically reduced in these cells, phosphorylation of MAPKs occurred normally after LPS stimulation. These studies convincingly demonstrate that the major Src-family kinases present in macrophages, Hck, Fgr, and Lyn are not obligatory in LPS-mediated signal transduction.
Materials and Methods
Generation of hck−/ −fgr− /−lyn− /− Mice.
hck −/− fgr −/− double mutant mice and lyn −/− single mutant mice were generated as previously described (Chan, V., F. Meng, A. DeFranco, and C. Lowell, manuscript submitted for publication and reference 28).
hck −/− fgr −/− double mutant and lyn −/− single mutant were interbred to generate hck −/− fgr −/− lyn −/− triple deficient mice. Genotyping of offspring was performed by PCR using specific primers and further confirmed by immunoblotting using specific polyclonal antibodies against Hck, Fgr, and Lyn. Primer sequences used to specifically amplify the wild-type hck gene were: hck forward 5′ GCT CCA TAG ATC CGT CGT GCC ATT TCC 3′ and hck reverse 5′ GTT GTT TGG TCC CAG CTT GCT GGA GG 3′. To amplify the hck mutant allele, primers, hck forward and 3′ neo, 5′ GCA TCG CCT TCT ATC GCC TTC TTG ACG 3′, were used. To specifically amplify the fgr wild-type allele, primers, fgr forward, 5′ CAA GGC CGG ACT TCG TCC GTC TTT CC 3′, and fgr reverse, 5′ GAG AGC CTT ACT GGA ATC CCT CTT TAG C 3′ were used. The fgr mutant allele was detected using primers, 5′ neo, 5′ CAG TCA TAG CCG AAT AGC CTC TCC ACC 3′, and fgr reverse. The lyn wild-type allele was amplified with lyn forward-2, 5′ CAT AGC CTG AGT TAG TTC CCT AGC 3′, and lyn reverse, 5′ TCA CAT ATG AAC ATG TGT GTG TAC ATG TC 3′. These primers amplify only the active lyn wild-type gene and do not react with the pseudogene (Chan et al., manuscript submitted for publication). The mutant lyn gene was amplified with the 3′ neo and lyn 6.2. 5′ AGC CAC CAT TGT CCA GAC TTC 3′. PCR reactions were carried out in a 20-μl vol containing 1× PCR buffer (Perkin-Elmer Cetus Instrs., Norwalk, CT), 200 μM dNTPs, 10% DMSO, 1× readiload (Research Genetics, Huntsville, AL), 0.4 μM primers, 2 U Taq DNA polymerase, and ∼100 ng tail biopsy DNA. Reaction conditions were: 94°C 45′′, 60°C 45′′, 74°C 60′′ for 35 cycles. The lyn PCR was done at 94°C 45′′, 55°C 45′′, 74°C 60′′. PCR products were resolved on a 2% agarose gel (1% regular, 1% Nu-Sieve GTG agarose; FMC Corp., Rockland, ME).
Isolation and Culture of Macrophages.
PEMs and BMDMs were obtained as described by Lowell et al. (28). BMDMs were cultured in a 37°C incubator with 5% CO2 in α-MEM (GIBCO BRL, Gaithersburg, MD) containing 20% L cell conditioned medium and 10% fetal calf serum for 7 d before use. PEMs were cultured in DMEM containing 10% fetal calf serum for 2 h and nonadherent cells were washed away before use. Macrophages were stimulated with IFN-γ (20 ng/ml) and different concentrations of LPS for indicated times.
Preparation of Cell Lysates and Immunoblotting.
After stimulation with LPS, PEMs and BMDMs were washed once with cold 1× PBS, then lysed in lysis buffer (150 mM NaCl, 10 mM Tris, pH 7.0, 10 mM EDTA, 1% Triton X-100, 0.5% deoxycholate, 1 mM Na2VO4, 1 mM PMSF, and 1 μg/ml each of leupeptin/pepstatin/aprotinin). The protein concentrations were determined using the Bio-Rad protein assay (Bio-Rad Labs., Richmond, CA). Total protein extracts (30 μg) were electrophoresed on 10% SDS-PAGE and transferred to nitrocellulose membranes. For immunoblotting with mAbs, antiphosphotyrosine mAb 4G10 (UBI, Lake Placid, NY), anti-Src mAb 327 (UBI), and anti-Yes mAb (gift from M. Sudol, Mount Sinai School of Medicine, NY), the nitrocellulose membrane was blocked with 2% BSA in TBST (150 mM NaCl, 25 mM Tris, pH 7.5, 0.2% Tween-20) for 1 h at room temperature. For immunoblotting with rabbit polyclonal antibodies, anti-Lck, anti-Fyn (both from Santa Cruz, Biotechnology, Santa Cruz, CA), anti-Hck, anti-Fgr, anti-Lyn, and antiBlk (produced against the unique domain of each protein; references 28, 29), the nitrocellulose membrane was blocked with 5% milk/10% goat serum in TBST for 1 h at room temperature. Membranes were incubated sequentially with primary antibodies followed by peroxidase-conjugated secondary antibodies for 1 h at room temperature with continuous shaking. Specific binding was visualized by the enhanced chemiluminescent detection system (ECL; Amersham Corp., Arlington Heights, IL).
Nitrite Assay.
PEMs and BMDMs were seeded on 96-well plates at 1 × 105 cells per well and cultured in 120 μl of appropriate media in the presence or absence of LPS and IFN-γ for 12 h. NO2 − released into supernatants was measured with Greiss reagent as described (30).
Quantitation of Cytokines and Herbimycin A Treatment.
The levels of IL-1 and TNF-α in the supernatants were determined by ELISA kits (IL-1; R&D Systems, Minneapolis, MN and TNF-α; Genzyme Corp., Cambridge, MA) using protocols described by the manufacturers. IL-6 was measured by ELISA using specific antibody pairs (PharMingen, San Diego, CA). In brief, 96-well plates were coated with 4 μg/ml purified rat anti–mouse IL-6 overnight at 4°C and blocked with PBS/10% fetal calf serum for 1 h at room temperature. 50 μl of cell culture supernatants, which were harvested from cells stimulated by LPS and IFN-γ for 12 h, were added to each well and incubated at room temperature for 2 h. After washing with PBS/0.2% Tween-20, biotin-conjugated rat anti–mouse IL-6 was added and incubated for 45 min. Wells were washed and then incubated with avidin-peroxidase for 30 min. After washing, ABTS substrate (Zymed Labs., South San Francisco, CA) diluted in 0.1 M citric acid buffer (pH 4.0) was added to each well and incubated for 30 min. The optical density at 405 nm was measured with a microtiter plate reader (Molecular Devices Corp., Menlo Park, CA). For herbimycin A treatment, adherent PEMs were treated with 10 μg/ml herbimycin A (Sigma Chem. Co., St. Louis, MO) for 4 h before stimulating with LPS and IFN-γ.
Cytotoxicity Assay.
Different numbers of PEMs and BMDMs were seeded on 96-flat well plates and stimulated with LPS (100 ng/ml) and IFN-γ (20 ng/ml) for 48 h. 104 P815 tumor cells prelabeled by incubation with 10 μCi of [3H]thymidine for 12 h were overlaid on the activated macrophages. After co-culture of tumor cells and stimulated macrophages for 24 h, 100 μl supernatant was used to determine [3H]thymidine release by liquid scintillation counting. The specific release was calculated based on the formula described by Jadus et al. (31).
ERK and JNK Kinase Assay.
PEMs and BMDMs that were prestimulated with LPS (10 ng/ml) and IFN-γ (20 ng/ml) were lysed in lysis buffer. Total protein lysates prepared from LPS/ IFN-γ–stimulated PEMs or BMDMs were immunoprecipitated (250 μg/assay) with ERK1/2 (antibody cross-reacts with both ERK isoforms) and JNK specific polyclonal antibodies, respectively (Santa Cruz Biotechnology), for 1 h at 4°C and incubated with 2% BSA preblocked protein A–Sepharose beads for another hour at 4°C with continuous shaking. After washing twice in lysis buffer and once in kinase buffer (20 mM Hepes, pH 7.0, 10 mM MgCl2, 0.5% Triton X-100, 10 mM MnCl2, and 0.1 mM Na2VO4), 10 μCi γ-[33P]ATP and 25 μg of the exogenous substrates myelin basic protein (Sigma) and c-Jun fusion protein (gift of J. Hambleton, University of California, San Francisco, CA) were added to the immunocomplexes of ERK1/2 and JNK, respectively, and incubated for 15 min at 30°C. The protein samples were boiled in 2× Laemmli buffer for 5 min and separated by 12% SDS-PAGE followed by autoradiography.
NF-κB Assay.
PEMs and BMDMs (5 × 106) were seeded onto 100-mm plates and stimulated with IFN-γ (20 ng/ml) and LPS (10 ng/ml) for 2 h. Preparation of nuclear extracts was performed as described by Kitchens et al. (32). Sense and antisense oligonucleotides, containing the NF-κB recognition sequences derived from the murine immunoglobulin κ enhancer, were synthesized, annealed and labeled with α-[32P]dCTP using Klenow fragment as described by Hambleton et al. (15). To assess for nonspecific binding, a double-stranded oligonucleotide containing a point mutation in the NF-κB–binding site that was incapable of binding NF-κB, was also prepared. Labeled oligonucleotides were incubated with nuclear extracts for 15 min, then NF-κB–bound oligonucleotides were resolved on a 4% TBE gel.
Results
Generation of hck− /−fgr− /−lyn− /− Mice.
To generate the hck −/− fgr −/− lyn −/−–deficient mice, hck −/− fgr −/− double mutant mice and lyn −/− single mutant mice were crossed. Genotyping of offspring was performed by PCR as shown in Fig. 1 A. Mice from this cross were interbred for several generations until triple mutant animals were obtained. The hck −/− fgr −/− lyn −/− mice are fertile and have no general physiologic phenotypes beyond that seen for the lyn −/− single mutant mice (Chan et al., manuscript submitted for publication and references 33, 34, 35). All single mutant animals used to generate the triple knockout line had been back-crossed at least two generations to C57BL/6 mice. To verify each gene disruption in the triple mutants, proteins corresponding to Hck, Fgr, and Lyn were examined using specific polyclonal antibodies. Expression of Hck, Fgr, and Lyn was only detected in wild-type macrophages but not in hck −/− fgr −/− lyn −/− homozygotes (Fig. 1 B). FACS® analysis of resident peritoneal macrophages, PEMs and BMDMs from hck −/− fgr −/− lyn −/− animals showed normal expression levels of CD14, Mac-1, Mac-2, F4/80, MHC class I, and CD29 (β1 integrin) surface molecules (data not shown).
Figure 1.


Genotype of offspring from heterozygous parents. (A) Genomic DNA was isolated from tail biopsies and analyzed by PCR. PCR primers were designed such that amplification of a larger fragment occurred from the mutant allele of all three genes while the smaller fragment indicates the wild-type allele. PCR was performed as described in Materials and Methods. DNA from wild-type (+/+), triple heterozygous (+/−), and triple homozygous (−/−) mice is shown. (B) hck −/− fgr −/− lyn −/− mice do not express Hck, Fgr, and Lyn proteins. Total cellular lysates were prepared from BMDMs of wild-type (+/+) or triple mutant (−/−) animals and examined for Hck, Fgr, and Lyn by immunoblotting as described in Materials and Methods. Hck, Fgr, and Lyn were also not detected in PEMs from triple mutant mice (data not shown).
Other Src-family Kinases Are Not Upregulated in Macrophages Derived from hck− /−fgr− /−lyn− /−Mice.
To address the possibility that other Src-family kinases might be upregulated or aberrantly expressed in macrophages derived from hck −/− fgr −/− lyn −/− mice, we examined the expression level of all other Src-family members in these cells by immunoblotting. The expression level of other Src-family kinases was examined using a panel of mAbs or polyclonal Abs specific for each family member. As shown in Fig. 2, no other Src-family kinases were detectable in hck −/− fgr −/− lyn −/− macrophages although each protein was easily seen in extracts prepared from brain tissue or lymphocytes. We conclude that hck −/− fgr −/− lyn −/− macrophages have no significant expression of any Src-family kinases.
Figure 2.

Expression of Src-family kinases in hck −/− fgr −/− lyn −/− macrophages. Total protein extracts (30 μg) obtained from BMDMs of wildtype (W) or triple mutant (T) mice were electrophoresed on 10% SDSPAGE and transferred to nitrocellulose as described in Materials and Methods. Membranes were blotted with different antibodies against Srcfamily kinases. Brain (B) and lymphocyte (L) extracts used as positive controls for antibody binding. Similar to these results, expression of other Src-family kinases was not observed in triple mutant PEMs (data not shown).
Production of Nitrite and Cytokines in hck− /−fgr− /−lyn− /− Mice upon Stimulation with LPS and IFN-γ.
Since there is extreme heterogeneity of macrophages (36) and their responses to LPS, we performed all our experiments on both inflammatory PEMs and BMDMs. To investigate the importance of Hck, Fgr, and Lyn in LPS-induced macrophage activation, we evaluated the LPS concentration-dependent induction of nitrite, IL-1, IL-6, and TNF-α in both wildtype and mutant cells. Over a wide range of LPS concentrations, there was no impairment of nitrite production in hck −/− fgr −/− lyn −/− macrophages compared to wild-type cells (Fig. 3). BMDMs from wild-type and triple mutant mice produced equivalent amounts of nitrite in response to LPS stimulation alone; PEMs did not respond to LPS-alone stimulation. To further prime macrophages for LPS responses, we stimulated cells with the combination of LPS and IFN-γ. With this costimulation, BMDMs produced more nitrite and manifested a left-shift in the LPS dose response curve; again, both wild-type and mutant macrophages responded equivalently. LPS/IFN-γ costimulation of PEMs induced abundant nitrite production in wild-type macrophages while triple mutant PEMs appeared hyperresponsive, especially at low doses of LPS (0.001–0.01 ng/ml), compared to wild-type mice. In the absence of LPS, IFN-γ alone failed to induce nitrite release from both PEMs and BMDMs.
Figure 3.

Nitrite secretion in response to LPS or LPS + IFN-γ stimulation. PEMs or BMDMs from wild-type or triple mutant mice were cultured in 96-well plates (105 cells/well) in the presence of LPS, at the indicated concentrations, with or without addition of IFN-γ (20 ng/ml). NO2 − release was determined with the Greiss reagent after 24 h of culture. Triplicate samples at each LPS concentration were averaged and plotted ± SD. Results shown are averaged data from three independent experiments. WT (triangles); WT + IFN-γ (squares); hck −/− fgr −/− lyn −/− (circles); hck −/− fgr −/− lyn −/−+ IFN-γ (diamonds).
Similarly, both types of triple mutant macrophages produced abundant amounts of IL-1, IL-6, and TNF-α at 12 h after LPS stimulation (Fig. 4). As seen with nitrite production, cytokine release from PEMs only occurred with LPS/ IFN-γ costimulation, while BMDMs showed a left-shift of the LPS dose-response curve with the addition of IFN-γ (data not shown). hck −/− fgr −/− lyn −/− PEMs demonstrated dramatically increased production of IL-1 compared to wildtype PEMs; triple mutant BMDMs also showed slightly increased IL-1 production compared to wild-type cells. The wild-type PEMs and BMDMs produced nearly the same amount of IL-1 after LPS/IFN-γ stimulation; note the change in scale between PEMs and BMDMs. Triple mutant PEMs also showed increased IL-6 production compared to wild-type cells; responses of BMDMs were equivalent. There were no differences in TNF-α production between triple mutant and wild-type PEMs or BMDMs. These data strongly support the conclusion that macrophages lacking Hck, Fgr, and Lyn can be readily activated by LPS. Indeed, PEMs from the triple mutant mice showed enhanced responses to LPS/IFN-γ stimulation compared to wild-type cells.
Figure 4.
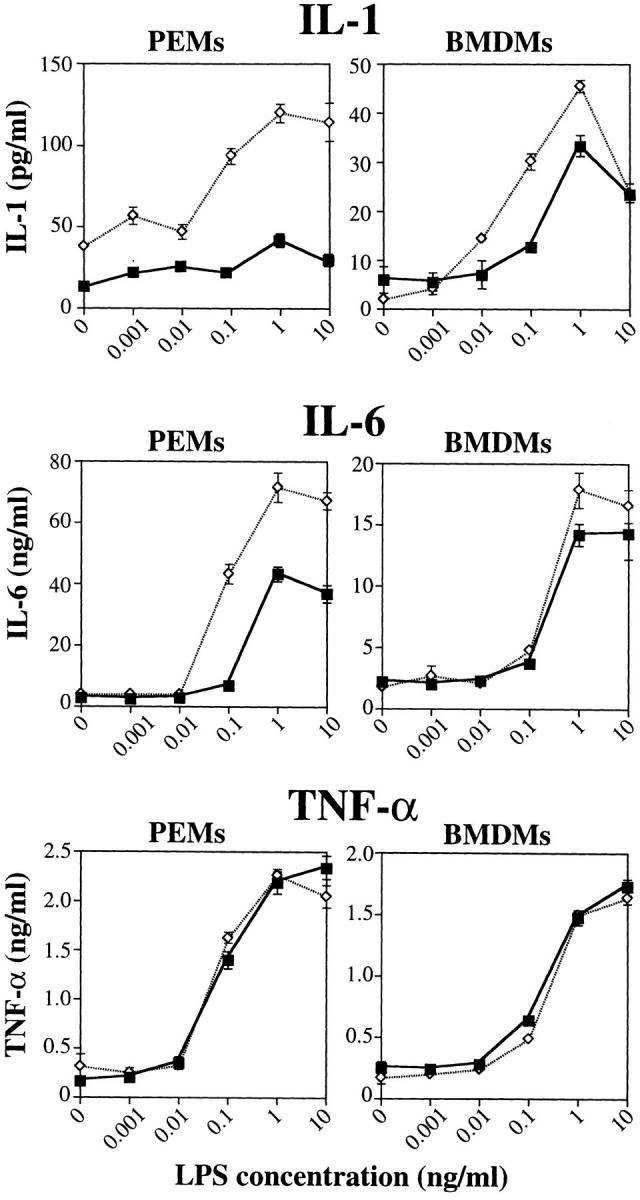
Secretion of IL-1, IL-6, and TNF-α in response to LPS and IFN-γ stimulation. PEMs or BMDMs from either wild-type or triple mutant mice were cultured in 96-well dishes (5 × 105 cells/well) and stimulated with LPS and IFN-γ (20 ng/ml) for 12 h. The concentration of IL-1, IL-6, and TNF-α in the culture media was determined by ELISA. Determinations were performed on triplicate samples and plotted ± SD. Results shown are averaged data from three independent experiments. WT + IFN-γ (squares); hck −/− fgr −/− lyn −/−+ IFN-γ (diamonds).
Partially Impaired Tumor Cytotoxicity in PEMs Derived from hck− /−fgr− /−lyn− /−–deficient Mice.
Another major functional response of LPS-stimulated macrophages is the development of tumoricidal activity. To test the tumoricidal ability of macrophages derived from the hck−/−fgr−/−lyn−/− mice, we assessed cytotoxicity against P815 mastocytoma cells after stimulation with LPS and IFN-γ. As demonstrated in Fig. 5, the stimulated macrophages killed tumor cells in a fashion proportional to the macrophage/tumor cell ratio; greater killing was seen at higher macrophage cell numbers in both wild-type and triple mutant mice. The extent of killing by PEMs derived from hck−/−fgr−/−lyn−/− mice was reproducibly less than wild-type PEMs at higher macrophage/ tumor cell ratios. This was not observed in BMDMs. This evidence demonstrates that tumoricidal activity could be induced in hck−/−fgr−/−lyn−/− macrophages by stimulation with LPS and IFN-γ.
Figure 5.

Analysis of tumor cytotoxicity. Different numbers of PEMs and BMDMs derived from wild-type and hck −/− fgr −/− lyn −/− mutant mice were plated on 96-well plates and activated with LPS (10 ng/ml) and IFN-γ (20 ng/ml) for 48 h and then overlaid with 104 [3H]thymidinelabeled P815 tumor cells. Cytotoxic activity against p815 cells was assessed by [3H]thymidine released into the culture supernatant after 24 h of coculture. Data is presented as % specific release ± SD of triplicate samples (100% was defined as the amount of [3H]thymidine in cells lysed with detergents). Specific release in the absence of LPS/IFN-γ stimulation is shown as NS. Experiment is representative of three independent experiments. Wild type (open bars); hck −/− fgr −/− lyn −/− (shaded bars).
hck− /−fgr− /−lyn− /− Macrophages Have Reduced Overall Tyrosine Phosphotyrosine Levels but Normal MAPK Phosphophorylation after LPS Stimulation.
To test the effects of loss of Hck, Fgr, and Lyn on the overall level of protein tyrosine phosphorylation, we assessed changes in tyrosine phosphorylation after LPS/IFN-γ stimulation, in both PEMs and BMDMs, using antiphosphotyrosine immunoblotting. As shown in Fig. 6, we observed that hck −/− fgr −/− lyn −/− macrophages showed dramatically decreased tyrosine phosphorylation of multiple proteins in both resting and LPS-stimulated cells. One protein with an apparent molecular mass of ∼42 kD was rapidly tyrosine phosphorylated upon LPS/ IFN-γ stimulation in both wild-type and triple mutant PEMs and BMDMs. This protein was one or both of the p42/p44 (ERK2/ ERK1) MAP kinase isoforms (confirmed by re-probing this blot with anti-ERK1/2, data not shown), as has been demonstrated previously (16, 17). Interestingly, hck −/− fgr −/− lyn −/− PEMs show slightly higher and more sustained tyrosine phosphorylation of the MAPKs compared to wild-type PEMs. In BMDMs, there was no difference in p42 phosphorylation between wild-type and triple mutant macrophages. p42 phosphorylation in PEMs was detectable after 15 min after LPS stimulation, reached a maximum by 60 min, and declined at 120 min.
Figure 6.

Protein-tyrosine phosphorylation after LPS/IFN-γ stimulation. PEMs and BMDMs from wild-type and hck −/− fgr −/− lyn −/− mice were stimulated with LPS (10 ng/ml) and IFN-γ (20 ng/ml) for the different time points indicated. Total cell lysates were prepared and separated on 10% SDS-PAGE and transferred to nitrocellulose as described in Materials and Methods. Filters were blotted with antiphosphotyrosine with 4G10, followed by peroxidase-conjugated secondary antibody and chemiluminescent detection.
Analysis of ERK1/2 and JNK Activity in LPS-Treated Macrophages.
To evaluate the role of Hck, Fgr, and Lyn in LPS-induced activation of MAPKs, we determined ERK1/2 and JNK kinase activity using defined substrates. ERK1/2 kinase activity was determined by phosphorylation of MBP. In both PEMs and BMDMs, ERK1/2 kinase activity was activated after LPS/IFN-γ treatment in hck −/− fgr −/− lyn −/− cells compared to wild-type cells (Fig. 7). No consistent difference in ERK1/2 activation was observed between wild-type and mutant cells. The activation of ERK1/2 was detected at 60 min and declined at 2 h after LPS and IFN-γ stimulation.
Figure 7.

Analysis of ERK1/2 and JNK activation after LPS/ IFN-γ stimulation. PEMs and BMDMs from wild-type and hck −/− fgr −/− lyn −/− mutant mice were stimulated with LPS (10 ng/ml) and IFN-γ (20 ng/ml) for the indicated time periods, then total cell lysates prepared. ERK1/2 and JNK proteins were immunoprecipitated (250 μg of cell lysates) using ERK1/2 and JNK polyclonal antibodies, respectively, and tested for kinase activity in the presence of γ-[33P]ATP using MBP and C-Jun fusion protein, respectively, as substrates. Phosphorylated proteins were resolved on 12% SDS-PAGE.
Activation of JNK kinase activity was carried out using a c-Jun/GST fusion protein as a exogenous substrate. JNK activity was detectable after 1 h stimulation with LPS and IFN-γ in both wild-type and mutant PEMs and BMDMs (Fig. 7). There was no consistent difference in JNK kinase activation between LPS-treated wild-type and hck −/− fgr −/− lyn −/− macrophages. In separate experiments, both JNK and ERK1/2 activation was observed within 15 min of LPS stimulation in wild-type and mutant cells (data not shown).
Effects of LPS on Activation of NF-κB DNA Binding Activity in hck− /−fgr− /−lyn− /− Macrophages.
Another major signaling pathway that is initiated in macrophages after LPS treatment is activation of NF-κB DNA binding activity by dissociation from Iκ-B (37). To assess whether the loss of Hck, Fgr, and Lyn affected NF-κB activation after LPS stimulation, we performed electrophoretic mobility shift assays (EMSA) using nuclear extracts from untreated and LPS-treated cells incubated with a double-stranded oligonucleotide containing an NF-κB recognition sequence (38). After LPS/IFN-γ stimulation for 2 h, we detected inducible NF-κB DNA binding activity in PEMs and BMDMs derived from both wild-type and hck −/− fgr −/− lyn −/− macrophages (Fig. 8). No binding was observed using an oligonucleotide containing point mutations in the NF-κB recognition sequence. The results demonstrate that the deficiency of Hck, Fgr, and Lyn did not effect NF-κB activation by LPS.
Figure 8.
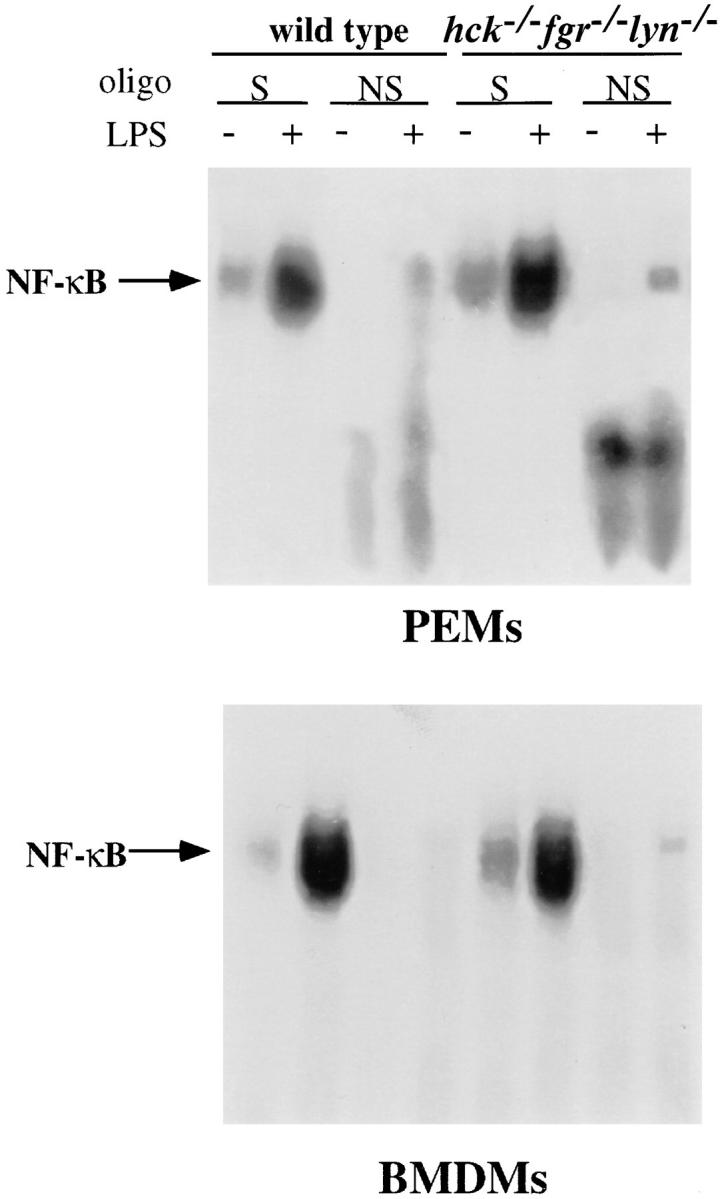
Activation of NF-κB binding activity by LPS/IFN-γ. PEM and BMDM (5 × 106 cells/assay) from wild-type and hck −/− fgr −/− lyn −/− mice were cultured with or without LPS (10 ng/ml) and IFN-γ (20 ng/ml) for 2 h. Nuclear extracts were prepared and incubated with a α-[32P]dCTP– labeled double-stranded oligonucleotide, corresponding to the murine immunoglobulin κ enhancer site. Protein-bound oligonucleotides were resolved by electrophoresis. Specific binding by NF-κB was confirmed by using an oligonucleotide containing mutations in the NF-κB binding motif. S, specific binding to wild-type oligonucleotide; NS, non-specific binding to oligonucleotide containing point mutations.
Herbimycin A Inhibits MAPK Phosphorylation and Nitrite Production in hck− /−fgr− /−lyn− /− Macrophages.
To investigate whether macrophage activation by LPS remained dependent on tyrosine kinase activity in hck −/− fgr −/− lyn −/− macrophages, we examined the effects of the protein tyrosine kinase inhibitor herbimycin A on LPS-induced tyrosine phosphorylation and secretion of nitrite. As shown in Fig. 9 A, pretreatment with herbimycin A significantly reduced LPSinduced tyrosine phosphorylation of the p42 (ERK1/2) protein in both wild-type and mutant cells. Similarly, nitrite production (Fig. 9 B) was reduced to the same extent in both wild-type and hck −/− fgr −/− lyn −/− BMDMs. This evidence demonstrates despite the lack of Hck, Fgr, and Lyn, the triple mutant macrophages remain sensitive to inhibition of LPS-induced activation by the protein tyrosine kinase inhibitor herbimycin A.
Figure 9.


Effects of herbimycin A on LPS inducible tyrosine phosphorylation and nitrite secretion. (A) BMDMs from wild-type or hck −/− fgr −/− lyn −/− mice were cultured in the presence of herbimycin A (10 μg/ml) for 4 h, then stimulated with LPS (10 ng/ml) and IFN-γ (20 ng/ml) for 30 min. Total cell lysates were prepared and electrophoresed on 10% SDS-PAGE, transferred to nitrocellulose and blotted with antiphosphotyrosine mAb 4G10. (B) BMDMs were pretreated with herbimycin A then stimulated for 12 h with LPS/IFN-γ. Nitrite release was measured using the Greiss reagent.
Discussion
Exposure of macrophages to bacterial LPS initiates a signal transduction cascade that leads to increased production of nitrite, secretion of proinflammatory cytokines, and acquisition of enhanced bactericidal/tumoricidal activity, the hallmarks of an activated macrophage. Therefore, regulation of this signaling pathway is critical for controlling the initial phases of the immune response to foreign organisms. A number of signaling pathways have been implicated in LPS responses in macrophages; however the most rapidly induced signaling responses are likely to involve tyrosine phosphorylation events. The predominant Src-family kinases expressed in macrophages, Hck, Fgr, and Lyn, have been implicated in a number of studies as being the initial signal transducers for tyrosine phosphorylation events after LPS stimulation (20–25). To directly test the role of these kinases in LPS signaling in macrophages, we generated hck −/− fgr −/− lyn −/− triple mutant mice by intercrossing single mutant animals, then tested LPS-induced functional responses and signaling properties in peritoneal exudate and BMDM from these mice. Much to our surprise, both triple mutant macrophage types showed no significant impairments in nitrite secretion, cytokine production, tumoricidal activation, or signal transduction after treatment with LPS. These data conclusively demonstrate that these Src-family kinases are not required for LPS-induced signal transduction or macrophage activation.
The primary surface receptor for LPS is CD14, which binds LPS with high affinity in the presence of LPS-binding protein. At low doses of LPS (<1 ng/ml) treatment of macrophages with anti-CD14 mAb completely blocks all LPS-induced signaling and macrophage functional activation (8, 16). At higher doses (>10 ng/ml) of LPS, anti-CD14 mAbs do not completely block LPS signaling or functional activation, indicating that CD14-independent mechanisms of LPS stimulation can occur. Moreover, activation of macrophages derived from CD14-deficient mice has also been observed at high LPS doses (7). The experiments done in this work were all conducted in the low dose range of LPS, hence the majority of signaling we examined was CD14 dependent. Since CD14 is a GPI-linked protein, and hence has no intracellular protein domains, it has been postulated that it serves only as a binding protein and that the LPS– CD14 complex then associates with another transmembrane protein that serves as the primary signal transduction unit (39). In analogy to many cytokine receptors, this heterodimeric LPS receptor complex would then activate the appropriate membrane associated tyrosine kinases to initiate the signaling cascade. Clearly, this process occurs normally in the absence of Hck, Fgr, and Lyn. Since triple mutant macrophages expressed normal levels of CD14, high-affinity binding of LPS is presumably not altered in the hck −/− fgr −/− lyn −/− cells. However, it is possible that the putative signal transducing subunit may have specificity for a wide range of kinases, or that CD14 can associate with a variety of signal transducing molecules, so that other non–Src-family kinases may be recruited to the membrane. Another possible explanation for the normal LPS responses in hck −/− fgr −/− lyn −/− mutants would be compensation by other Srcfamily kinases that become aberrantly expressed in the triple mutant cells; however, immunoblot analysis revealed that no other Src-family kinases were expressed in the triple mutant cells (Fig. 2). Src has been reported to be expressed in human monocytes (40), however we did not detect it in our immunoblots. Moreover, LPS-induced nitrite secretion and cytokine production occur normally in src −/− macrophages (Lowell, C.A., unpublished observations). Nevertheless, we can not rule out that very low expression of known Src-family kinases, or the presence of other undescribed Src-family kinases, may be contributing to LPS- induced responses in our hck −/− fgr−/−lyn−/− macrophages. One such kinase may be the Yrk kinase; a Src-family kinase cloned from avian cells for which a mammalian homologue has not yet been described (41).
Based on previous studies, macrophage activation by LPS is closely related to activation of Src-family kinases in macrophages, in particular Hck and Lyn, and correlates with the association of Lyn with CD14 and PI3-kinase (21, 23). However, as shown in Figs. 3 and 4, a deficiency of these kinases does not impair nitrite secretion or cytokine release over a wide range of LPS concentrations. Indeed, with thioglycolate-elicited PEM, we observed just the opposite, the triple mutant macrophages tended to be hyperresponsive to LPS activation. The hyperresponsiveness of hck −/− fgr −/− lyn −/− PEMs was correlated with the observations that LPS-induced tyrosine phosphorylation of the p42/p44 ERK proteins was slightly higher and more prolonged in triple mutant cells compared to wild-type (Fig. 6), although we were unable to demonstrate increased enzymatic activation of these kinases. Analysis of hck −/− fgr −/− double mutants and lyn −/− single mutant mice has shown that the hyperresponsiveness of PEMs is the result of the loss of Lyn activity (data not shown and reference 28). Thus, it appears that rather than serving as a positive regulator of LPS signaling, it is more likely that the Lyn kinase can serve as a negative regulator of macrophage activation. A negative regulatory function of Lyn is also observed in lyn −/− B-cells (Chan et al., manuscript submitted for publication and reference 34). Cross-linking of the B cell antigen receptor on lyn −/− cells produces a hyperproliferative response that is correlated with enhanced activation of ERK1/ ERK2. The hyperresponsiveness of immune cells from lyn −/− mice may contribute to the development of autoimmunity in these animals (33, 35). lyn −/− B-cells also failed to show normal downregulation of proliferation after co–cross-linking of the antigen receptor with the FcγRIIB receptor (Chan et al., manuscript submitted for publication and 34). The FcγRIIB receptor is known to downregulate antigen receptor signaling by recruiting the tyrosine phosphatase, SHP-1, into the receptor complex (42), suggesting that Lyn may be required for activation of this tyrosine phosphatase. Alternatively, Lyn may be required for activation of the p145SHIP inositol phosphatase, which has also been implicated in downregulation B cell and mast cell activation through the FcγRIIB receptor (43). Whether Lyn is acting to regulate these tyrosine phosphatases and whether they, in turn, function to downregulate LPS-induced macrophage activation remains to be determined. Of the cytokines we tested, it appeared that hck −/− fgr −/− lyn −/− macrophages showed the greatest overproduction of IL-1. Indeed, we found that PEMs from the triple mutant mice spontaneously secreted IL-1 in the presence of IFN-γ alone and when cultured without any stimuli at all (Fig. 4 and data not shown). Since IL-1 production has been shown to induce expression of IL-6 by inflammatory macrophages elicited with turpentine injection (44), it is possible that the higher levels of IL-6 production may be secondary to continuous IL-1 secretion. In contrast to PEMs, triple mutant BMDMs do not spontaneously secrete IL-1; hence some aspect of the inflammatory stimulus caused by the peritoneal injection of thioglycolate used to elicit macrophage influx into the abdominal cavity is contributing to the hyperresponsiveness of these cells.
Most of the experiments shown use macrophages that are stimulated with LPS plus IFN-γ. We found that PEMs failed to respond to LPS-alone stimulation (as assessed by nitrite and cytokine release) while BMDMs showed less robust responses. Induction of macrophage tumor cell cytotoxicity also requires costimulation of cells with LPS plus IFN-γ (31). The mechanism by which IFN-γ primes macrophages for augmented responses to LPS is unknown, however it is unlikely to be mediated by increased signaling through Src-family kinases since the triple mutant BMDMs showed equivalent enhancement of LPS responses with IFN-γ costimulation compared to wild-type cells.
The induction of tumor cell cytotoxicity in hck −/− fgr −/− lyn −/− macrophages by LPS/IFN-γ was completely normal in BMDMs but reproducibly showed a 50% decrease in PEMs (Fig. 5). Given that other aspects of macrophage activation and signal transduction caused by LPS were increased in triple mutant PEMs, the cause for the modest impairment in tumoricidal capability is unclear. Previous work has suggested that hck −/− fgr −/− neutrophils and macrophages have defects related to integrin-mediated adhesion (45). Since integrin-dependent adhesion also contributes to macrophage cytotoxicity (46), it is possible that impairments in cell adhesion may account for the slightly diminished tumoricidal capacity of triple mutant PEMs.
Activation of the pleiotropic transcription factor NF-κB is also a well-described signaling pathway initiated by LPS treatment of macrophages (10). Activation of NF-κB DNA binding activity requires phosphorylation of serine residues of both the p65 and p50 subunits (47). Although the signaling pathway from membrane CD14 that leads to NF-κB activation is not clearly defined, tyrosine phosphorylation events may be important, since herbimycin A will block LPS-induced NF-κB activation (48). An indirect effect of herbimycin A on inhibition of NF-κB activation has also been suggested (49). Activation of NF-κB binding activity occurred normally in LPS/IFN-γ–stimulated triple mutant macrophages (Fig. 8), demonstrating that these Src-family kinases are not essential in initiating this LPS-induced signal transduction pathway.
The major conclusion from this work is that despite the biochemical evidence suggesting that Hck, Fgr, and Lyn are involved in LPS signal transduction, macrophages that are devoid of these kinases, and express no other detectable Src-family kinases, have normal (or even enhanced) LPS functional responses. Activation of the major mediators of LPS signal transduction—MAP kinases, JNK, and NFκB—occurs essentially normally in the absence of Src-family kinases. This begs the question of what is (are) the other kinase(s) responsible for in CD14-mediated LPS signal transduction. Based on the observation that herbimycin A suppresses nitrite production and p42/44 activation equivalently in both wild-type and triple mutant macrophages (Fig. 9 A), a tyrosine kinase is likely to be involved in some step of the pathway, though not necessarily at the initial step. It is unlikely that the p72syk kinase is involved, since LPS responses are also normal in syk −/− mutant macrophages (Crowley, M., and A. DeFranco, unpublished observations). An attractive candidate for the kinase responsible for initiating LPS signaling may be the ceramide activated protein kinase, since LPS has been shown to activate this enzyme (50). Moreover, ceramide responses are defective in the LPS non-responder macrophages cultured from C3H/ HeJ mice (18). Use of the hck −/− fgr −/− lyn −/− macrophages may dramatically facilitate biochemical and/or genetic approaches to isolation of the responsible kinases involved in LPS signaling since Src-family members are not expressed in these cells. Finally, this work also has major implications in designing therapeutics for endotoxic shock; it is very likely that compounds that inhibit Src-family kinase function will not serve as successful drugs for septic shock.
Acknowledgments
We wish to thank J. Hambleton, V. Chan, S. Weinstein, M. Crowley, and A. DeFranco for help with reagents, assays, and critical review of this work.
This work was supported by grants from the National Institutes of Health (T32 A107334-09 to F. Meng and DK50267 and HL54476 to C.A. Lowell).
Footnotes
1 Abbreviations used in this paper: BMDM, bone marrow–derived macrophage; EMSA, electrophoretic mobility shift assay; ERK, extracellular signal-regulated kinase; JNK, c-Jun NH2-terminal kinase; MAPK, mitogen-activated protein kinase; MBP, myelin basic protein; PEM, thioglycolate-elicited peritoneal macrophage; PTK, protein tyrosine kinase.
References
- 1.Dinarello CA. Biologic basis for interleukin-1 in disease. Blood. 1996;87:2095–2147. [PubMed] [Google Scholar]
- 2.Akira S, Taga T, Kishimoto T. Interleukin-6 in biology and medicine. Adv Immunol. 1993;54:1–78. doi: 10.1016/s0065-2776(08)60532-5. [DOI] [PubMed] [Google Scholar]
- 3.Tracey KJ, Cerami A. Tumor necrosis factor: a pleiotropic cytokine and therapeutic target. Annu Rev Med. 1994;45:491–503. doi: 10.1146/annurev.med.45.1.491. [DOI] [PubMed] [Google Scholar]
- 4.Kielian TL, Blecha F. CD14 and other recognition molecules for lipopolysaccharide: a review. Immunopharmacology. 1995;29:187–205. doi: 10.1016/0162-3109(95)00003-c. [DOI] [PubMed] [Google Scholar]
- 5.Morrison DC, Ryan JL. Endotoxins and disease mechanisms. Annu Rev Med. 1987;38:417–432. doi: 10.1146/annurev.me.38.020187.002221. [DOI] [PubMed] [Google Scholar]
- 6.Ferrero E, Jiao D, Tsuberi BZ, Tesio L, Rong GW, Haziot A, Goyert SM. Transgenic mice expressing human CD14 are hypersensitive to lipopolysaccharide. Proc Natl Acad Sci USA. 1993;90:2380–2384. doi: 10.1073/pnas.90.6.2380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Haziot A, Ferrero E, Kontgen F, Hijiya N, Yamamoto S, Silver J, Stewart CL, Goyert SM. Resistance to endotoxin shock and reduced dissemination of gram-negative bacteria in CD14-deficient mice. Immunity. 1996;4:407–414. doi: 10.1016/s1074-7613(00)80254-x. [DOI] [PubMed] [Google Scholar]
- 8.Weinstein SL, June CH, DeFranco AL. Lipopolysaccharide-induced protein tyrosine phosphorylation in human macrophages is mediated by CD14. J Immunol. 1993;151:3829–3838. [PubMed] [Google Scholar]
- 9.Arditi M, Zhou J, Torres M, Durden DL, Stins M, Kim KS. Lipopolysaccharide stimulates the tyrosine phosphorylation of mitogen-activated protein kinases p44, p42, and p41 in vascular endothelial cells in a soluble CD14dependent manner. Role of protein tyrosine phosphorylation in lipopolysaccharide-induced stimulation of endothelial cells. J Immunol. 1995;155:3994–4003. [PubMed] [Google Scholar]
- 10.Ulevitch RJ, Tobias PS. Receptor-dependent mechanisms of cell stimulation by bacterial endotoxin. Annu Rev Immunol. 1995;13:437–457. doi: 10.1146/annurev.iy.13.040195.002253. [DOI] [PubMed] [Google Scholar]
- 11.Daniel-Issakani S, Spiegel AM, Strulovici B. Lipopolysaccharide response is linked to the GTP binding protein, Gi2, in the promonocytic cell line U937. J Biol Chem. 1989;264:20240–20247. [PubMed] [Google Scholar]
- 12.Chen TY, Lei MG, Suzuki T, Morrison DC. Lipopolysaccharide receptors and signal transduction pathways in mononuclear phagocytes. Curr Top Micro Immunol. 1992;181:169–188. doi: 10.1007/978-3-642-77377-8_6. [DOI] [PubMed] [Google Scholar]
- 13.Shapira L, Takashiba S, Champagne C, Amar S, Van Dyke TE. Involvement of protein kinase C and protein tyrosine kinase in lipopolysaccharide-induced TNF-α and IL-1β production by human monocytes. J Immunol. 1994;153:1818–1824. [PubMed] [Google Scholar]
- 14.Han J, Lee JD, Bibbs L, Ulevitch RJ. A MAP kinase targeted by endotoxin and hyperosmolarity in mammalian cells. Science (Wash DC) 1994;265:808–811. doi: 10.1126/science.7914033. [DOI] [PubMed] [Google Scholar]
- 15.Hambleton J, Weinstein SL, Lem L, DeFranco AL. Activation of c-Jun N-terminal kinase in bacterial lipopolysaccharide-stimulated macrophages. Proc Natl Acad Sci USA. 1996;93:2774–2778. doi: 10.1073/pnas.93.7.2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Han J, Lee JD, Tobias PS, Ulevitch RJ. Endotoxin induces rapid protein tyrosine phosphorylation in 70Z/3 cells expressing CD14. J Biol Chem. 1993;268:25009–25014. [PubMed] [Google Scholar]
- 17.Weinstein SL, Sanghera JS, Lemke K, DeFranco AL, Pelech SL. Bacterial lipopolysaccharide induces tyrosine phosphorylation and activation of mitogen-activated protein kinases in macrophages. J Biol Chem. 1992;267:14955–14962. [PubMed] [Google Scholar]
- 18.Barber SA, Perera PY, Vogel SN. Defective ceramide response in C3H/HeJ (Lpsd) macrophages. J Immunol. 1995;155:2303–2305. [PubMed] [Google Scholar]
- 19.Novogrodsky A, Vanichkin A, Patya M, Gazit A, Osherov N, Levitzki A. Prevention of lipopolysaccharide-induced lethal toxicity by tyrosine kinase inhibitors. Science (Wash DC) 1994;264:1319–1322. doi: 10.1126/science.8191285. [DOI] [PubMed] [Google Scholar]
- 20.Henricson BE, Carboni JM, Burkhardt AL, Vogel SN. LPS and Taxol activate Lyn kinase autophosphorylation in Lps(n), but not in Lps(d), macrophages. Mol Med. 1995;1:428–435. [PMC free article] [PubMed] [Google Scholar]
- 21.Stefanova, I., M.L. Corcoran, E.M. Horak, L.M. Wahl, J.B. Bolen, and I.D. Horak. 1993. Lipopolysaccharide induces activation of CD14-associated protein tyrosine kinase p53/ 56lyn. J. Biol. Chem. 268:20725–20728. [PubMed]
- 22.Boulet I, Ralph S, Stanley E, Lock P, Dunn AR, Green SP, Phillips WA. Lipopolysaccharide- and interferon-γ-induced expression of Hck and Lyn tyrosine kinases in murine bone marrow-derived macrophages. Oncogene. 1992;7:703–710. [PubMed] [Google Scholar]
- 23.Herrera-Velit P, Reiner NE. Bacterial lipopolysaccharide induces the association and coordinate activation of p53/56lynand phosphatidylinositol 3-kinase in human monocytes. J Immunol. 1996;156:1157–1165. [PubMed] [Google Scholar]
- 24.Beaty CD, Franklin TL, Uehara Y, Wilson CB. Lipopolysaccharide-induced cytokine production in human monocytes: role of tyrosine phosphorylation in transmembrane signal transduction. Eur J Immunol. 1994;24:1278–1284. doi: 10.1002/eji.1830240606. [DOI] [PubMed] [Google Scholar]
- 25.Ziegler SF, Wilson CB, Perlmutter RM. Augmented expression of a myeloid-specific protein tyrosine kinase gene (hck) after macrophage activation. J Exp Med. 1988;168:1801–1810. doi: 10.1084/jem.168.5.1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.English BK, Ihle JN, Myracle A, Yi T. Hck tyrosine kinase activity modulates tumor necrosis factor production by murine macrophages. J Exp Med. 1993;178:1017–1022. doi: 10.1084/jem.178.3.1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lowell CA, Soriano P. Knockouts of Src-family kinases: stiff bones, wimpy T cells, and bad memories. Genes Dev. 1996;10:1845–1857. doi: 10.1101/gad.10.15.1845. [DOI] [PubMed] [Google Scholar]
- 28.Lowell CA, Soriano P, Varmus HE. Functional overlap in the src-gene family: inactivation of Hck and Fgr impairs natural immunity. Genes Dev. 1994;8:387–398. doi: 10.1101/gad.8.4.387. [DOI] [PubMed] [Google Scholar]
- 29.Law DA, Chan VWF, Datta SK, DeFranco AL. B-cell antigen receptor motifs have redundant signalling capabilities and bind the tyrosine kinases PTK72, Lyn and Fyn. Curr Biol. 1993;3:645–657. doi: 10.1016/0960-9822(93)90062-s. [DOI] [PubMed] [Google Scholar]
- 30.Ding AH, Nathan CF, Stuehr DJ. Release of reactive nitrogen intermediates and reactive oxygen intermediates from mouse peritoneal macrophages. Comparison of activating cytokines and evidence for independent production. J Immunol. 1988;141:2407–2412. [PubMed] [Google Scholar]
- 31.Jadus MR, Good RW, Crumpacker DB, Yannelli JR. Effects of interleukin 4 upon human tumoricidal cells obtained from patients bearing solid tumors. J Leukoc Biol. 1991;49:139–151. doi: 10.1002/jlb.49.2.139. [DOI] [PubMed] [Google Scholar]
- 32.Kitchens RL, Ulevitch RJ, Munford RS. Lipopolysaccharide (LPS) partial structures inhibit responses to LPS in a human macrophage cell line without inhibiting LPS uptake by a CD14-mediated pathway. J Exp Med. 1992;176:485–494. doi: 10.1084/jem.176.2.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hibbs ML, Tarlinton DM, Armes J, Grail D, Hodgson G, Maglitto R, Stacker SA, Dunn AR. Multiple defects in the immune system of Lyn-deficient mice, culminating in autoimmune disease. Cell. 1995;83:301–311. doi: 10.1016/0092-8674(95)90171-x. [DOI] [PubMed] [Google Scholar]
- 34.Wang JY, Koizumi T, Watanabe T. Altered antigen receptor signaling and impaired fas-mediated apoptosis of B-cells in lyn-deficient mice. J Exp Med. 1996;184:831–838. doi: 10.1084/jem.184.3.831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nishizumi H, Taniuchi I, Yamanashi Y, Kitamura D, Ilic D, Mori S, Watanabe T, Yamamoto T. Impaired proliferation of peripheral B cells and indication of autoimmune disease in lyn-deficient mice. Immunity. 1995;3:549–560. doi: 10.1016/1074-7613(95)90126-4. [DOI] [PubMed] [Google Scholar]
- 36.Naito M, Umeda S, Yamamoto T, Moriyama H, Umezu H, Hasegawa G, Usuda H, Shultz LD, Takahashi K. Development, differentiation, and phenotypic heterogeneity of murine tissue macrophages. J Leukoc Biol. 1996;59:133–138. doi: 10.1002/jlb.59.2.133. [DOI] [PubMed] [Google Scholar]
- 37.Henkel T, Machleidt T, Alkalay I, Kronke M, BenNeriah Y, Baeuerle PA. Rapid proteolysis of IκB-α is necessary for activation of transcription factor NF-κB. Nature (Lond) 1993;365:182–185. doi: 10.1038/365182a0. [DOI] [PubMed] [Google Scholar]
- 38.Hambleton J, McMahon M, DeFranco AL. Activation of Raf-1 and mitogen-activated protein kinase in murine macrophages partially mimics lipopolysaccharide-induced signaling events. J Exp Med. 1995;182:147–154. doi: 10.1084/jem.182.1.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee JD, Kravchenko V, Kirkland TN, Han J, Mackman N, Moriarty A, Leturcq D, Tobias PS, Ulevitch RJ. Glycosyl-phosphatidylinositol-anchored or integral membrane forms of CD14 mediate identical cellular responses to endotoxin. Proc Natl Acad Sci USA. 1993;90:9930–9934. doi: 10.1073/pnas.90.21.9930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.De Nichilo MO, Yamada KM. Integrin αvβ5dependent serine phosphorylation of paxillin in cultured human macrophages adherent to vitronectin. J Biol Chem. 1996;271:11016–11022. doi: 10.1074/jbc.271.18.11016. [DOI] [PubMed] [Google Scholar]
- 41.Sudol M, Greulich H, Newman L, Sarkar A, Sukegawa J, Yamamoto T. A novel Yes-related kinase, Yrk, is expressed at elevated levels in neural and hematopoietic tissues. Oncogene. 1993;8:823–831. [PubMed] [Google Scholar]
- 42.D'Ambrosio D, Hippen KL, Minskoff SA, Mellman I, Pani G, Siminovitch KA, Cambier JC. Recruitment and activation of PTP1C in negative regulation of antigen receptor signaling by Fcγ RIIB1. Science (Wash DC) 1995;268:293–297. doi: 10.1126/science.7716523. [DOI] [PubMed] [Google Scholar]
- 43.Ono M, Bolland S, Tempst P, Ravetch JV. Role of the inositol phosphatase SHIP in negative regulation of the immune system by the receptor FcγRIIB. Nature (Lond) 1996;383:263–266. doi: 10.1038/383263a0. [DOI] [PubMed] [Google Scholar]
- 44.Zheng H, Fletcher D, Kozak W, Jiang M, Hofmann KJ, Conn CA, Soszynski D, Grabiec C, Trumbauer ME, Shaw A. Resistance to fever induction and impaired acute-phase response in interleukin-1β-deficient mice. Immunity. 1995;3:9–19. doi: 10.1016/1074-7613(95)90154-x. [DOI] [PubMed] [Google Scholar]
- 45.Lowell CA, Fumagalli L, Berton G. Deficiency of Src family kinases p59/61hck and p58c-fgrresults in defective adhesion-dependent neutrophil functions. J Cell Biol. 1996;133:895–910. doi: 10.1083/jcb.133.4.895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pak CC, Fidler IJ. Molecular mechanisms for activated macrophage recognition of tumor cells. Semin Cancer Biol. 1991;2:189–195. [PubMed] [Google Scholar]
- 47.Hayashi T, Sekine T, Okamoto T. Identification of a new serine kinase that activates NFκB by direct phosphorylation. J Biol Chem. 1993;268:26790–26795. [PubMed] [Google Scholar]
- 48.Ishikawa Y, Mukaida N, Kuno K, Rice N, Okamoto S, Matsushima K. Establishment of lipopolysaccharide-dependent nuclear factor kappa-B activation in a cellfree system. J Biol Chem. 1995;270:4158–4164. [PubMed] [Google Scholar]
- 49.Mahon TM, O'Neill LA. Studies into the effect of the tyrosine kinase inhibitor herbimycin A on NF-κB activation in T lymphocytes. Evidence for covalent modification of the p50 subunit. J Biol Chem. 1995;270:28557–28564. doi: 10.1074/jbc.270.48.28557. [DOI] [PubMed] [Google Scholar]
- 50.Joseph CK, Wright SD, Bornmann WG, Randolph JT, Kumar ER, Bittman R, Liu J, Kolesnick RN. Bacterial lipopolysaccharide has structural similarity to ceramide and stimulates ceramide-activated protein kinase in myeloid cells. J Biol Chem. 1994;269:17606–17610. [PubMed] [Google Scholar]