

Abstract
We investigated the relationship between PS1 and γ-secretase processing of amyloid precursor protein (APP) in primary cultures of neurons. Increasing the amount of APP at the cell surface or towards endosomes did not significantly affect PS1-dependent γ-secretase cleavage, although little PS1 is present in those subcellular compartments. In contrast, almost no γ-secretase processing was observed when holo-APP or APP-C99, a direct substrate for γ-secretase, were specifically retained in the endoplasmic reticulum (ER) by a double lysine retention motif. Nevertheless, APP-C99-dilysine (KK) colocalized with PS1 in the ER. In contrast, APP-C99 did not colocalize with PS1, but was efficiently processed by PS1-dependent γ-secretase. APP-C99 resides in a compartment that is negative for ER, intermediate compartment, and Golgi marker proteins. We conclude that γ-secretase cleavage of APP-C99 occurs in a specialized subcellular compartment where little or no PS1 is detected. This suggests that at least one other factor than PS1, located downstream of the ER, is required for the γ-cleavage of APP-C99. In agreement, we found that intracellular γ-secretase processing of APP-C99-KK both at the γ40 and the γ42 site could be restored partially after brefeldin A treatment. Our data confirm the “spatial paradox” and raise several questions regarding the PS1 is γ-secretase hypothesis.
Keywords: presenilin 1; amyloid peptide; γ-secretase; ER retention; APP processing
Introduction
Amyloid precursor protein (APP)* and presenilins (PSs) are key proteins in the pathogenesis of Alzheimer's disease (Selkoe, 1999). PSs are multimembrane-spanning domain proteins, essentially located in the ER, the intermediate compartment, and the cis-Golgi region (Walter et al., 1996; Culvenor et al., 1997; Annaert et al., 1999; Kim et al., 2000). Some evidence for the presence of minute amounts of PS at the cell surface has been provided (Ray et al., 1999). APP is a type I transmembrane protein processed by secretases, resulting in the generation of various fragments, most notoriously the amyloid peptide (Aβ). Aggregation of this peptide is considered to be a central event in the process leading to Alzheimer's disease (Selkoe, 1999). The β-secretase BACE (β-site amyloid cleaving enzyme) has been identified recently. It is an aspartyl protease cleaving APP at the NH2 terminus of the amyloid peptide sequence (Vassar et al., 1999). The α-secretase cleaves in the Aβ sequence itself, precluding further Aβ production. Several members of the ADAM family (a disintegrin and metalloprotease), i.e., TACE, MDC9, and ADAM10, have been implied in α-secretase processing of APP (Buxbaum et al., 1998; Koike et al., 1999; Lammich et al., 1999). Finally, presenilin 1 (PS1)-dependent γ-secretase cleavage of APP releases the COOH terminus of the amyloid peptide (De Strooper et al., 1998). PS-dependent γ-secretase activities are also involved in the processing of other integral membrane proteins like Notch and APLP-1 (De Strooper et al., 1999; Naruse et al., 1998; Song et al., 1999; Struhl and Greenwald, 1999). This γ-secretase cleavage is a novel type of proteolytic processing, occurring in the hydrophobic environment of the cell membrane. In the case of Notch, the cleavage results in the release of its intracellular domain that regulates gene transcription in a process that has been called “regulated intramembrane proteolysis” (Annaert and De Strooper, 1999; Brown et al., 2000).
Several lines of evidence have led to the hypothesis that PS is the γ-secretase. First, γ-secretase cleavage of APP and Notch is inactivated partially in PS1-deficient cells and completely in PS1/PS2 double–deficient cells (De Strooper et al., 1998, 1999; Song et al., 1999; Struhl and Greenwald, 1999; Herreman et al., 2000; Zhang et al., 2000). Moreover, mutagenesis of one or two aspartyl residues located in transmembrane domain 6 or 7 results in dominant negative–like effects on γ-secretase cleavage, suggesting that these aspartates contribute to the active site of PS (Wolfe et al., 1999). Some similarities with the prepilin peptidases has been taken as further evidence that the PS are proteases (Steiner et al., 2000). However, the strongest argument in favor for this idea comes from the observation that several compounds that inhibit γ-secretase bind specifically to PS (Esler et al., 2000; Li et al., 2000; Seiffert et al., 2000). Nevertheless, direct proof that the PS can hydrolyze peptide bounds is still lacking.
Remarkably, both Notch and APP apparently first need proteolytical trimming of their extracellular domain by TACE and α-/β-secretase, respectively, to become a substrate for PS/γ-secretase (De Strooper et al., 1998; Brou et al., 2000; Mumm et al., 2000; Struhl and Adachi, 2000). These proteases are located downstream in the biosynthetic pathway and therefore both Notch and APP first have to leave the ER to become a substrate for PS/γ-secretase. Despite significant advances in our understanding of γ-secretase processing, it remains unclear how APP, after its cleavage by α- or β-secretase, becomes exposed to PS. Moreover, Aβ appears to be produced mainly in the endosomal compartment (Koo and Squazzo, 1994; Hartmann et al., 1997), whereas PS is abundantly present in the ER and intermediate compartment (Walter et al., 1996; Culvenor et al., 1997; Annaert et al., 1999; Kim et al., 2000). This problem can be summarized as “the spatial paradox” (Annaert and De Strooper, 1999) and we now tackle this problem further by analyzing the processing of a series of APP-trafficking mutants in primary cortical neurons derived from PS1+/+ or PS1−/− mice. We analyzed the consequences of restricting APP trafficking to certain subcellular compartments for the generation of total Aβ and Aβ42. Constructs tested: (a) APP with deleted cytosolic tail (APP-Δct); (b) APP with a double lysine ER retention motif in its cytoplasmic tail (APP-KK); (c) APP with a cytoplasmic tail from the LDL receptor (APP-LDL); (d) APP truncated at the β-secretase cleavage site (APP-C99); and (e) APP-C99 with a double lysine (KK) motif. The APP-Δct is poorly reinternalized and stays for longer times at the cell surface (Tienari et al., 1996). The KK motif acts an ER-retrieval signal (Jackson et al., 1993; Gaynor et al., 1994) and therefore APP-KK colocalizes abundantly with endogenous PS1 (Peraus et al., 1997; Annaert et al., 1999). The APP-LDL recycles between the endosomal system and the cell surface (Annaert et al., 1999). APP-C99 is a direct substrate for γ-secretase and after addition of the KK ER retention motif (APP-C99-KK) we anticipated creating an excellent substrate for PS1/γ-secretase. Overall, our data confirm the spatial paradox in the PS-is-γ-secretase hypothesis and provide evidence that the transfer of APP-C99 in a compartment downstream of the ER is needed in addition to processed PS1 to obtain γ-secretase activity in primary cultures of neurons.
Results
Different APP-trafficking mutants (Fig. 1) were expressed in primary cortical neurons derived from PS1+/+ and PS1−/− littermate embryos. Metabolically labeled full-length APP and secretase-cleaved APP fragments were immunoprecipitated and analyzed by phosphorimaging as described in Materials and methods. For all constructs, protein expression levels are very similar in PS1+/+ and in PS1−/− cells (Fig. 2 A). Both full-length wild-type and APP-LDL chimera run as a doublet (Fig. 2 A) of ∼120 kD, corresponding to mature- and immature-glycosylated APP. The majority for both the APP-KK and APP-Δct recombinant proteins is, however, the immature protein. This is expected for APP-KK, as the dilysine motif actively retains the protein in pre-Golgi compartments. The stronger immature APP-Δct band could reflect the rapid processing of the mature form by α-secretase at the cell surface (Tienari et al., 1997). Next, we analyzed proteolytic fragments derived from the different APP mutants. Secretion of the soluble ectodomain after α- or β-secretase cleavage generated from the different trafficking mutants was not significantly altered by PS1 deficiency, although the APP-Δct, for instance, produced roughly twice as many APPs (Fig. 2). β-Cleaved COOH-terminal stubs (APP-CTF) derived from APP-WT accumulated strongly in PS1−/− neurons (Fig. 2 B), confirming previous data (De Strooper et al., 1998). Interestingly, the same relative accumulation was observed for APP-CTF derived from the APP-LDL chimera. Also, total Aβ secretion was severely decreased in PS1−/− neurons transduced for APP-WT and APP-LDL (Fig. 2 B). Although the secretion of Aβ42 derived from APP-LDL is reduced by 80% in PS1+/+ neurons, in line with previous observations (Koo and Squazzo, 1994; Perez et al., 1999), the additional relative decrease caused by PS1−/− deficiency remained unaltered when compared with APP-WT (Fig. 2, B and C). Together, these data suggest that in the case of the APP-LDL chimera, the enhanced recycling in the endosomal limb does not directly influence PS1-dependent γ-secretase processing.
Figure 1.

APP-trafficking mutants. APP is schematically represented (APP-WT). The different relevant proteolytic fragments are indicated at the top. The ectodomain is detected by pAb207, the amyloid peptide region 1–16 is detected by pAb B7, region 1–5 by mAb 3D6, region 17–24 is detected by mAb4G8, and the last 20 amino acids of the COOH-terminal cytoplasmic tail are detected by pAb B11/4 and pAb 6687. α-, β-, and γ-secretase cleavage sites are indicated. In the APP-C99 construct, a supplementary Asp and Ala residue (DA) has been added to obtain cleavage by the signal peptidase at the β-secretase site (see Lichtenthaler et al., 1999). For further details see Materials and methods.
Figure 2.
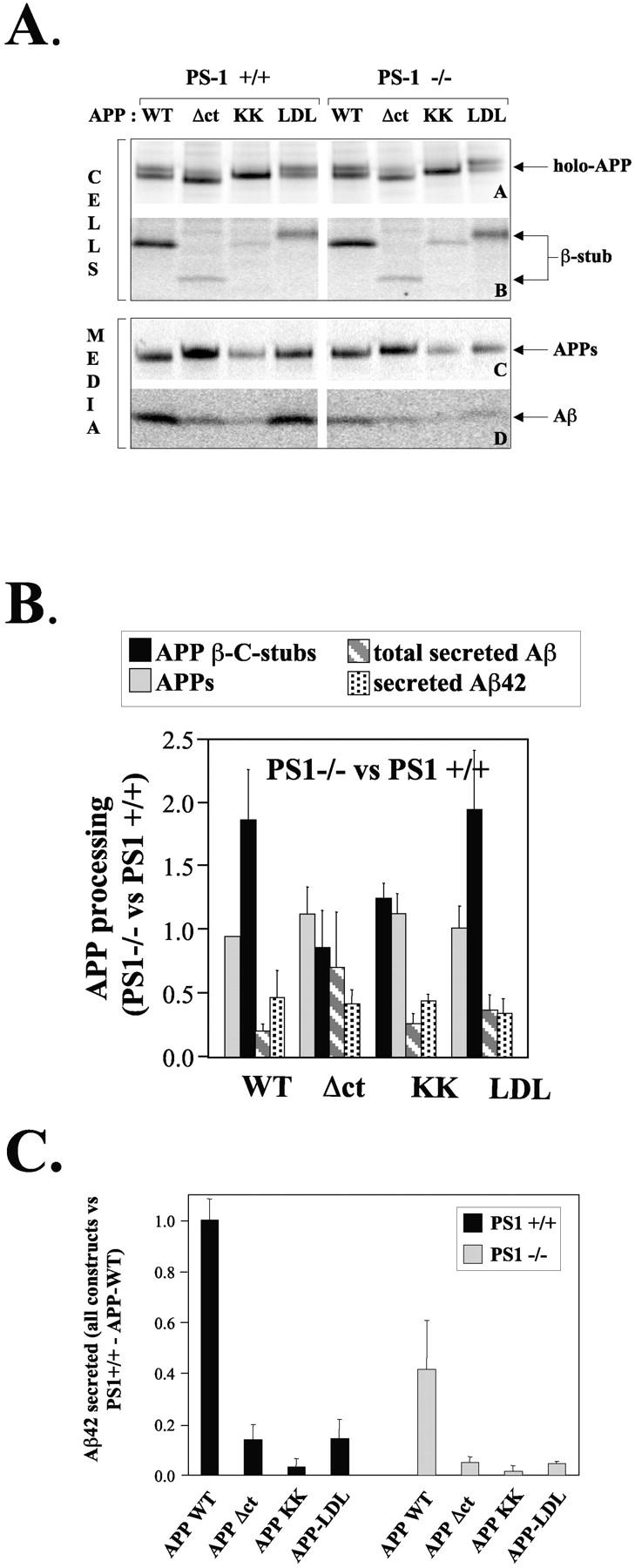
Processing of APP trafficking mutants in PS1+/+ and PS1−/− neurons. (A) PS1+/+ or PS1−/− primary neuronal cultures were transduced with pSFV bearing APP-WT, APP-Δct, APP-KK, or APP-LDL as described, and metabolically labeled with [35S]methionine for 4 h at 37°C. Cell extracts (two top panels) and culture media (two bottom panels) were immunoprecipitated using the appropriate antibodies and analyzed on 10% or 4–12% NuPage gels. The position of the various APP fragments is indicated. Note that the APP-Δct holo-forms and COOH-terminal β-fragments migrate faster because the cytoplasmic domain is deleted, whereas the APP-LDL fragments migrates slower due to the large LDL receptor cytoplasmic domain. The COOH-terminal α-stub is not detected here because antibody B7 does not react with that fragment. (B) APP fragments from three independent experiments were quantified using PhosphorImaging and normalized to the level of expression of the APP holo-forms (De Strooper et al., 1995, 1998; Annaert et al., 1999). Data obtained in the PS1−/− neurons are compared with the data obtained in the PS1+/+ neurons. This shows the relative effect of the absence of PS1 on each APP fragment separately. Since APP-Δct is processed by β-secretase to a limited extent only, the results displayed in Fig. 3 are more significant for conclusions regarding the level of γ-secretase processing of this construct. (C) The secretion of Aβ42 was analyzed by ELISA. All results are normalized to the level secreted by PS1+/+ neurons transfected with APP-WT. Dark bars and light bars represent data obtained in PS1+/+ and PS1−/− neurons, respectively.
The reduction of β-cleaved APP-CTF in the APP-Δct–transduced wild-type neurons is accompanied by an 80% decrease in secretion of total Aβ and Aβ42 (Fig. 2, A and C). Moreover, no relative accumulation of β-cleaved APP-CTF in PS1−/− neurons was observed. All this can be explained by a predominant α-secretase cleavage of APP-Δct at or near the plasma membrane, as reflected by the increased secretion of APPs cleaved at the α-secretase site and the concomitant fivefold increase in p3 secretion (Fig. 3) . Interestingly, secretion of p3 is dramatically inhibited in the absence of PS1 (Fig. 3, bottom), indicating that α-stubs generated near or at the cell surface are still substrates for PS1-dependent γ-secretase processing. Therefore, the data on APP-Δct and APP-LDL clearly demonstrate that PS1 is needed for normal γ-secretase processing in the late Golgi region, at the cell surface, and in the endosomal compartments.
Figure 3.

Secretion of p3 in PS1+/+ and −/− neurons expressing APP-Δct. Neurons were transduced and metabolically labeled as above. Cell extracts (top) and culture media (bottom) were immunoprecipitated using pAb207 (holo-APP) or mAb4G8 (αAPPs and Aβ/p3). The intermediate band running between Aβ and p3 in cells transduced with APP-WT corresponds to the truncated Aβ generated by β-secretase cleavage at the Glu11 position (Creemers et al., 2000).
In contrast to the other mutants, the levels of APPs and of the COOH-terminal β-stubs generated from APP-KK are very low, confirming the low α- and β-secretase activity in the ER (Fig. 2, A and B). Accordingly, the total Aβ (Fig. 2 B) and Aβ42 (Fig. 2 C) secretion was also strongly reduced by 90%. However, when overexpressed in PS1−/− neurons, secretion of Aβ peptides was inhibited to the same extent as observed for peptides derived from APP-WT (Fig. 2, B and C).
To circumvent the need for “preactivation” of APP-KK by α- or β-secretase, we generated APP-C99-KK, corresponding to APP truncated at the β-secretase site (Lichtenthaler et al., 1999) and containing the KK-ER retention motif. When expressed in neurons, APP-C99-KK migrated with the same apparent molecular weight (10 kD) as the β-stub coming from APP-WT or the APP-C99 without KK motif (Fig. 4 A; Lichtenthaler et al., 1999). APP-C99, like APP-WT, can be processed by α-secretase as reflected by the generation of α-APPCTF (Fig. 4 A). APP-C99-KK, in contrast, is either not processed or processed very little by α- or β-secretase, confirming the specific retention of APP-C99-KK in the ER and cis-Golgi region. In line with the prediction that APP-C99 does not require α- or β-secretase cleavage to become a substrate for γ-secretase, an estimated sevenfold increase in total Aβ secretion (Fig. 4 A) is observed. This effect is also observed with Aβ42, as detected by ELISA (Fig. 4 B). The γ-secretase cleavage of APP-C99 is strongly inhibited in the absence of PS1 (Fig. 4 C). Most surprisingly, however, was the observation that APP-C99-KK, which was also expected to be a substrate for PS1-γ-secretase because it is retained in the ER, turned out to yield little if any Aβ peptides (Fig. 4, A and B). It is unlikely that the KK motif directly interfered with the recognition of C99 by the γ-secretase complex, since the cytoplasmic tail of APP can be removed without affecting γ-secretase cleavage (Fig. 3). Moreover, C99KK becomes a substrate for γ-secretase under conditions specified below. Alternatively, the low Aβ secretion from neurons expressing APP-C99-KK might be explained by intracellular retention of newly formed Aβ peptides. Again, this could be ruled out, as no increased amounts of Aβ could be immunoprecipitated from cell extracts (result not shown, but see Fig. 7 below).
Figure 4.
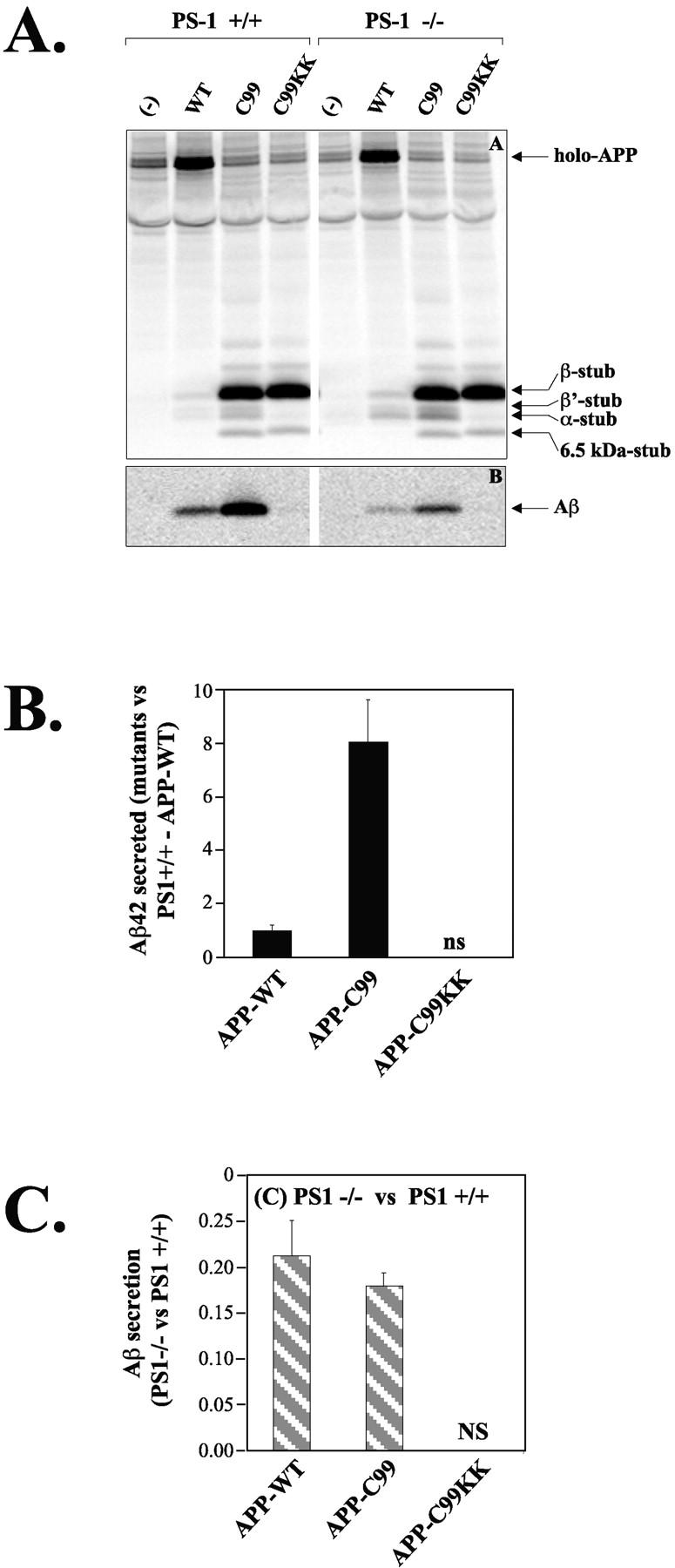
γ-secretase processing of APP-C99 trafficking mutants in PS1+/+ and PS1−/− neurons. (A) Cells extracts (top) and culture media (bottom) of neurons expressing APP-WT, APP-C99, or APP-C99-KK were immunoprecipitated using antibodies B11/4 against the APP COOH-terminal domain (top), or B7 against the amyloid region. Arrows on the gels indicate the position of the various APP fragments. Notice the striking absence of Aβ generation with APP-C99-KK. The 6.5-kD stub seen with APP-C99 and APP-C99-KK is PS1-independent and probably not physiologically relevant, since it is never observed with APP-WT. (B) Aβ42 levels were measured by ELISA as in Fig. 2 B. Results are expressed relatively to those obtained for PS1+/+ neurons transfected with APP-WT. NS, nonsignificant (values below detection limit). (C) Aβ-fragments were quantified using PhosphorImaging and normalized to the level of expression of the APP holo-forms (n = 3; mean ± SEM). Data obtained in the PS1−/− neurons are compared with the data obtained in the PS1+/+ neurons. This shows the relative effect of the absence of PS1 on Aβ production from APP-WT and from APP-C99. The Aβ signals obtained with APP-C99-KK are below detection limit.
Figure 7.

BFA treatment partially restores γ-secretase processing of APP-C99-KK. Neurons were transduced with SFV-APP-C99 or -APP-C99-KK and treated with or without 10 μM BFA for 4 h. Culture media and cell extracts were immunoprecipitated using antibody B7 and separated on 10% NuPage gels. Detection of secreted and intracellular Aβ was done by Western blotting using the W0-2 mAb raised against the NH2 terminus of the Aβ sequence (Ida et al., 1996). Neurons transduced with SFV-APP-C99-KK were treated with BFA as above. Cell extracts were sequentially immunoprecipitated using antibody FCA42, specific for Aβ peptides ending at residue 42 and FCA40, specific for Aβ peptides ending at residue 42 (Barelli et al., 1997), and separated on 10% NuPage gels. After blotting, Aβ peptides were revealed using the WO-2 mAb as above.
Using confocal scanning microscopy, we confirmed that APP-C99-KK was effectively retained in the ER, as demonstrated by its colocalization with the ER marker protein BIP (Fig. 5 , A–C), and to a more limited extent with the intermediate compartment marker ERGIC-53 (Fig. 5, G–I). Importantly, APP-C99-KK colocalized abundantly with PS1 (Fig. 5, D–F), indicating that the simple presence of PS1 is not sufficient for γ-secretase processing to occur. In this respect, it is important to note that we have demonstrated previously that PS1 in the ER is already processed towards NH2- and COOH-terminal fragments (Annaert et al., 1999). We next investigated the subcellular localization of APP-C99 in primary cortical neurons. APP-C99 is clearly not distributed in the ER (Fig. 6 , A–C), as deduced from the complete lack of colocalization with BIP. Similarly, little if any colocalization was observed with ERGIC-53 marking the intermediate compartment (arrowheads in Fig. 6, D–F, including horizontal section [arrow]). Most surprisingly, however, was the observation that APP-C99 immunoreactivity also essentially did not distribute into the Golgi apparatus as demonstrated by GM130 staining (Fig. 6, G–I). Finally, and despite APP-C99 being a good γ-secretase substrate (Fig. 4), this fragment does not colocalize with PS1 (arrowheads and vertical section in Fig. 6, J–L). This is in clear contrast to the colocalization of PS1 with the inactive γ-secretase substrate APP-C99-KK (see also Fig. 5).
Figure 5.

APP-C99-KK colocalizes with PS1 in the endoplasmic reticulum. Hippocampal neurons transduced with SFV-APP-C99-KK were fixed 6 h postinfection and stained with an antibody against APP (A with pab 6687; D and G with mab 3D6) and BIP (B), PS1 (E), or ERGIC-53 (H). C, F, and I represent the merged pictures. APP-C99-KK abundantly colocalized with the ER-marker protein BIP and with PS1. Little if any colocalization was observed with ERGIC-53, a resident protein of the intermediate compartment. Detection of primary antibodies was done with Alexa 488– and Alexa 546–conjugated goat anti–mouse or goat anti–rabbit secondary antibodies (1/1,000 diluted). Bar, 10 μm.
Figure 6.
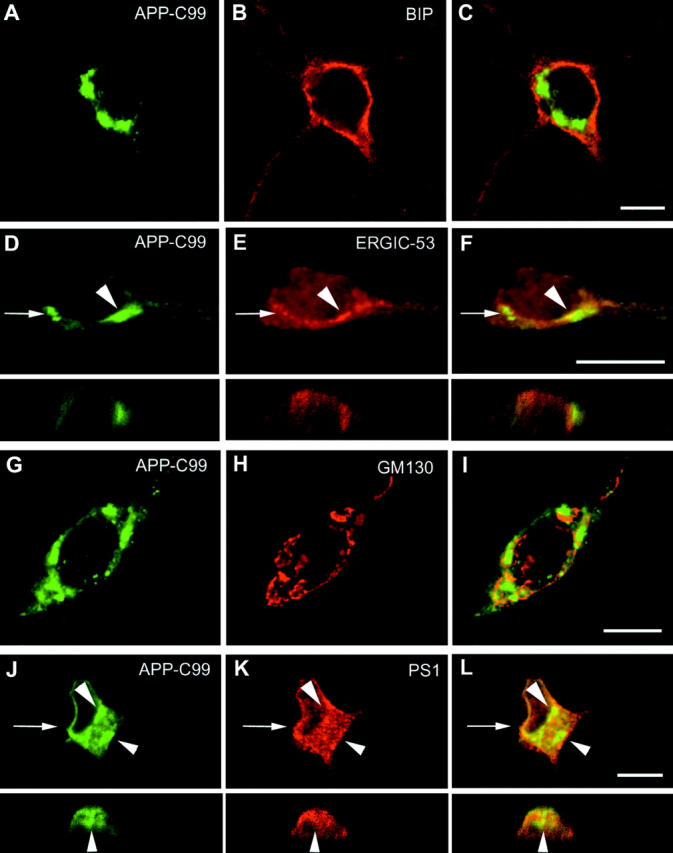
APP-C99 does not colocalize with PS1 and is essentially present in a BIP, ERGIC-53, and GM130 negative compartment. SFV-APP-C99 transduced hippocampal neurons were fixed and the subcellular localization of APP-C99 (A and G with pAb 6687; D and J with mAb 3D6) was compared with established marker proteins of the ER (BIP in B), the intermediate compartment (ERGIC-53 in E), the Golgi apparatus (GM130 in H), and finally with PS1 (K). C, F, I, and L show merged pictures. For D–F and J–L, the arrows point to the position of the corresponding vertical sections. Immunodetection was done as in Fig. 6. APP-C99 immunoreactivity was mainly concentrated in discrete spots that did not colocalize with the ER (C for BIP and arrowheads in L for PS1), and the Golgi region (merged panel I). Occasionally minor colocalization with ERGIC-53 could be observed (arrowheads in D–F). Bar, 10 μm.
To test the hypothesis that a component of a more downstream subcellular compartment is needed to process APP-C99-KK in the ER, we treated neurons expressing APP-C99 or APP-C99-KK with brefeldin A (BFA), a drug that causes a rapid redistribution of the Golgi apparatus into the ER (Lippincott-Schwartz et al., 1989). This treatment resulted in a drastic reduction in APP-C99–derived Aβ secretion, whereas no difference was observed in the case of APP-C99-KK (Fig. 7 A). However, addition of BFA resulted in a strong increase in intracellular Aβ generated from APP-C99-KK, towards levels indistinguishable from those obtained in BFA-treated APP-C99–transduced cells. This suggests that some unknown factors required for γ-secretase activity are redistributed from the Golgi region to the ER, which appears to be sufficient to partially restore Aβ production from APP-C99-KK. It has been suggested that Aβ generated in the endoplasmic reticulum consists mainly of Aβ ending at residue 42 (Chyung et al., 1997; Cook et al., 1997; Hartmann et al., 1997; Wild-Bode et al., 1997). Remarkably, although we detect little intracellular Aβ under our experimental conditions (Fig. 7, A and B), treatment with BFA induces the generation of Aβ peptides, ending at residue 40 as well as at residue 42 (Fig. 7 B).
Discussion
Previous studies (Koo and Squazzo, 1994; Hartmann et al., 1997; Tienari et al., 1997) have shown that Aβ can be generated at the cell surface and in the endosomes of neurons, where little, if any, PS1 is present (Annaert et al., 1999). The first aim of the current study was to investigate to what extent γ-secretase activity in these subcellular compartments depends indeed on PS1. To this end we expressed APP-Δct and APP-LDL, two previously characterized trafficking mutants of APP (Peraus et al., 1997; Tienari et al., 1997; Annaert et al., 1999), in neurons. APP-Δct lacks the endocytosis signals in the cytoplasmic domain of APP and is therefore only slowly internalized. Compared with APP-WT, APP-Δct is processed mainly by an α-secretase activity at the cell surface (Sisodia, 1992; Tienari et al., 1997; Annaert et al., 1999). This results in the replacement of Aβ production by a roughly equal production of p3, the product of consecutive α- and γ-secretase activity (Figs. 2 and 3), confirming that the APP-Δct construct indeed behaves as predicted. Therefore, p3 production from this substrate reflects mainly γ-secretase cleavage of the α-APPCTF at the cell surface. Expression of this construct in PS1-deficient neurons resulted in a strong decrease of p3, demonstrating that γ-secretase cleavage at the cell surface depends on PS1 (Fig. 3). Since αAPPs secretion is not affected in the absence of PS1 (Fig. 3), we rule out the possibility that the decreased p3 production is caused by decreased α-secretase activity in these PS1-deficient cells.
APP-LDL expression selectively increased Aβ40 secretion, as predicted from its preferential localization in the endosomes (Koo and Squazzo, 1994; Peraus et al., 1997; Perez et al., 1999). Again, in the absence of PS1, Aβ generation from APP-LDL strongly decreased, demonstrating that γ-secretase processing of APP in the endosomes depends on PS1. It should be noted that we have not been able to detect any endogenous PS1 immunoreactivity at the cell surface or in the endosomes of the neuronal cells used in the current study (Annaert et al., 1999).
We next investigated whether we could increase γ-secretase processing of APP in the ER and the intermediate compartment, which contain the bulk of PS1 immunoreactivity in these neurons. Although several authors have provided evidence that the ER is a production site of this peptide, in particular Aβ42 (Chyung et al., 1997; Cook et al., 1997; Hartmann et al., 1997; Wild-Bode et al., 1997), we and others have not been able to confirm these observations (Annaert et al., 1999; Iwata et al., 2001; Maltese et al., 2001). As shown in Fig. 2, a severe decrease in both Aβ40 and Aβ42 secretion is observed when APP is retained in the ER by means of the double lysine retention motif (APP-KK). Since the retention of APP in the ER implies that cleavage by α- and β-secretase (residing in the TGN, cell surface, and endosomes) becomes strongly compromised, and since proteolytic trimming of the APP ectodomain is a prerequisite for further γ-secretase processing (Struhl and Adachi, 2000), it seems logical that the APP-KK mutation results in a strongly decreased γ-secretase cleavage and inhibition of Aβ generation. It is by consequence difficult to interpret the data obtained with APP-KK in regard to γ-secretase sensitivity. Therefore, we generated the APP-C99 construct, which is a direct substrate for γ-secretase (Lichtenthaler et al., 1999), and added to it the double lysine ER retention motif. As shown in Fig. 5, this construct is indeed retained in the ER, where it codistributes abundantly with PS1. Although we expected increased Aβ production, no significant amount of Aβ could be precipitated from the wild-type neurons (Fig. 4; Iwata et al., 2001; Maltese et al., 2001). This result was even more significant, as neurons expressing APP-C99 secreted about seven times more total Aβ, or specifically Aβ42, than those expressing APP-WT. The possibility that the Aβ generated from APP-C99-KK is retained in the cells was ruled out, since no significant accumulation of cells associated Aβ could be demonstrated (Fig. 7 A, lane 9, and B, lanes 2 and 5)The possibility that the KK motif directly interfered with γ-secretase cleavage was ruled out by two observations. First, it is clear from the results with APP-Δct that the cytoplasmic domain of APP is not involved in the processing of APP by γ-secretase. Second, treatment of the cell cultures with BFA could restore the processing of C99-KK, clearly demonstrating that the inhibitory effect of the KK mutations is a trans, and not a cis phenomenon (Fig. 7). It should be pointed out here that PS1 in the ER is already proteolytically processed by the “presenilinase” and present as PS1-CTF and PS1-NTF (Annaert et al., 1999), thus in the putative active conformation. It follows from these data that the simple colocalization of a substrate (APP-C99-KK) with its putative protease (PS1) is not sufficient for proteolytic cleavage.
We conclude that Aβ production, including Aβ42, can not occur directly in the ER and requires at least one factor derived from a post-ER compartment. This conclusion is corroborated by two important pieces of evidence from the current work. First, APP-C99, which is a good substrate for γ-secretase, is mainly localized in a PS1-, BIP-, and ERGIC53-negative subcellular compartment (Fig. 6). Although the characterization of this compartment requires further experimentation, this result suggests that at least the proteolytic cleavage of the APP-C99 substrate occurs in a non-ER compartment. The second, more important argument comes from the experiments using BFA. BFA is a drug that causes the redistribution of post-ER compartments into the ER (Lippincott-Schwartz et al., 1989). Treatment with this drug was sufficient to restore partially γ-secretase processing of APP-C99-KK proving that at least one non-ER component is needed to activate γ-secretase in the ER (Fig. 7). BFA treatment results in γ-secretase processing of APP at position Aβ42, something which could possibly be anticipated based on current knowledge (Chyung et al., 1997; Cook et al., 1997; Hartmann et al., 1997; Wild-Bode et al., 1997). Surprisingly, however, cleavage at position Aβ40 is also observed (Fig. 7 B), which is believed to occur mainly in the late compartments of the secretory and in the endosomal limbs of the subcellular trafficking pathways (Koo and Squazzo, 1994; Hartmann et al., 1997; Tienari et al., 1997).
Summarizing, our data indicate that efficient γ-secretase cleavage of APP can occur in compartments that contain little, if any, PS1, and that no or little γ-secretase activity is observed in compartments where abundant (maturated) PS1 is residing. This is a surprising result since most of the data currently available imply that PS1 is closely involved in γ-secretase type enzymatic activity (De Strooper et al., 1998, 1999; Steiner et al., 1999, 2000; Struhl and Greenwald, 1999; Wolfe et al., 1999; Esler et al., 2000; Herreman et al., 2000; Li et al., 2000; Zhang et al., 2000). The spatial paradox between PS1 localization and γ-secretase activity can be explained by two hypotheses: (a) the minute amounts of PS1 in post–cis-Golgi compartments are the active enzymes. This implies that in addition to activation by presenilinase, additional proteins from these compartments are needed to make PS1 enzymatically active; and (b) PS1 is actually not the γ-secretase but acts, probably in concert with other proteins in the ER and intermediate compartment, to dispatch APP or γ-secretase to the subcellular compartment where cleavage will occur. In this hypothesis, BFA treatment results in the relocalization of the catalytic subunit(s) of γ-secretase to the ER.
In support for the first hypothesis, evidence for the presence of minute amounts of PS1 in other compartments than the ER has been provided. In some studies, the specificity of the antibodies used can be questioned (Dewji and Singer, 1997), but in polarized cell types like MDCK cells (Georgakopoulos et al., 1999) or in specialized cell membranes like growth cones or lamellipodia (Schwarzman et al., 1999), some PS1 seems to be located near or at the cell surface. Ray et al. (1999) provided evidence that PS1 can travel in complex with Notch to the cell surface, albeit that this conclusion was based on a single type of experimental approach, i.e., cell surface biotinylation. Furthermore, the hypothesis that PS travels together with its substrates from the ER to the cell surface and becomes activated there is probably not tenable if one takes into account that the relative amount of PS1 to its substrates is extremely low (Thinakaran et al., 1998). If PS1 needs to accompany every substrate that it will cleave from the ER to the cell surface, it becomes, given the limited amounts of PS available in any cell type, difficult to understand why strong overexpression of APP does not saturate the system and does not lead to inefficient γ-secretase processing.
Although it remains impossible to exclude that minute amounts of PS1 in the endosomes or at the cell surface are the active enzymes, it is clear that this hypothesis raises several new questions that need to be addressed. In addition, the function of the “inactive” pool of PS1 in the ER also remains to be explained. From the BFA experiments it follows that at least one other component of a non-ER compartment is needed to obtain activated PS1. This component is not the elusive protease called “presenilinase” responsible for PS maturation, and most probably not nicastrin, since this protein is present in the ER (Yu et al., 2000).
The second hypothesis, that PS1 is needed for the correct trafficking of γ-secretase and its substrates, finds some theoretical support in the analogy with the sterol regulatory element–binding protein (SREBP)–SCREB cleavage–activating protein (SCAP) complex and the regulation of the proteolytical cleavage of SREBP (Brown et al., 2000). SCAP is, like PS1, an ER resident, multitransmembrane domain–containing protein and regulates the trafficking of the membrane-bound transcription factor SREBP to post-ER compartments. Upon cholesterol depletion, SREBP travels to the cis-Golgi, where a site 1 protease cleaves SREBP in its luminal domain. The remaining NH2-terminal, membrane-bound fragment then becomes a substrate for a site 2 protease that cleaves in the transmembrane domain of SREBP (Rawson et al., 1997; Nohturfft et al., 1999). The coupling of vesicular protein transport and proteolysis provides a stringent control on the activation of the system and it could be envisaged that PS1, like SCAP, provides a similar control on APP (and Notch) processing. However, it should be pointed out that in case of SREBP processing, SCAP regulates its luminal cleavage and that the site 2 intramembraneous cleavage occurs by default. In case of PS1, its role is limited to the regulation of the intramembraneous γ-secretase cleavage (De Strooper et al., 1998).
In conclusion, our data indicate a complex relationship between APP trafficking and its processing by γ-secretase. Moreover, they directly question the exact role of PS1 in γ-secretase processing and demonstrate that at least one cofactor (in the first hypothesis) or the protease (in the second hypothesis) is located in a compartment downstream of the ER. In the future, reconstitution of γ-secretase processing of APP, by mixing extracts of purified ER fractions and post-ER compartments, should allow us to further define at a molecular level this highly intriguing proteolytic system.
Materials and methods
Neuronal cell culture
Primary cultures of mixed cortical neurons derived from wild-type and PS1 knock-out littermates were generated as described previously (De Strooper et al., 1998). In brief, total brain of 14-d-old embryos was dissected in HBSS medium (GIBCO BRL), trypsinized, and plated on dishes (Nunc) precoated with poly-l-lysine (Sigma-Aldrich). Cultures were maintained in neurobasal medium (GIBCO BRL) with B27 supplement (GIBCO BRL) and 5 μM cytosine arabinoside to prevent glial cell proliferation.
Semliki forest virus (SFV) constructs
The following modifications were made to the cDNA coding for human APP695 (Fig. 1): (a) deletion of the last 43 amino acids of the cytoplasmic tail (APP-Δct) as described in Tienari et al. (1996); (b) addition of a di-lysine motif (QM mutated to KK, APP-KK) by site-directed mutagenesis (Stratagene); (c) exchange of its cytoplasmic domain with that of the LDL receptor (APP-LDL); (d) full deletion of the APP ectodomain until the Asp1 amino acid of the amyloid peptide region (β-secretase site). An additional AspAla (DA) was cloned by site-directed mutagenesis (Stratagene) between the last amino acid of the signal sequence (Ala) and the first of the Aβ region (Lichtenthaler et al., 1999) (APP-C99). Site-directed mutagenesis was used to add the di-lysine motif (APP-C99-KK). Recombinant SFV containing the APP mutants were generated as described previously (De Strooper et al., 1995).
Transduction and metabolic labeling of neuronal cell cultures
Recombinant SFV was diluted 10-fold in neurobasal medium and added to the culture dishes. After a 1-h incubation at 37°C, fresh neurobasal medium was added. After 2 h, cells were metabolically labeled in methionine-free MEM (GIBCO BRL) supplemented with B27 and 100 μCi/ml [35S]methionine (ICN Biomedicals). 10 μM BFA (Epicentre) was added when indicated. After 4 h, conditioned medium and cells were recovered. Cells were lysed in IP buffer (20 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1% Triton X-100, 1% sodium deoxycholate and 0.1% SDS) and cleared by centrifugation. Labeled protein was immunoprecipitated using 25 μl protein G Sepharose and specific antibodies as indicated below and in Fig. 1. Immunoprecipitates were finally solubilized in 25 μl Nu-Page sample buffer (Invitrogen) and electrophoresed on 4–12% or 10% acrylamide NuPage Bis-Tris gels under reducing conditions and MES in the running buffer (Invitrogen). Results were analyzed using a PhosphorImager (Molecular Dynamics) and ImagQuaNT4.1. All data were normalized to the value obtained for the corresponding APP holoform to normalize for variations between culture dishes. The following antibodies were used (Fig. 1). Rabbit pAb B7 recognizes the first 17 amino acids of Aβ, B11 the last 20 amino acids of the APP COOH-terminal domain (De Strooper et al., 1995), goat pAb 207 the APP ectodomain (provided by Dr. M. Savage, Cephalon, Inc., West Chester, PA), mAb 4G8 (Senetek) amino acids 17 to 24 of Aβ, and mab W0-2 the NH2-terminal region of Aβ (provided by Dr. Tobias Hartmann and Konrad Beyreuther, University of Heidelberg, Heidelberg, Germany) (Ida et al., 1996). Antibodies FCA40 and FCA42 specifically precipitate Aβ peptides ending at residue 40 (γ40) or at 42 (γ42) and were provided by Dr. F. Checler (Institut de Pharmacologie Moléculaire et Cellulaire, Valbonne, France) (Barelli et al., 1997).
Quantification of the Aβ42 peptide by ELISA
Levels of Aβ42 in conditioned media and cell lysates were quantified by a sandwich ELISA test (De Strooper et al., 1998), according to published protocols (Vanderstichele et al., 2000). In brief, samples were dried by speed vacuum (Savant), resuspended in 300 μl of sample diluent, and incubated on 96-well ELISA plates precoated with mAb 3D6 against Aβ. After washing, samples were incubated with a biotin-labeled anti-Aβ42 antibody mAb 21F12 that only recognizes the final two amino acids of the Aβ42 sequence, followed by streptavidine-HRP. After adding the HRP substrate, samples were measured spectrophotometrically using a Victor2 (Wallac) with a 450-nm filter. The Aβ42 concentration in the samples was calculated based on the Aβ42 standards sigmoid curve equation, and using Prism 3.0 (GraphPad Software).
Confocal microscopy
Hippocampal neurons grown on poly-l-lysine–coated glass coverslips in the presence of a glial feeder layer (Goslin and Banker, 1991; De Strooper et al., 1995) were transduced with SFV-APP-C99 or -C99KK. 4 h after transduction, cycloheximide (100 μg/ml) was added to block further protein synthesis. After 6 h, neurons were fixed in 120 mM phosphate buffer containing 4% paraformaldehyde (pH 7.3) and 4% sucrose, permeabilized in ice-cold methanol and acetone, and immunostained (Annaert et al., 1999). Recombinant APP-C99 was detected with pAb 6687 (gift of Dr. C. Haass, University of München, Germany) or mAb 3D6 (gift of Dr. Seubert, Elan Pharmaceuticals, San Francisco, CA). mAbs to BIP were purchased from Sigma. PAb B17.2 was used to detect endogenous PS1 (Annaert et al., 1999). The intermediate compartment and Golgi region were identified using anti-ERGIC-53, provided by Dr. J. Saraste (University of Bergen, Norway), and anti-GM130 (Transduction Laboratories), respectively. Alexa 488– and Alexa 546–conjugated secondary antibodies were from Molecular Probes. BioRad MRC1024 confocal microscope and Adobe Photoshop® 5.2 were used for final processing (Annaert et al., 1999).
Acknowledgments
The authors acknowledge Dr. P. Seubert (Elan Pharmaceuticals), Dr. K. Beyreuther and Dr. T. Hartmann (University of Heidelberg, Germany), Dr. C. Haass (University of München, Germany), Dr. F. Checler, Dr. M. Savage (Cephalon, West Chester, PA), and Dr. J. Saraste (University of Bergen, Bergen, Norway) for providing us with their valuable antibodies. We also thank Dr. P. Tienari (EMBL, Heidelberg, Germany) for providing us with the APP-ΔCT construct.
This work was financially supported by the F.W.O.-Vlaanderen, the K.U. Leuven, the Flanders interuniversity Institute for Biotechnology, and the Bayer Alzheimer Disease Research Network (BARN). P. Cupers and W. Annaert are holders of a Fonds voor Wetenschappelijik Onderzoek postdoctoral fellowship.
Footnotes
Abbreviations used in this paper: Aβ, amyloid peptide; APP, amyloid precursor protein; BFA, brefeldin A; CTF, COOH-terminal fragment; KK, dilysine; PAb, polyclonal antibody; PS, presenilin; SCAP, SCREB cleavage–activating protein; SFV, semliki forest virus; SCREB, sterol regulatory element–binding protein.
References
- Annaert, W., and B. De Strooper. 1999. Presenilins: molecular switches between proteolysis and signal transduction. Trends Neurosci. 22:439–443. [DOI] [PubMed] [Google Scholar]
- Annaert, W.G., L. Levesque, K. Craessaerts, I. Dierinck, G. Snellings, D. Westaway, P.S. George-Hyslop, B. Cordell, P. Fraser, and B. De Strooper. 1999. Presenilin 1 controls γ-secretase processing of amyloid precursor protein in pre-golgi compartments of hippocampal neurons. J. Cell Biol. 147:277–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barelli, H., A. Lebeau, J. Vizzanova, P. Delaere, N. Chevalier, C. Drouot, P. Marambaud, K. Ancolio, J.D. Buxbaum, O. Khorkova, et al. 1997. Characterization of new polyclonal antibodies specific for 40 and 42 amino acid long amyloid β peptides: their use to examine the cell biology of presenilins and the immunohistochemistry of sporadic Alzheimer's disease and cerebral amyloid angiopathy cases. Mol. Med. 3: 695–707. [PMC free article] [PubMed] [Google Scholar]
- Brou, C., F. Logeat, N. Gupta, C. Bessia, O. LeBail, J.R. Doedens, A. Cumano, P. Roux, R.A. Black, and A. Israel. 2000. A novel proteolytic cleavage involved in Notch signaling: the role of the disintegrin-metalloprotease TACE. Mol. Cell. 5:207–216. [DOI] [PubMed] [Google Scholar]
- Brown, M.S., J. Ye, R.B. Rawson, and J.L. Goldstein. 2000. Regulated intramembrane proteolysis: a control mechanism conserved from bacteria to humans. Cell. 100:391–398. [DOI] [PubMed] [Google Scholar]
- Buxbaum, J.D., K.N. Liu, Y. Luo, J.L. Slack, K.L. Stocking, J.J. Peschon, R.S. Johnson, B.J. Castner, D.P. Cerretti, and R.A. Black. 1998. Evidence that tumor necrosis factor α converting enzyme is involved in regulated α-secretase cleavage of the Alzheimer amyloid protein precursor. J. Biol. Chem. 273:27765–27767. [DOI] [PubMed] [Google Scholar]
- Chyung, A.S.C., B.D. Greenberg, D.G. Cook, R.W. Doms, and V.M. Lee. 1997. Novel β-secretase cleavage of β-amyloid precursor protein in the endoplasmic reticulum/intermediate compartment of NT2N cells. J. Cell Biol. 138:671–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook, D.G., M.S. Forman, J.C. Sung, S. Leight, D.L. Kolson, T. Iwatsubo, V.M. Lee, and R.W. Doms. 1997. Alzheimer's A β(1-42) is generated in the endoplasmic reticulum/intermediate compartment of NT2N cells. Nat. Med. 3:1021–1023. [DOI] [PubMed] [Google Scholar]
- Creemers, J.W., D.I. Dominguez, E. Plets, L. Serneels, N.A. Taylor, G. Multhaup, K. Craessaerts, W. Annaert, and B. De Strooper. 2000. Processing of β-secretase (Bace) by furin and other members of the proprotein convertase family. J. Biol. Chem. 8:8. [DOI] [PubMed] [Google Scholar]
- Culvenor, J.G., F. Maher, G. Evin, F. Malchiodi-Albedi, R. Cappai, J.R. Underwood, J.B. Davis, E.H. Karran, G.W. Roberts, K. Beyreuther, and C.L. Masters. 1997. Alzheimer's disease-associated presenilin 1 in neuronal cells: evidence for localization to the endoplasmic reticulum-Golgi intermediate compartment. J. Neurosci. Res. 49:719–731. [DOI] [PubMed] [Google Scholar]
- De Strooper, B., M. Simons, G. Multhaup, F. Van Leuven, K. Beyreuther, and C.G. Dotti. 1995. Production of intracellular amyloid-containing fragments in hippocampal neurons expressing human amyloid precursor protein and protection against amyloidogenesis by subtle amino acid substitutions in the rodent sequence. EMBO J. 14:4932–4938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Strooper, B., P. Saftig, K. Craessaerts, H. Vanderstichele, G. Guhde, W. Annaert, K. Von Figura, and F. Van Leuven. 1998. Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature. 391:387–390. [DOI] [PubMed] [Google Scholar]
- De Strooper, B., W. Annaert, P. Cupers, P. Saftig, K. Craessaerts, J.S. Mumm, E.H. Schroeter, V. Schrijvers, M.S. Wolfe, W.J. Ray, A. Goate, and R. Kopan. 1999. A presenilin-1-dependent γ-secretase-like protease mediates release of Notch intracellular domain. Nature. 398:518–522. [DOI] [PubMed] [Google Scholar]
- Dewji, N.N., and S.J. Singer. 1997. Cell surface expression of the Alzheimer disease-related presenilin proteins. Proc. Natl. Acad. Sci. USA. 94:9926–9931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esler, W.P., W.T. Kimberly, B.L. Ostaszewski, T.S. Diehl, C.L. Moore, J.Y. Tsai, T. Rahmati, W. Xia, D.J. Selkoe, and M.S. Wolfe. 2000. Transition-state analogue inhibitors of γ-secretase bind directly to presenilin-1. Nat. Cell Biol. 2:428–434. [DOI] [PubMed] [Google Scholar]
- Gaynor, E.C., S. te Heesen, T.R. Graham, M. Aebi, and S.D. Emr. 1994. Signal-mediated retrieval of a membrane protein from the Golgi to the ER in yeast. J. Cell Biol. 127:653–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgakopoulos, A., P. Marambaud, S. Efthimiopoulos, J. Shioi, W. Cui, H.C. Li, M. Schutte, R. Gordon, G.R. Holstein, G. Martinelli, P. Mehta, V.L. Friedrich, Jr., and N.K. Robakis. 1999. Presenilin-1 forms complexes with the cadherin/catenin cell-cell adhesion system and is recruited to intercellular and synaptic contacts. Mol. Cell. 4:893–902. [DOI] [PubMed] [Google Scholar]
- Goslin, K., and G. Banker. 1991. Rat hippocampal neurons in low-density culture. Culturing Nerve Cells. G. Banker and K. Goslin, editors. MIT Press, Cambridge, MA. 251–281.
- Hartmann, T., S.C. Bieger, B. Bruhl, P.J. Tienari, N. Ida, D. Allsop, G.W. Roberts, C.L. Masters, C.G. Dotti, K. Unsicker, and K. Beyreuther. 1997. Distinct sites of intracellular production for Alzheimer's disease A β40/42 amyloid peptides. Nat. Med. 3:1016–1020. [DOI] [PubMed] [Google Scholar]
- Herreman, A., L. Serneels, W. Annaert, D. Collen, L. Schoonjans, and B. De Strooper. 2000. Total inactivation of γ-secretase activity in presenilin-deficient embryonic stem cells. Nat. Cell Biol. 2:461–462. [DOI] [PubMed] [Google Scholar]
- Ida, N., T. Hartmann, J. Pantel, J. Schroder, R. Zerfass, H. Forstl, R. Sandbrink, C.L. Masters, and K. Beyreuther. 1996. Analysis of heterogeneous A4 peptides in human cerebrospinal fluid and blood by a newly developed sensitive Western blot assay. J. Biol. Chem. 271:22908–22914. [DOI] [PubMed] [Google Scholar]
- Iwata, H., T. Tomita, K. Maruyama, and T. Iwatsubo. 2001. Subcellular compartment and molecular subdomain of β-amyloid precursor protein relevant to the Abeta 42-promoting effects of Alzheimer mutant presenilin 2. J. Biol. Chem. 216:21678–21685. [DOI] [PubMed] [Google Scholar]
- Jackson, M.R., T. Nilsson, and P.A. Peterson. 1993. Retrieval of transmembrane proteins to the endoplasmic reticulum. J. Cell Biol. 121:317–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, S.H., J.J. Lah, G. Thinakaran, A. Levey, and S.S. Sisodia. 2000. Subcellular localization of presenilins: association with a unique membrane pool in cultured cells. Neurobiol. Dis. 7:99–117. [DOI] [PubMed] [Google Scholar]
- Koike, H., S. Tomioka, H. Sorimachi, T.C. Saido, K. Maruyama, A. Okuyama, A. Fujisawa-Sehara, S. Ohno, K. Suzuki, and S. Ishiura. 1999. Membrane-anchored metalloprotease MDC9 has an α-secretase activity responsible for processing the amyloid precursor protein. Biochem. J. 343:371–375. [PMC free article] [PubMed] [Google Scholar]
- Koo, E.H., and S.L. Squazzo. 1994. Evidence that production and release of amyloid β-protein involves the endocytic pathway. J. Biol. Chem. 269:17386–17389. [PubMed] [Google Scholar]
- Lammich, S., E. Kojro, R. Postina, S. Gilbert, R. Pfeiffer, M. Jasionowski, C. Haass, and F. Fahrenholz. 1999. Constitutive and regulated α-secretase cleavage of Alzheimer's amyloid precursor protein by a disintegrin metalloprotease. Proc. Natl. Acad. Sci. USA. 96:3922–3927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Y.M., M. Xu, M.T. Lai, Q. Huang, J.L. Castro, J. DiMuzio-Mower, T. Harrison, C. Lellis, A. Nadin, J.G. Neduvelil, et al. 2000. Photoactivated γ-secretase inhibitors directed to the active site covalently label presenilin 1. Nature. 405:689–694. [DOI] [PubMed] [Google Scholar]
- Lichtenthaler, S.F., G. Multhaup, C.L. Masters, and K. Beyreuther. 1999. A novel substrate for analyzing Alzheimer's disease γ-secretase. FEBS Lett. 453:288–292. [DOI] [PubMed] [Google Scholar]
- Lippincott-Schwartz, J., L.C. Yuan, J.S. Bonifacino, and R.D. Klausner. 1989. Rapid redistribution of Golgi proteins into the ER in cells treated with brefeldin A: evidence for membrane cycling from Golgi to ER. Cell. 56:801–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maltese, W.A., S. Wilson, Y. Tan, S. Suomensaari, S. Sinha, R. Barbour, and L. McConlogue. 2001. Retention of the Alzheimer's amyloid precursor protein fragment C99 in the endoplasmic reticulum prevents formation of amyloid β-peptide. J. Biol. Chem. 23:20267–20279. [DOI] [PubMed] [Google Scholar]
- Mumm, J.S., E.H. Schroeter, M.T. Saxena, A. Griesemer, X. Tian, D.J. Pan, W.J. Ray, and R. Kopan. 2000. A ligand-induced extracellular cleavage regulates γ-secretase-like proteolytic activation of Notch1. Mol. Cell. 5:197–206. [DOI] [PubMed] [Google Scholar]
- Naruse, S., G. Thinakaran, J.J. Luo, J.W. Kusiak, T. Tomita, T. Iwatsubo, X. Qian, D.D. Ginty, D.L. Price, D.R. Borchelt, P.C. Wong, and S.S. Sisodia. 1998. Effects of PS1 deficiency on membrane protein trafficking in neurons. Neuron. 21:1213–1221. [DOI] [PubMed] [Google Scholar]
- Nohturfft, A., R.A. DeBose-Boyd, S. Scheek, J.L. Goldstein, and M.S. Brown. 1999. Sterols regulate cycling of SREBP cleavage-activating protein (SCAP) between endoplasmic reticulum and Golgi. Proc. Natl. Acad. Sci. USA. 96:11235–11240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peraus, G.C., C.L. Masters, and K. Beyreuther. 1997. Late compartments of amyloid precursor protein transport in SY5Y cells are involved in β-amyloid secretion. J. Neurosci. 17:7714–7724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez, R.G., S. Soriano, J.D. Hayes, B. Ostaszewski, W. Xia, D.J. Selkoe, X. Chen, G.B. Stokin, and E.H. Koo. 1999. Mutagenesis identifies new signals for β-amyloid precursor protein endocytosis, turnover, and the generation of secreted fragments, including aβ42. J. Biol. Chem. 274:18851–18856. [DOI] [PubMed] [Google Scholar]
- Rawson, R.B., N.G. Zelenski, D. Nijhawan, J. Ye, J. Sakai, M.T. Hasan, T.Y. Chang, M.S. Brown, and J.L. Goldstein. 1997. Complementation cloning of S2P, a gene encoding a putative metalloprotease required for intramembrane cleavage of SREBPs. Mol. Cell. 1:47–57. [DOI] [PubMed] [Google Scholar]
- Ray, W.J., M. Yao, J. Mumm, E.H. Schroeter, P. Saftig, M. Wolfe, D.J. Selkoe, R. Kopan, and A.M. Goate. 1999. Cell surface presenilin-1 participates in the γ-secretase-like proteolysis of notch. J. Biol. Chem. 274:36801–36807. [DOI] [PubMed] [Google Scholar]
- Schwarzman, A.L., N. Singh, M. Tsiper, L. Gregori, A. Dranovsky, M.P. Vitek, C.G. Glabe, P.H. St George-Hyslop, and D. Goldgaber. 1999. Endogenous presenilin 1 redistributes to the surface of lamellipodia upon adhesion of jurkat cells to a collagen matrix. Proc. Natl. Acad. Sci. USA. 96:7932–7937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seiffert, D., J.D. Bradley, C.M. Rominger, D.H. Rominger, F. Yang, J.E. Meredith, Jr., Q. Wang, A.H. Roach, L.A. Thompson, S.M. Spitz, et al. 2000. Presenilin-1 and -2 are molecular targets for γ-secretase inhibitors. J. Biol. Chem. 275:34086–34091. [DOI] [PubMed] [Google Scholar]
- Selkoe, D.J. 1999. Translating cell biology into therapeutic advances in Alzheimer's disease. Nature. 399:A23–A31. [DOI] [PubMed] [Google Scholar]
- Sisodia, S.S. 1992. β-amyloid precursor protein cleavage by a membrane-bound protease. Proc. Natl. Acad. Sci. USA. 89:6075–6079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song, W., P. Nadeau, M. Yuan, X. Yang, J. Shen, and B.A. Yankner. 1999. Proteolytic release and nuclear translocation of Notch-1 are induced by presenilin-1 and impaired by pathogenic presenilin-1 mutations. Proc. Natl. Acad. Sci. USA. 96:6959–6963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner, H., K. Duff, A. Capell, H. Romig, M.G. Grim, S. Lincoln, J. Hardy, X. Yu, M. Picciano, K. Fechteler, et al. 1999. A loss of function mutation of presenilin-2 interferes with amyloid β-peptide production and notch signaling. J. Biol. Chem. 274:28669–28673. [DOI] [PubMed] [Google Scholar]
- Steiner, H., M. Kostka, H. Romig, G. Basset, B. Pesold, J. Hardy, A. Capell, L. Meyn, M.L. Grim, R. Baumeister, K. Fechteler, and C. Haass. 2000. Glycine 384 is required for presenilin-1 function and is conserved in bacterial polytopic aspartyl proteases. Nat. Cell Biol. 2:848–851. [DOI] [PubMed] [Google Scholar]
- Struhl, G., and A. Adachi. 2000. Requirements for presenilin-dependent cleavage of notch and other transmembrane proteins. Mol. Cell. 6:625–636. [DOI] [PubMed] [Google Scholar]
- Struhl, G., and I. Greenwald. 1999. Presenilin is required for activity and nuclear access of Notch in Drosophila. Nature. 398:522–525. [DOI] [PubMed] [Google Scholar]
- Thinakaran, G., J.B. Regard, C.M. Bouton, C.L. Harris, D.L. Price, D.R. Borchelt, and S.S. Sisodia. 1998. Stable association of presenilin derivatives and absence of presenilin interactions with APP. Neurobiol. Dis. 4:438–453. [DOI] [PubMed] [Google Scholar]
- Tienari, P.J., B. De Strooper, E. Ikonen, M. Simons, A. Weidemann, C. Czech, T. Hartmann, N. Ida, G. Multhaup, C.L. Masters, F. Van Leuven, K. Beyreuther, and C.G. Dotti. 1996. The β-amyloid domain is essential for axonal sorting of amyloid precursor protein. EMBO J. 15:5218–5229. [PMC free article] [PubMed] [Google Scholar]
- Tienari, P.J., N. Ida, E. Ikonen, M. Simons, A. Weidemann, G. Multhaup, C.L. Masters, C.G. Dotti, and K. Beyreuther. 1997. Intracellular and secreted Alzheimer β-amyloid species are generated by distinct mechanisms in cultured hippocampal neurons. Proc. Natl. Acad. Sci. USA. 94:4125–4130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderstichele, H., E. Van Kerschaver, C. Hesse, P. Davidsson, M.A. Buyse, N. Andreasen, L. Minthon, A. Wallin, K. Blennow, and E. Vanmechelen. 2000. Standardization of measurement of β-amyloid(1-42) in cerebrospinal fluid and plasma. Amyloid. 7:245–258. [DOI] [PubMed] [Google Scholar]
- Vassar, R., B.D. Bennett, S. Babu-Khan, S. Kahn, E.A. Mendiaz, P. Denis, D.B. Teplow, S. Ross, P. Amarante, R. Loeloff, et al. 1999. β-secretase cleavage of Alzheimer's amyloid precursor protein by the transmembrane aspartic protease BACE. Science. 286:735–741. [DOI] [PubMed] [Google Scholar]
- Walter, J., A. Capell, J. Grunberg, B. Pesold, A. Schindzielorz, R. Prior, M.B. Podlisny, P. Fraser, P.S. Hyslop, D.J. Selkoe, and C. Haass. 1996. The Alzheimer's disease-associated presenilins are differentially phosphorylated proteins located predominantly within the endoplasmic reticulum. Mol. Med. 2:673–691. [PMC free article] [PubMed] [Google Scholar]
- Wild-Bode, C., T. Yamazaki, A. Capell, U. Leimer, H. Steiner, Y. Ihara, and C. Haass. 1997. Intracellular generation and accumulation of amyloid β-peptide terminating at amino acid 42. J. Biol. Chem. 272:16085–16088. [DOI] [PubMed] [Google Scholar]
- Wolfe, M.S., W. Xia, B.L. Ostaszewski, T.S. Diehl, W.T. Kimberly, and D.J. Selkoe. 1999. Two transmembrane aspartates in presenilin-1 required for presenelin endoproteolysis and γ-secretase activity. Nature. 398:513–517. [DOI] [PubMed] [Google Scholar]
- Yu, G., M. Nishimura, S. Arawaka, D. Levitan, L. Zhang, A. Tandon, Y.Q. Song, E. Rogaeva, F. Chen, T. Kawarai, et al. 2000. Nicastrin modulates presenilin-mediated notch/glp-1 signal transduction and βAPP processing. Nature. 407:48–54. [DOI] [PubMed] [Google Scholar]
- Zhang, Z., P. Nadeau, W. Song, D. Donoviel, M. Yuan, A. Bernstein, and B.A. Yankner. 2000. Presenilins are required for γ-secretase cleavage of β-APP and transmembrane cleavage of Notch-1. Nat. Cell Biol. 2:463–465. [DOI] [PubMed] [Google Scholar]