

Abstract
The ouabain-like sodium pump inhibitor in mammals (so-called “endogenous ouabain”) has been considered a subtle structural isomer of ouabain. Its structural investigation, however, has long been hindered by the paucity of sample material. Our recent purification of endogenous ouabain (3 μg) from bovine hypothalamus allowed the measurement of its 1H-NMR. The obtained spectrum as well as reexamination of past microscale structural studies on endogenous ouabain led us to identify the purified material as ouabain in an unusual manner. It turned out that the structural analysis had been complicated by a facile ouabain–borate complexation in borosilicate glassware. In retrospect, it is not surprising that the polyhydroxylated ouabain molecule serves as a polydentate ligand to inorganic species. In its physiological environment, ouabain may exist as some unknown complex. The chemical species giving rise to the reported biological activities of hypothalamic inhibitory factor preparations remain to be clarified.
Search for the endogenous ligands of Na+,K+-ATPase has been an enticing yet puzzling research subject for the last several decades (1–4). Putative endogenous sodium pump inhibitors have been detected many times in various mammalian tissues and plasma. However, purification, structural analysis, and physiological characterization of such compounds have been fraught with difficulties. Part of the reason for this problem is that the enzyme assays are susceptible to many nonspecific interferences, which sometimes lead to false-positive results (1). Recognition of this problem led to the employment of multiple assay systems, which greatly reduced the risk of falsely detecting putative physiologic Na+,K+-ATPase inhibitors. Nevertheless, progress is still hindered by the extreme paucity of material available from tissues.
Among the putative endogenous inhibitors of Na+,K+-ATPase are a group of compounds that are considered to be related to ouabain, a plant-origin cardiac glycoside (5). These compounds have been extracted from various animal tissues, and in several cases sufficient material, albeit submicrogram-to-low microgram quantity, was purified to allow further chemical and physiological characterizations. The molecular mass appeared to be identical to ouabain by MS, and the retention time of reversed-phase HPLC (RP-HPLC) was the same as ouabain. Moreover, two compounds, ouabain-like compound (OLC) from human plasma (6) and “adrexin C” from bovine adrenal (7), were indistinguishable from plant ouabain by multiple biochemical and physiological criteria.
On the other hand, a factor from bovine hypothalamus (hypothalamic inhibitory factor, HIF) showed different physiological profiles from ouabain in various assays, such as inhibition of ouabain-insensitive isoform (rat α1) of Na+,K+-ATPase (8), inhibition of sarcoplasmic reticulum Ca2+-ATPase, and inhibition of “backdoor” phosphorylation of purified Na+,K+-ATPase (9), binding kinetics to kidney epithelial cells and purified enzyme (10, 11), lipid bilayer permeability (11), and reversible inotropic effect on cardiac myocytes (12). For the structural characterization of HIF, further purification was carried out by using affinity chromatography combined with RP-HPLC (13). It turned out that the purified HIF was identical to ouabain by LC/MS. Furthermore, glycosidase treatment and acid hydrolysis showed HIF to be an α-l-rhamnoside, as is ouabain; also, des-rhamnosyl HIF, the aglycone, was indistinguishable from ouabagenin by LC/MS.
These findings led to attempts to differentiate HIF and plant ouabain by using nanogram-scale chemical derivatization. Naphthoylation of HIF (300 ng) followed by RP-HPLC showed that the major derivatization product from HIF eluted slightly earlier than ouabain pentanaphthoate, the major product from ouabain naphthoylation; moreover, CD spectroscopy revealed that this HIF derivative showed no distinct CD, whereas ouabain pentanaphthoate showed a clear positive CD couplet. Although the molecular ion peak of this HIF derivative was not clear, the product was described as HIF pentanaphthoate (13). Subsequent microscale derivatization of OLC (400 ng, gift from Upjohn Laboratories) also gave the same RP-HPLC and CD (“zero-CD”) profiles as those of HIF pentanaphthoate (14). This led to the conclusion that the ouabain-like sodium pump inhibitor is an ouabain isomer, often referred to as endogenous ouabain (2, 4, 5), even though the physiological discrepancies between HIF and other compounds still remained to be clarified (2): the term endogenous ouabain has been used to distinguish this compound from plant ouabain (2, 4, 5), although its true origin has not yet been clarified.
Based on the chromatographic and spectroscopic information from the naphthoylation studies on endogenous ouabain, a search for the isomer of ouabain pentanaphthoate with the same HPLC and CD (zero-CD) profile was initiated. Two lines of structural evidence, the fact that the HIF aglycone was indistinguishable from ouabagenin by LC/MS and that the sugar moiety of HIF was α-l-rhamnoside, suggested that HIF could be an α-l-rhamnoside position isomer of ouabain. This led to the computation of CD spectra of all possible sugar position isomers of ouabain pentanaphthoate and determination of the validity of calculations by preparing as many synthetic isomers as possible (15). The series of comparison between theoretical and experimental data demonstrated that theoretical CD calculation can fairly accurately reproduce experimental CD couplets of ouabain pentanaphthoate isomers despite their substantial conformational flexibility. However, the theoretically calculated 18 isomers and experimentally prepared 13 isomers showed that all searched isomers have intense positive couplets (15). With no conceivable candidate among sugar position isomers of ouabain, our study came to the point of reexamining the results obtained from the previous nanogram-scale naphthoylation of HIF and OLC (13, 14).
More recently, we succeeded in the purification of 3 μg of HIF (“3-μg HIF”) with a modified HIF purification protocol that omitted cation and anion exchangers and substituted an affinity column by using anti-digoxin antibody 26-10 (16, 17) for immobilized Na+,K+-ATPase to improve the recovery of HIF. The 1H-NMR of the purified HIF as well as the reexamination of the past nanogram-scale naphthoylation study led to identify this 3-μg HIF as ouabain in an unusual manner. The past microscale structural analyses on endogenous ouabain (13, 14) are reevaluated in light of our current findings, and the implications of the current findings with respect to endogenous ouabain research are discussed.
MATERIALS AND METHODS
Materials.
Ouabain octahydrate and sodium tetraborate decahydrate were from Acros Organics (Pittsburgh), and all other chemicals and HPLC solvents were from Aldrich. Anti-digoxin antibody 26-10 (16, 17) was a gift from M. N. Margolies at Harvard Medical School (Boston). Electrospray ionization (ESI) MS was measured by using Micromass Q-Tof at Micromass (Manchester, U.K.) and the Suntory Institute for Bioorganic Research (Osaka). NMR spectra [1H, correlated spectroscopy, heteronuclear multiple quantum correlation, heteronuclear multiple bond correlation, and rotating-frame Overhauser effect spectroscopy (ROESY)] were recorded on a Bruker DMX500 or a Bruker DRX300WB distortionless enhancement by polarization transfer (DEPT) [13C, 11B] spectrometer. NMR samples were dissolved in 10% acetonitrile-d3 in deuterium oxide. The methyl group of acetonitrile-d3 was used as an internal standard for the 1H- and 13C-NMR chemical shift. Boron trifluoride diethyl etherate (BF3⋅OEt2) was used as an external standard for the chemical shift of 11B-NMR. ROESY of ouabain borate was measured with a spin-lock time of 100 ms.
Anti-Digoxin Antibody 26-10 Affinity Column.
Cyanogen bromide-activated Sepharose 4B was suspended in 1 mM HCl. The gel was washed with the same HCl solution on a glass filter. After the HCl solution was suction-removed, the gel was immediately transferred to the solution of antibody 26-10 (16, 17), which had been dialyzed against 0.1 M sodium bicarbonate (pH 8.3) containing 0.5 M sodium chloride. The coupling reaction was done at 4°C with gentle shaking for 3 days. The obtained antibody–Sepharose resin was stored in 0.1 M PBS (pH 7.2) containing 0.1% sodium azide.
Purification of HIF (3-μg HIF).
Methanol extract of bovine hypothalamus was purified by gel filtration (Sephadex LH-20), and reversed-phase LC (Mitsubishi CHP20P gel) as reported (13). Na+,K+-ATPase inhibitory activity was monitored by the 86Rb+ uptake assay by using human erythrocytes (9). The dried material from the reversed-phase LC (CHP20P) was dissolved in 0.1 M PBS buffer (pH 7.2), loaded onto the antibody 26-10 affinity column. and incubated at 4°C overnight. The bound HIF was eluted with 40% methanol in water. The dried material was reconstituted in water and purified by semipreparative RP-HPLC (Waters μBondapak C18, 10 μm, 125Å, 19 × 150 mm, 0–30% acetonitrile, 6 ml/min). The purified material was subjected to RP-HPLC [Alltech Alltima C18, 5 μm, 4.6 × 250 mm, 5–30% acetonitrile containing 0.05% trifluoroacetic acid (TFA) over 20 min]. The elution was monitored by a Hewlett-Packard Photodiode Array Detector (Series 1100), and the peak with butenolide UV profile (λmax 220 nm) was collected into a disposable borosilicate glass tube (10 × 75 mm), and the solution was dried in vacuo. The purified HIF (3 μg), which was quantified by the UV absorption at 220 nm, was submitted to the 1H-NMR measurement, in which signal was averaged by overnight accumulation.
Preparation of Ouabain Tetrahedral-Borate.
Method A. Ouabain aqueous solution (1 μg or less in 100 μl of water) was placed in a new borosilicate vial (a disposable vial, 1 dram), kept at ambient temperature overnight, and lyophilized to give a white powder. ESIMS showed distinct peaks at m/z 633.24 (ouabain + 11BO-H + Na)+ and at 632.26 (ouabain + 10BO-H + Na)+. ESIMS/MS of the peak at m/z 633.24 gave a dominant fragment peak at m/z 487.24 (ouabagenin + 11BO-H + Na)+. It should be noted that a new disposable glass vial (1 dram) could contain soluble borate enough to convert as much as ≈20 μg of ouabain into its borate complex, although this depends on the kind of borosilicate glass. Rinsing a new vial removes most of the borate and other ionic species; however, plastic vials should be used for long-term storage of low microgram or submicrogram quantities of ouabain. Small amounts of ouabain that have already been dried in borosilicate glassware can be efficiently dissolved in 10% acetonitrile in water, and ouabain can be recovered by RP-HPLC using 0.1% trifluoroacetic acid (TFA)-containing eluents.
Method B.
Sodium tetraborate (2.6 mg, 6.8 μmol) was added to ouabain octahydrate (5 mg, 6.9 μmol) in water (1 ml), and the solution was lyophilized. 1H- and 13C-NMR assignments are shown in Table 1. For the 1H-NMR measurement in acidic pH (Fig. 4A), the obtained material (0.2 mg) was dissolved in 10% acetonitrile-d3 in deuterium oxide containing 0.1% TFA.
Table 1.
NMR data for ouabain–borate complexes
| Tetrahedral borate
|
No. | |||||
|---|---|---|---|---|---|---|
| Major isomer
|
Minor isomer
|
Trigonal borate
|
||||
| 13C | 1H | 13C | 1H | 13C | 1H | |
| 76.4 | 3.46 | 74.1 | 4.77 | 1 | 71.3 | 5.07 |
| 39.1 | 2.00, 1.63 | 33.2 | 2.05, 1.81 | 2 | 32.1 | 2.19, 2.05 |
| 71.1 | 3.56 | 71.1 | 3.95 | 3 | 71.5 | 4.09 |
| 44.1 | 1.85, 1.62 | 34.6 | 2.04, 1.59 | 4 | 34.1 | 2.22, 1.71 |
| 75.3 | — | 74.2 | — | 5 | 76.2 | — |
| 31.1 | 1.63, 1.40 | 36.4 | 1.77, 1.42 | 6 | 33.7 | 1.41** |
| 22.0 | 1.80, 1.40 | 24.0 | 1.81, 1.19 | 7 | 23.2 | 1.83, 1.26 |
| 36.3 | 2.20 | 40.0 | 1.64 | 8 | 39.9 | 1.81 |
| 52.4 | 1.43 | 45.1 | 1.55 | 9 | 46.5 | 1.64 |
| 44.9 | — | 41.3 | — | 10 | 45.3 | — |
| 67.2 | 3.80 | 67.8 | 3.62 | 11 | 68.8 | 3.95 |
| 47.7 | 1.65, 1.42 | 49.7 | 1.61, 1.38 | 12 | 49.3 | 1.67, 1.42 |
| 50.5 | — | 50.7 | — | 13 | 50.9 | — |
| 87.5 | — | 86.0 | — | 14 | 86.0 | — |
| 32.8 | 2.11, 1.71 | 32.3 | 2.06, 1.67 | 15 | 32.5 | 2.12, 1.71 |
| 27.0 | 2.14, 1.74 | 27.4 | 2.12, 1.74 | 16 | 27.5 | 2.15, 1.77 |
| 51.1 | 2.86 | 50.4 | 2.81 | 17 | 50.4 | 2.84 |
| 17.0 | 0.88 | 17.0 | 0.79 | 18 | 17.1 | 0.86 |
| 61.3 | 3.92 | 65.5 | 4.27, 3.83 | 19 | 59.1 | 4.05, 3.79 |
| 179.5* | — | 179.1* | — | 20 | 179.0* | — |
| 75.9 | 4.95 | 75.8 | 4.92 | 21 | 75.8 | 4.93 |
| 117.1 | 5.91 | 117.1 | 5.88 | 22 | 117.3* | 5.90 |
| 179.0* | — | 179.0* | — | 23 | 178.9 | — |
| 98.8 | 4.83 | 98.4 | 4.80 | 1′ | 98.8 | 4.75 |
| 71.4 | 3.79 | 71.5 | 3.86 | 2′ | 71.2 | 3.82 |
| 71.1 | 3.67 | 71.1 | 3.85 | 3′ | 71.1 | 3.72 |
| 73.0 | 3.32 | 73.3 | 3.30 | 4′ | 73.1 | 3.33 |
| 69.6 | 3.68 | 69.2 | 3.77 | 5′ | 69.6 | 3.68 |
| 17.5 | 1.19 | 17.5 | 1.17 | 6′ | 17.5 | 1.19 |
Assignments in the same column may be reversed.
The methylene protons are overlapped.
Figure 4.

1H-NMR spectra of ouabain borate dissolved in acidic solvent (containing 0.1% TFA) (A), 3-μg HIF, same as Fig. 1A (B), and ouabain trigonal-borate (C). All measurements were carried out in 10% acetonitrile-d3 in D2O. ∗ indicates the peaks arising from either mechanical noise or because of unidentified impurities. The spectrum in C also contains signals from a small amount of ouabain.
Preparation of Ouabain Trigonal-Borate.
Ouabain octahydrate (10 mg, 14 μmol) was dissolved in water (1 ml). Boric acid (1 mg, 16 μmol) was added, and the solution was lyophilized. 1H- and 13C-NMR assignments are shown in Table 1.
Naphthoylation Protocol.
The “glassware-treated” ouabain prepared by using method A (containing 200 ng ouabain) was added to 1-(2-naphthoyl)-imidazole (2 mg in 200 μl of anhydrous acetonitrile) and 1,8-diazabicyclo[5,4,0]undec-7-ene (20 μl of 2% anhydrous acetonitrile solution). The mixture was stirred at room temperature for 2 hr and quenched with water (0.5 ml). Acetonitrile was removed under reduced pressure, and the white aqueous suspension was loaded onto a Waters Sep-Pak C18 cartridge. The cartridge was washed successively with 20%, 40%, and 50% acetonitrile in water (10 ml, 10 ml, and 5 ml, respectively). The naphthoylated products were eluted with acetonitrile (6 ml) and then subjected to RP-HPLC on a Vydac 218TP column (C18, 4.6 × 250 mm, 300 Å, 5 μm) by using isocratic elution with acetonitrile/water (82:18) at 1 ml/min. The elution was monitored with a Shimadzu RF-551 fluorescence detector (λex 234 nm, λem 360 nm).
The naphthoylation of glycerol (2 mg) was also done with the same protocol except that the amounts of reagents were scaled up accordingly.
RESULTS
1H-NMR of Purified HIF (3-μg HIF).
The modified HIF purification protocol with the anti-digoxin antibody affinity column yielded 3 μg of HIF, which allowed 1H-NMR measurement. The obtained spectrum appeared to be a mixture of two different compounds, which is indicated by the two peaks in the methyl proton region around 0.85 ppm (Fig. 1A). Although some of the HIF peaks (Fig. 1A) appeared to match the signals of ouabain (Fig. 1B), we had no clue about the rest of the HIF signals at that point. The answer to this puzzling 1H-NMR profile of HIF, however, came from the subsequent reexamination of past nanogram-scale naphthoylation studies on HIF and OLC (13, 14).
Figure 1.

1H-NMR spectra of 3-μg HIF (A), ouabain (B), an ouabain sample (10 μg) that had been stored in a borosilicate vial as an aqueous solution (C), and ouabain tetrahedral-borates (D). A and B were measured in 10% acetonitrile-d3 in D2O, whereas C and D were measured in D2O. ∗ indicates the peaks arising from either mechanical noise or because of unidentified impurities.
The Zero-CD Product from Endogenous Ouabain.
At that point, ouabain was also included in the scope of our research as a viable candidate for endogenous ouabain, because all microscale structural analyses, except for the nanogram-scale naphthoylation (13, 14), pointed to ouabain. In addition, the CD calculation study (15) had made suspect the “zero-CD” product obtained from the past naphthoylation of endogenous ouabain (13, 14). We subsequently, in retrospect accidentally, encountered the elusive zero-CD product in a reaction mixture resulting from a nanogram-scale ouabain naphthoylation. An aqueous solution of ouabain (≈200 ng) had been stored overnight in a borosilicate vial and lyophilized before this naphthoylation reaction. Surprisingly, this ouabain naphthoylation did not afford the expected major product, ouabain pentanaphthoate, but instead gave a product that coeluted with the HIF pentanaphthoate obtained earlier (14) (Fig. 2). This product gave the same zero-CD profile as HIF pentanaphthoate and OLC pentanaphthoate(13, 14), whereas its UV showed the presence of the naphthoate group (data not shown). ESIMS of this zero-CD product gave a distinct (M+H)+ peak at m/z 555.1834. The UV/CD spectra and the calculated elemental composition (C36H27O6) suggested that the product could be glycerol trinaphthoate, which would have an intense naphthoate band but no CD. Synthetic glycerol trinaphthoate indeed reproduced the RP-HPLC retention time and the zero-CD profile encountered earlier in the HIF and OLC studies (13, 14). The glycerol found in this study presumably arises from an incidental contamination during the nanogram-level handling of the sample. This unexpected finding, however, solved our long-standing enigma regarding the zero-CD product from endogenous ouabain naphthoylation.
Figure 2.

Fluorescence-detected HPLC traces for the naphthoylation product (retention time ≈10 min) from the ouabain sample that had been stored in a borosilicate vial as an aqueous solution (see text) (A), the naphthoylation product from HIF (14) (B), and coinjection of A and B (C).
Structural Characterization of Glassware-Treated Ouabain.
The presence of glycerol in the ouabain sample, however, does not explain why ouabain pentanaphthoate was not obtained in the reaction above, where a large excess of the naphthoylation reagent was used. Spectroscopic analyses of the ouabain sample that had been stored in a borosilicate vial not only solved this additional mystery but also led us eventually to identify the structure of the 3-μg HIF. ESIMS of this glassware-treated sample gave an intense peak corresponding to the ouabain 11B-borate complex with one sodium (m/z 633.24) together with its smaller 10B-isotope peak (m/z 632.26). 1H-NMR showed two distinct sets of signals with a ratio of ≈3:2 as clearly seen in the high field (around 0.9 ppm) and low field (around 5.9 ppm) regions (Fig. 1C). The two sets of peaks matched neither the ouabain signals (Fig. 1B) nor the signals of the 3-μg HIF (Fig. 1A) (The difference between glassware-treated ouabain and 3-μg HIF is discussed below.) Because ESIMS showed that the ratio between ouabain and borate is 1:1 (ouabain “mono”-borate), the two sets of NMR signals were assumed to arise from two coordination isomers of ouabain borate. For further characterization by NMR, a milligram-scale chemical preparation of ouabain borate was carried out (method B above). The chemically prepared ouabain borate reproduced the 1H-NMR profile of the glassware-treated ouabain (Fig. 1D). The NMR signals of the major and minor isomers were separately assigned to the framework of ouabain by using 1H, 13C, DEPT (90° and 135°), correlated spectroscopy, heteronuclear multiple quantum correlation, heteronuclear multiple bond correlation (4, 8, and 12 Hz), and ROESY (100 ms) (Table 1).
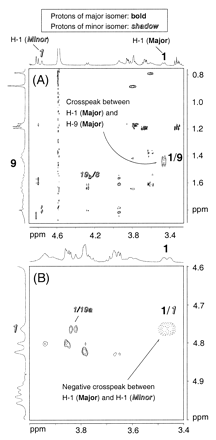
The most notable structural difference between the two isomers is in ring A (Fig. 3). The chemical shift of H-1 in the major isomer is 3.46 ppm, whereas the same proton in the minor isomer appears at 4.77 ppm (Table 1). In addition, a ROESY (100 ms) cross-peak, which is in a positive phase (NOE peak) (18, 19), is observed between H-1 and H-9 of the major isomer (see the supplemental data on the PNAS web site, www.pnas.org). These results suggest that ring A of the major isomer is distorted into a boat-like form, where H-1 is placed in a shielded axial-like position and sterically closer to H-9. Moreover, negative ROESY crosspeaks were observed between major and minor isomers, e.g., between H-1 (major) and H-1 (minor), which indicate the occurrence of an exchange between the two coordination isomers (18, 19) (see the supplemental data on the PNAS web site, www.pnas.org).
Figure 3.

Structures of the two coordination isomers of ouabain tetrahedral-borates.
The borate coordination site in each isomer was determined by ESIMS/MS and 11B-NMR. ESIMS/MS of the ouabain borate ion at m/z 633.24 gave a dominant fragment ion at m/z 487.24, corresponding to ouabagenin borate with sodium. Therefore, borate is attached to the aglycone moiety. Furthermore, 11B-NMR showed a borate peak at 0.78 ppm (broad singlet) from BF3⋅OEt2 indicating the presence of tetrahedral borate; tetrahedral and trigonal borates appear around 0 ppm and 20 ppm, respectively, in 11B-NMR (chemical shift reference: BF3⋅OEt2) (20). These ESIMS/MS and 11B-NMR results show that tetrahedral borate is attached to three hydroxyls on the aglycone moiety. The only possible combinations of three hydroxyls that can accommodate the tetrahedral borate group are 1,11,19- and 1,5,19-hydroxyl groups. The major isomer observed in the NMR analysis fits the 1,11,19-borate, in which the coordinated borate stabilizes the ring A boat conformation (Fig. 3). On the other hand, NOE peaks in the minor isomer are consistent with the 1,5,19-borate, in which borate formation fixes ring A into its chair conformation (Fig. 3).
Because of this “cyclic borate protection” at 1,11,19- or 1,5,19-hydroxyl groups, the nanogram-scale naphthoylation of the glassware-treated ouabain had not yielded the anticipated ouabain 1,19,2′,3′,4′-pentanaphthoate.
Structure of the 3-μg HIF.
Subsequent study found that the two interconverting complexes between ouabain and tetrahedral borate, as seen above (Fig. 3), do not survive acidic pH and decompose into a mixture of ouabain and its trigonal borate complex, which exhibit the 1H-NMR signals observed in the previously obtained 3-μg HIF spectrum (Fig. 1A). 1H-NMR comparison between ouabain borate in acidic pH (Fig. 4A) and the 3-μg HIF (Fig. 4B) showed that both contain the same two species although the ratio between the two species are slightly different: the doublet at 1.2 ppm and peaks around 3.6 ppm in Figs. 1A and 4B are background impurities. It turns out that the 1H-NMR spectrum of 3-μg HIF (Fig. 5B) is simply a summation of the signals of ouabain (Fig. 5A) and the ouabain-trigonal borate (Fig. 5C), which can be prepared from ouabain and boric acid (see Materials and Methods). Thus, 3-μg HIF is confirmed to be ouabain, which has been, however, partially converted into its trigonal borate after purification in a disposable borosilicate glass tube; trigonal borate formed because of TFA used in the HPLC eluents.
Figure 5.

Expanded 1H-NMR of ouabain, same as Fig. 1B (A), 3-μg HIF, same as Figs. 1A and 4B (B), and ouabain trigonal-borate, same as Fig. 4C (C). ∗ indicates the peaks arising from either mechanical noise or because of unidentified impurities. The spectrum in C also contains signals from a small amount of ouabain.
DISCUSSION
The structural analysis of the 3-μg HIF was initially hindered by the unexpected borate complexation, which had occurred in a disposable borosilicate glass tube. The confusing 1H-NMR profile, however, eventually led to the identification of the purified molecule as ouabain. At the final HPLC purification stage, the peak corresponding to HIF was collected in a borosilicate tube, which resulted in converting almost half of the purified HIF (3 μg) into its trigonal borate as seen in the obtained 1H-NMR. Trigonal borate, instead of tetrahedral borates (Fig. 3), had formed because the TFA from HPLC eluents kept the sample at acidic pH. The HIF samples used for the past structural studies (13, 14), however, were purified without the use of TFA at the final RP-HPLC step. Therefore, the past HIF samples probably consisted of the tetrahedral borate complex formed in the borosilicate glassware.
Although ouabain has already been purified from bovine adrenals, in which its structure was confirmed by UV, MS, and 1H-NMR (21), its relation to the endogenous ouabain research has been obscured because of the widely accepted notion that endogenous ouabain is a structural isomer of plant ouabain (13, 14). This notion is based on the zero-CD product from the nanogram-scale naphthoylation of HIF and OLC. The assignment of this zero-CD product to HIF pentanaphthoate has been questioned because of the ambiguous fast atom bombardment MS result of this compound (3), and our recent theoretical and experimental CD study also resulted in making us suspect of the past assignment of this product (15). The subsequent reexamination of the nanogram-scale naphthoylation study accidentally clarified that the zero-CD product was a byproduct of glycerol contamination. Hence, there is no structural data that indicate the presence of ouabain isomer in mammalian tissues and plasma, and the notion from our past naphthoylation study (13, 14) has to be retracted. The finding of ouabain in mammals still leaves many questions about its origin, which is, however, beyond the scope of the current study.
In addition to the glycerol contamination, the borate complexation had complicated the past nanogram-scale naphthoylation study on endogenous ouabain. As pointed out above, the HIF and OLC samples used for the past naphthoylation (13, 14) were “protected” by the borate group, which prevented the formation of ouabain pentanaphthoate, the major product from ouabain naphthoylation. The conspicuous glycerol trinaphthoate as well as the absence of ouabain pentanaphthoate misled the entire naphthoylation study on endogenous ouabain. On the other hand, the past LC/MS study on HIF was not affected by this borate complexation (13), indicating that the complex did not survive the HPLC condition (pH 4.7) used in this analysis. In general, organic borate esters are stable in basic conditions, in which they exist as tetrahedral borates, whereas, in acidic pH, they are converted into less stable trigonal borates: cyclic borate has been used for the protection of a catechol group in organic synthesis, in which protection and deprotection were carried out in basic and acidic pH, respectively (22). Therefore, even if the HIF sample had formed borate adducts before the LC/MS analysis, the acidic buffer transformed the complex into trigonal borate, which is easily hydrolyzed in the HPLC conditions used. Attempts to determine the borate coordination site in ouabain trigonal borate has been hindered by this chemical instability. In addition, the trigonal-borate coordination site is hard to determine because the trigonal borate group has only two attachment points to ouabain, and ouabain has many permutations of diols that could accommodate the trigonal borate.
The current finding of ouabain borate suggests that the polyhydroxylated ouabain molecule serves as a polydentate ligand to many other inorganic species. In addition to the prevalent inorganic species, many trace elements, such as boron and vanadium, are known to be present in the body in microgram quantities or more, but their physiological roles are still at a speculative stage (23). On the other hand, many inorganic salts and reagents are used in the detection, purification, and biological studies of endogenous ouabain. The dynamic isomeric interconversion, as seen in ouabain borate (Fig. 3), as well as the complexation itself should affect solubility, chromatographic profiles, and the affinity to biopolymers, including the sodium pump and the antibodies used for the detection of endogenous ouabain. Many previous contradictory results regarding the detection, purification, and biological studies of endogenous ouabain need to be reviewed in the light of such possibilities (24–34).
The biological discrepancies between HIF and the other ouabain-like sodium pump inhibitors, such as OLC and adrexin C, still remain unresolved. The previous HIF purification protocols indicate that the HIF samples for biological studies (9, 12, 35, 36) were relatively crude compared with the one used for structural studies (13, 14), where repetitive RP-HPLC was used for purification. Therefore, the HIF preparations that showed the distinct biological properties could have contained not only ouabain, but also other chemical, i.e., inorganic and/or organic, species. It is still possible that some unidentified species with high-affinity binding and unique kinetic interactions with the sodium pump might have been responsible for the biological activity known as HIF.
Another possibility that has to be considered is whether inorganic complexation was the cause for the reported biological properties of HIF. Our preliminary biochemical study showed that the glassware treatment (method A in Materials and Methods) of ouabain affected the outcome of the coupled enzyme assay by using the rat kidney Na+,K+-ATPase (rat α1 isoform), which had been used previously for the distinction between HIF and ouabain (8): IC50 of glassware-treated ouabain, ranging from 10−5 M to 10−7 M, was somewhat inconsistent but was clearly lower than that of ouabain (≈10−3 M). These differences from ouabain, however, may not be due to the sodium pump inhibition, because ouabain and glassware-treated ouabain appeared identical (low affinity) when the activity of the same Na+,K+-ATPase (rat α1 isoform) was directly measured by the amount of released phosphate.
In conclusion, the ouabain-like sodium pump inhibitor isolated from mammalian brain in previous studies (13, 14) was in fact ouabain. Under certain conditions, however, it was converted into its borate and/or other inorganic derivatives; the form in vivo adopted by ouabain is unknown. The current findings have clarified the ambiguities associated with past structural studies on HIF and OLC. It is necessary to be aware of the possible occurrence of inorganic complexation during the detection, purification, and biological studies of endogenous ouabain when dealing with low microgram or sub-microgram quantities of material.
Supplementary Material
Acknowledgments
We thank Drs. John Decatur and Masaru Hashimoto, CU, for help in the NMR measurements and discussions, respectively, and Drs. Brian Chait, Rong Wang, (Rockefeller University, New York) and LeRoy B. Martin, III (Micromass, Beverly, MA) for the MS. This work is supported in part by National Institutes of Health Grant HL 52282.
ABBREVIATIONS
- HIF
hypothalamic inhibitory factor from bovine hypothalamus
- OLC
ouabain-like compound from human plasma
- RP-HPLC
reversed-phase high performance liquid chromatography
- ESI
electrospray (ionization)
- ROESY
rotating-frame nuclear Overhauser effect spectroscopy
- TFA
trifluoroacetic acid
References
- 1.Goto A, Yamada K, Yagi N, Yoshioka M, Sugimoto T. Pharmacol Rev. 1992;44:377–399. [PubMed] [Google Scholar]
- 2.Hamlyn J M, Hamilton B P, Manunta P. J Hypertens. 1996;14:151–167. doi: 10.1097/00004872-199602000-00002. [DOI] [PubMed] [Google Scholar]
- 3.Doris P A. Miner Electrolyte Metab. 1996;22:303–310. [PubMed] [Google Scholar]
- 4.de Wardener H E. J Hypertens Suppl. 1996;14:S9–18. [PubMed] [Google Scholar]
- 5.Goto A, Yamada K. Curr Opin Nephrol Hypertens. 1998;7:189–196. doi: 10.1097/00041552-199803000-00008. [DOI] [PubMed] [Google Scholar]
- 6.Hamlyn J M, Blaustein M P, Bova S, DuCharme D W, Harris D W, Mandel F, Mathews W R, Ludens J H. Proc Natl Acad Sci USA. 1991;88:6259–6263. doi: 10.1073/pnas.88.14.6259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tamura M, Konishi F, Sakakibara M, Inagami T. In: The Sodium Pump, Structure Mechanism, Hormonal Control and Its Role in Disease. Bamberg E, Schoner W, editors. New York: Springer; 1994. pp. 763–766. [Google Scholar]
- 8.Ferrandi M, Minotti E, Salardi S, Florio M, Bianchi G, Ferrari P. Am J Physiol. 1992;263:F739–F748. doi: 10.1152/ajprenal.1992.263.4.F739. [DOI] [PubMed] [Google Scholar]
- 9.Carilli C T, Berne M, Cantley L C, Haupert G T., Jr J Biol Chem. 1985;260:1027–1031. [PubMed] [Google Scholar]
- 10.Cantiello H F, Chen E, Ray S, Haupert G T., Jr Am J Physiol. 1988;255:F574–F580. doi: 10.1152/ajprenal.1988.255.4.F574. [DOI] [PubMed] [Google Scholar]
- 11.Anner B M, Rey H G, Moosmayer M, Meszoely I, Haupert G T., Jr Am J Physiol. 1990;258:F144–F153. doi: 10.1152/ajprenal.1990.258.1.F144. [DOI] [PubMed] [Google Scholar]
- 12.Hallaq H A, Haupert G T., Jr Proc Natl Acad Sci USA. 1989;86:10080–10084. doi: 10.1073/pnas.86.24.10080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tymiak A A, Norman J A, Bolgar M, DiDonato G C, Lee H, Parker W L, Lo L-C, Berova N, Nakanishi K, Haber E, Haupert G T., Jr Proc Natl Acad Sci USA. 1993;90:8189–8193. doi: 10.1073/pnas.90.17.8189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhao N, Lo L-C, Berova N, Nakanishi K, Tymiak A A, Ludens J H, Haupert G T., Jr Biochemistry. 1995;34:9893–9896. doi: 10.1021/bi00031a010. [DOI] [PubMed] [Google Scholar]
- 15.Dong, J.-G., Guo, J., Akritopoulou-Zanze, I., Kawamura, A., Nakanishi, K. & Berova, N. Chirality, in press. [DOI] [PubMed]
- 16.Mudgett-Hunter M, Margolies M N, Ju A, Haber E. J Immunol. 1982;129:1165–1172. [PubMed] [Google Scholar]
- 17.Mudgett-Hunter M, Anderson W, Haber E, Margolies M N. Mol Immunol. 1985;22:477–488. doi: 10.1016/0161-5890(85)90132-4. [DOI] [PubMed] [Google Scholar]
- 18.Henning J, Limbach H H. J Magn Reson. 1982;49:322–328. [Google Scholar]
- 19.Bothner-By A A, Stephens R L, Lee J-m. J Am Chem Soc. 1984;106:811–813. [Google Scholar]
- 20.Kidd R G. In: NMR of Newly Accessible Nuclei. Laszlo P, editor. Vol. 2. New York: Academic; 1983. pp. 49–77. [Google Scholar]
- 21.Schneider R, Wray V, Nimtz M, Lehmann W D, Kirch U, Antolovic R, Schoner W. J Biol Chem. 1998;273:784–792. doi: 10.1074/jbc.273.2.784. [DOI] [PubMed] [Google Scholar]
- 22.Greene T W, Wuts P G M. Protective Groups in Organic Synthesis. New York: Wiley; 1991. p. 173. [Google Scholar]
- 23.Nielsen F H. FASEB J. 1991;5:2661–2667. [PubMed] [Google Scholar]
- 24.Krackle G R. J Lab Clin Med. 1983;101:105–113. [PubMed] [Google Scholar]
- 25.Cloix J F, Crabos M, Grichois M L, Meyer P. Can J Physiol Pharmacol. 1987;65:1522–1527. doi: 10.1139/y87-240. [DOI] [PubMed] [Google Scholar]
- 26.Wechter W J, Benaksas E J. Prog Drug Res. 1990;34:231–260. doi: 10.1007/978-3-0348-7128-0_6. [DOI] [PubMed] [Google Scholar]
- 27.Hamlyn J M, Manunta P. J Hypertens. 1992;10, Suppl. 7:S99–S111. [PubMed] [Google Scholar]
- 28.Gomez-Sanchez E P, Foecking M F, Sellers D, Blankenship M S, Gomez-Sanchez C E. Am J Hypertens. 1994;7:647–650. doi: 10.1093/ajh/7.7.647. [DOI] [PubMed] [Google Scholar]
- 29.Doris P A, Jenkins L A, Stocco D M. Hypertension. 1994;23:632–638. doi: 10.1161/01.hyp.23.5.632. [DOI] [PubMed] [Google Scholar]
- 30.Doris P A. Am J Physiol. 1994;266:H360–H364. doi: 10.1152/ajpheart.1994.266.1.H360. [DOI] [PubMed] [Google Scholar]
- 31.Lewis L K, Yandle T G, Lewis J G, Richards A M, Pidgeon G B, Kaaja R J, Nicholls M G. Hypertension. 1994;24:549–555. doi: 10.1161/01.hyp.24.5.549. [DOI] [PubMed] [Google Scholar]
- 32.Pidgeon G B, Lewis L K, Richards A M, Nicholls M G. Hypertension. 1994;24:385. doi: 10.1161/01.hyp.24.5.549. [DOI] [PubMed] [Google Scholar]
- 33.Di Bartolo V, Balzan S, Pieraccini L, Ghione S, Pegoraro S, Biver P, Revoltella R, Montali U. Life Sci. 1995;57:1417–1425. doi: 10.1016/0024-3205(95)02104-q. [DOI] [PubMed] [Google Scholar]
- 34.Ferrandi M, Manunta P, Balzan S, Hamlyn J, Bianchi G, Ferrari P. Hypertension. 1997;30:886–896. doi: 10.1161/01.hyp.30.4.886. [DOI] [PubMed] [Google Scholar]
- 35.Haupert G T, Jr, Carilli C T, Cantley L C. Am J Physiol. 1984;247:F919–F924. doi: 10.1152/ajprenal.1984.247.6.F919. [DOI] [PubMed] [Google Scholar]
- 36.Janssens, S. P., Kachoris, C., Parker, W. L., Hales, C. A. & Haupert, G. T., Jr. (1993) J. Cardiovasc. Pharmacol.22, Suppl. 2, S42–S46. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}