

Abstract
In recent years, there has been a surge in the number of studies exploring the relationship between proteins’ equilibrium dynamics and structural changes involved in function. An emerging concept, supported by both theory and experiments, is that under native state conditions proteins have an intrinsic ability to sample conformations that meet functional requirements. A typical example is the ability of enzymes to sample open and closed forms, irrespective of substrate, succeeded by the stabilization of one form (usually closed) upon substrate binding. This ability is structure-encoded, and plays a key role in facilitating allosteric regulation, which suggests complementing the sequence-encodes-structure paradigm of protein science by structure-encodes-dynamics-encodes-function. The emerging connection implies an evolutionary role in selecting/conserving structures based on their ability to achieve functional dynamics, and in turn, selecting sequences that fold into such ‘apt’ structures.
Introduction
Proteins sample an ensemble of conformations under equilibrium conditions. This ensemble is broadly distributed in the denatured state, and it becomes narrowly distributed - mainly confined to the neighborhood of the folded state - under native state conditions. Of interest are those conformations accessible near the global energy minimum, also called substates when separated by low energy barriers. Because proteins perform their function under these conditions, interconversions between these conformations are potentially functional.
The fact that folded proteins are not static, but undergo ‘wigglings and jigglings’ as put forth by Feynman, is now well-established. We have indeed come a long way since the idea was first put forward in the pioneering molecular dynamics (MD) simulations of proteins by McCammon, Karplus, Wolynes, Levitt, van Gunsteren and others in the late 1970s and early ‘80s. With recent advances in experiments and theory, increased evidence is now being provided for the biological functionality of these apparently random motions. Experiments now permit us to visualize the structural flexibility and heterogeneity of biomolecules and assess their relevance to catalysis or signaling [1•;2••]. On the theoretical side, novel coarse-grained models and methods are providing insights into structure-dynamics relations on a global, rather than local, scale [3;4•]. The rapidly accumulating data lead to the emergence of concepts such as the pre-disposition or intrinsic ability of proteins to undergo conformational changes required for function, and a possible evolutionary pressure for selecting such structures, while also raising new questions with regard to old concepts.
New Questions on Old Concepts
The first question concerns the conformational changes observed between the substrate-bound- and -unbound forms of a given enzyme, a phenomenon broadly referred to as ‘induced fit’ after the original proposition of Koshland [5]. The question is, to what extent these conformational changes are literally ‘induced’ by substrate. Would it be possible for the substrate to drive a change if the structure was not pre-disposed to undergo the change? Does the substrate simply stabilize the ‘fittest’ conformations that already exist in the unbound state, following the redistribution of a pre-existing population [6] originally proposed by Weber [7]? Does it essentially select the lowest-energy-cost pathways away from the original minimum to optimize its interaction with the enzyme?
The second question relates to allosteric changes in conformations usually occurring on a large scale (quaternary changes) stated to be ‘driven’ by local phenomena like ATP binding or hydrolysis, ligand binding, phosphorylation etc. Again, would it be possible to elicit cooperative responses, if these were not already energetically favored by the structure? How similar are the experimentally known allosteric changes, and those theoretically predicted to be naturally sampled by the particular structure?
Third, how can we reconcile the ensemble of conformations accessible near folded state and the two-state transition observed in many allosteric proteins in accord with the Monod-Wyman-Changeux (MWC) model [8]? Does all-or-none transition reflect passages between the most probable substates? Are there intermediates or sequence of events not detectable within experimental time frames? Do simulations reveal such features for small allosteric proteins or signaling proteins, which lack the regularity and symmetry that enhances cooperativity in multimeric quaternary structures? Given that certain key sites and interactions modulate the propagation of signals in many proteins, can we think of a structure-encoded network of interactions, and thereby a sequence of events, reminiscent of the Koshland-Neméthy-Filmer (KNF) model [9]?
Here, we will focus on the nature and functional significance of proteins’ intrinsic dynamics. First, we present recent data that point to the pre-existence, or accessibility, of functional conformations even in the absence of activity or triggering events. Second, we emphasize the fact that these motions are not random, but uniquely defined by the 3-dimensional structure. In a sense, proteins appear to have optimally evolved to achieve their functional dynamics. Third, we call attention to the ability of structures to define, not only mechanisms of concerted motions, but also pathways of communication that ensure rapid propagation of perturbations and allosteric responses.
Pre-existing dynamics of enzymes revealed by recent experiments
Conformational variability in the presence of different substrates
Structural changes occur at multiple levels, ranging from concerted rearrangements of intact subunits or domain movements, to intrinsic disorder on a local scale. This ability to assume a well-defined ensemble of substates, near the ‘folded’ state indeed emerges as a consequence of the internal degrees of freedom that permit the structure to relax/rearrange without altering the fold. Figure 1A–B illustrates different conformations assumed by two well-studied enzymes in the presence of different substrates, (A) cyclin-dependent kinases (CDKs), and (B) HIV-1 reverse transcriptase (RT). CDK exhibits changes at the active loop conformation (cyan) and its N-lobe reorients relative to the C-lobe; RT undergoes global changes (e.g. movement of the thumb subdomain (red) on p66 subunit).
Figure 1. Experimental evidence for conformational diversity of folded proteins and comparison with theoretical predictions.
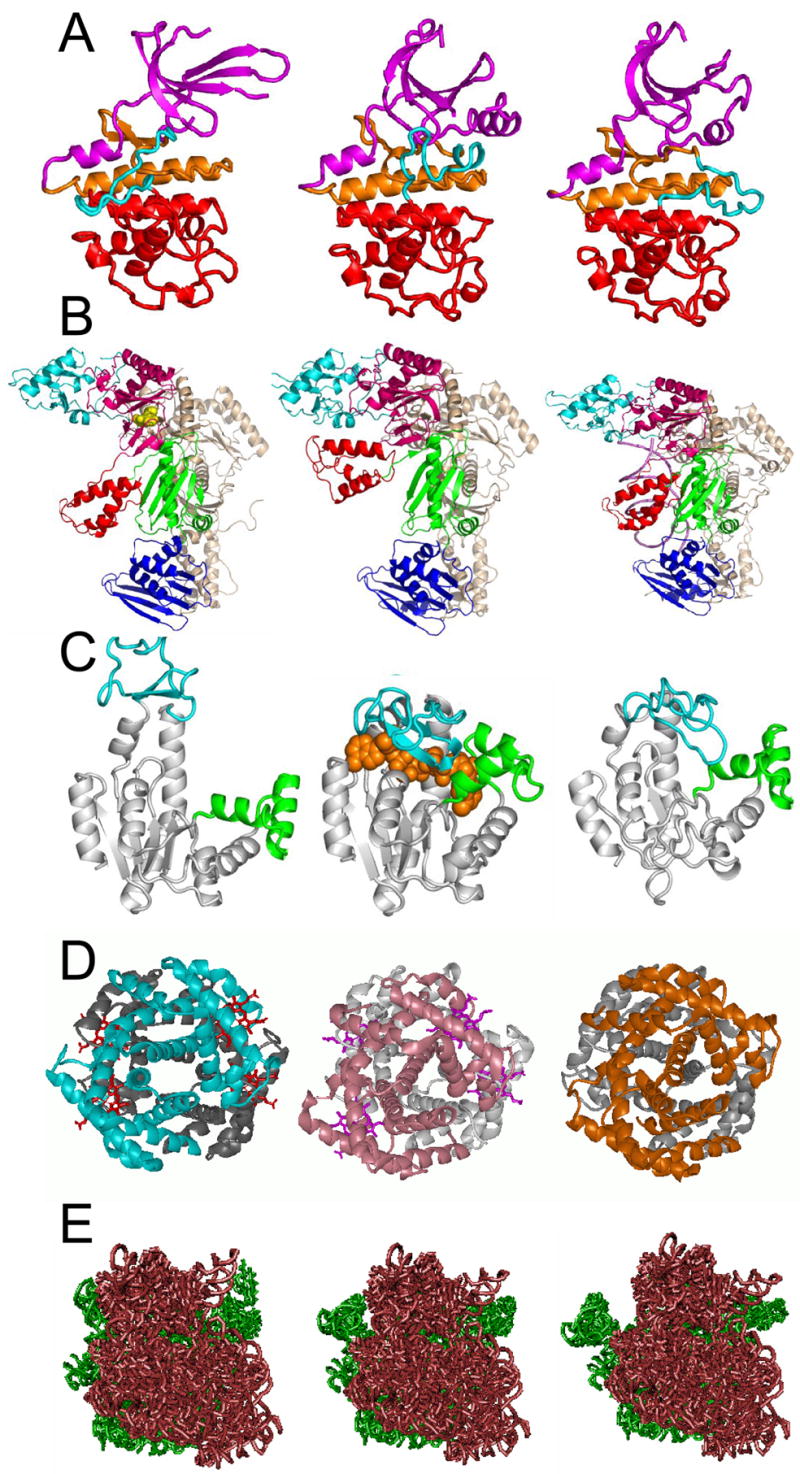
(A) Three conformations of cyclin dependent kinases (CDKs) adopted in the free form (middle), and in the presence of two different substrates, an inhibitor (INK4; left) and its activator (cylin; right). The corresponding Protein Data Bank (PDB) codes are 1bi7, 1hcl and 1fin, in reading order. Colors refer to N-lobe (purple), C-lobe (red), hinge residues (orange) and activation loop (cyan). Both activation and inhibition involve conformational changes in and around the catalytic cleft. The activation loop (cyan) rotates towards the substrate (not shown). (B) Alternative conformations of HIV-1 RT. RT is composed of two subunits, p66 and p51 (wheat); the p66 subunit consists of two domains, polymerase and RNase H (blue); and the polymerase domain contains four subdomains, thumb (red), fingers (blue), palm (pink), connection (green). Comparison of the inhibitor-bound (nevirapine; space filling form in yellow; left), unliganded (middle) and DNA-bound (right) forms a (PDB codes 1rth, 1dlo and 2hmi) shows domain movements. (C) The left two diagrams display the free (left) and substrate-bound (middle) forms of adenylate kinase (AK) (respective PDB codes: 4ake and 1ake), and the diagram on the right is a reconfigured form of the free enzyme computed by deforming the unbound structure along the lowest frequency ANM mode (mode 1)[18]. AK contains three domains: core (white), lid (cyan), and AMP-binding (green) domains. The substrate is shown in orange, space-filling representation. In the substrate-bound form as well as the model on the right, the lid approaches the core. (D) Comparison of the T (tense, unliganded) (left) and R2 (relaxed, CO-bound) (middle) forms of hemoglobin (Hb) (respective PDB codes: 1a3n and 1bbb), and the model (right) calculated [19] by deforming the T form along ANM mode 2. The model approximates the experimentally observed torsion of the α2β2 dimer (front, colored) with respect to the α1β1 dimer (bottom, gray). (E) Comparison of the ribosome structure experimentally determined (middle), and two conformers (left and right) sampled by fluctuations along ANM mode 3 [62], approximating the ratchet-like rearrangement of the 70S subunit (green) with respect to the 30S (maroon), suggested by experimental data (see also the original work of [63]).
Such conformational variability observed in substrate- or ligand-bound forms is not restricted to enzymes [6], as illustrated in the seminal work of Frauenfelder and coworkers for myoglobin [10]. A recent significant study shows that the antibody SPE7 binds a range of antigens upon adopting different binding-site conformations in the free state [11•]. The crystal structure of the encounter complex on ligand-binding pathway reveals that the antibody becomes selective after a ‘postbinding’ conformational switch that stabilizes specific complexes, while others are dissociated [12••]. This study thus unravels a functional combination of prebinding equilibrium and post-binding (induced) fit that provides a discriminatory mechanism in ligand recognition [12••].
Accessibility of functional motions in the absence of substrate
While structural changes assumed in the presence of different substrates are common in proteins, what is new is their accessibility in the absence substrate. Also, motions involved in biophysical (e.g. substrate recognition and binding), biochemical (e.g. catalysis) or biological (e.g. signaling, transcription) activities are observed to be sampled even prior to activity.
The linkage between pre-existing dynamics and catalytic function is highlighted in recent reviews [1•;13]. Motions characteristic of catalytic function were detected by Kern lab, for example, in the free states of prolyl cis-trans isomerase cyclophilin A, CypA [14•], and peptidylprolyl isomerase (Pin1) catalytic domain [15], leading to the conclusion that particular catalytic residues were ‘primed for catalysis’[15]. Recent small angle X-ray scattering measurements also show that the D236A mutant of ATCase almost equally populates the T and R states [16], supporting the view of accessibility of alternative functional forms, detectable upon perturbing the structure at a critical site. These observations conform to the MWC model, where the R-T equilibrium is stated to be ‘an intrinsic property of allosteric oligomers accessible in the absence of ligand, with the ligand stabilizing the conformation to which it binds with higher affinity’[2••].
Molecular basis of pre-existing dynamics: Insights from theory and simulations
The pre-existence of conformational motions is not unfamiliar to theoretical and computational biologists. Yet, their relevance to function is being established only recently.
Two major groups of studies have been undertaken toward this goal. The first uses analytical approaches (e.g. normal mode analysis (NMA) with coarse-grained (e.g. elastic network) [3;4•]. NMA readily provides a hierarchy of accessible modes of motion uniquely defined by the given architecture. The low frequency modes among them are usually cooperative (collectively involving large segments/domains/subunits). They also are most readily accessible, since they are, by definition, along the direction of the lowest ascent (smallest curvature/force constant) away from the original energy minimum approximated by a harmonic well. As a result, they conceivably present a favorable, or intrinsically preferred, mechanism for dissipating the energy increase arising from external perturbations.
Two classical examples illustrating the relevance of low frequency modes to functional changes in structure are shown in Figure 1, panels C and D. Panel C displays the open (left) and closed (middle) forms of E. coli adenylate kinase (AK), both determined by X-ray crystallography. The right diagram, on the other hand, displays an alternative conformation predicted by the anisotropic network model (ANM [17]) upon deforming the unliganded structure (left) along the slowest mode accessible in this substrate-free state [18]. Thus the closed form is readily approached by the open form upon movement along the direction of the first (energetically easiest) mode of motion. A similar phenomenon is observed in hemoglobin (Hb), a prime example of an allosteric protein (panel C). In this case, the tense (T) (left) structure is reconfigured to approximate the liganded (R2 (middle)) form, by moving along the 2nd slowest mode direction (right) [19]. Similar results for other systems support the view of the intrinsic tendency of the unbound proteins to approach their bound conformations via low frequency modes [20–22]. An application to ribosome, illustrated in panel E, also points to the functional relevance of slow modes. The low frequency modes predicted by coarse-grained NMA can now be readily retrieved and viewed for all PDB structures using recently developed webservers (e.g. [17]).
The second group of studies is based on MD simulations, accelerated by advanced algorithms such as replica exchange, steered MD or simplified models and force fields. While simulations require significantly longer computing time and may suffer from convergence problems, their main advantage is in exploring substates that may not be accessible via NMA. Recent simulations clearly demonstrate the energetic grounds for the ability of AK to fluctuate between open and closed forms [23•]. Likewise, unrestrained MD with implicit solvent shows the ability of HIV-1 protease to fluctuate between close, semi-open and fully open states, while predominantly populating the semi-open state [24•]. Another extensive MD study of a series of protein-protein complexes showed the existence of an ensemble of conformers prior to binding, the stabilization of selected conformers–those distinguished by shape complementarity- to form complexes, succeeded by further structural rearrangements[25•]. Simulations prove particularly useful when combined with NMA or coarse-grained models [26–31].
A key question, yet to be answered is how the internal dynamics of enzymes influences reaction rates. Theoretical studies on CypA dynamics indicate that a transfer of energy from first hydration shell and solvent-exposed regions into the protein interior all the way to the active site, through a network of vibrations, promotes catalysis by lowering the energy barrier along the reaction coordinate [32]. Similar observations were made for DHFR [1•;32]. A link between collective mechanics and interactions at the catalytic site is suggested by the co-localization of the global hinge site with the catalytic site, shown for a representative set of enzymes using elastic network models [33]. Another recent ANM study showed the coupling between domain motions and loop rearrangements at the catalytic site of triosephosphate isomerase [34]. These studies also drew attention to the conservation of key residues that concert cooperative motions [1•;32;33;35;36].
While the coupling between dynamics and function is recognized, quantitative assessment of the correlation between intrinsic dynamics and reaction rates remains to be clarified. The above described changes are physical events in the neighborhood of the original state. Catalytic reaction rates are controlled by chemical events farther along the reaction coordinate; their quantitative assessment requires consideration of free energy paths and barrier at transition state; and in this respect, the contribution of conformational motions to catalysis has been challenged [37]. The effect of distant mutations on DHFR catalytic reaction has been explained quantitatively in terms of the changes in the pre-organization of polar residues, arising as a consequence of the change in energy landscape in the presence of mutations [38].
Allosteric changes in conformations: Communication via concerted motions
Many allosteric biomolecules are multimeric and symmetric, which enhances the cooperativity of their conformational changes, leading to an all-or-none transition in accord with the MWC model [2]. Yet, allostery appears to be featured by a broad range of proteins [39•] including single domain proteins, and intermediates or sequential events are also observed, especially in some small proteins and ligand-gated ion channels.
The prominent feature in all allosteric proteins appears to be the accessibility of functional conformers, and the efficient communication between distant residues (e.g. ligand-binding and catalytic residues). In nicotinic acetylcholine receptor (nAChR), for example, signal transduction occurs between nACh binding and pore opening sites separated by 50Å. Such distant couplings may be readily accounted for by low frequency modes. The NMA of nAChR indeed showed that pore opening is ensured by the lowest frequency mode intrinsically favored by the structure irrespective of neurotransmitter binding [40]. This mode entails a global twist of the quaternary structure, similar to the pore opening mechanism identified for a series of potassium channels [41].
Low frequency modes are most effective in spreading signals, due to their cooperative nature and accessibility with minimal energy requirement. Yet, local effects may also play a critical role in mediating signals. The assessment of signal transduction mechanism may therefore necessitate the consideration of multiple modes, including those in the high frequency regime and mode-mode couplings [1•;32;42;43]. For example, a loop (β4-α4) has been pointed out in CheY to mediate the rotational isomerization of substrate-binding tyrosine (Y106) succeeding the phosphorylation event 9.5Å apart [26]. Subsequent simulations showed, however, that the loop is stabilized by the isomerization of Y106 in an active conformation [44•], consistent with the population shift model, and MWC mechanism. Yet, a recently solved CheY shows the same loop in an intermediate conformation, inviting attention to the accessibility of intermediate conformations [45].
Signal transduction along well-defined pathways mediated by collective motions emerges now as a common feature of allosteric proteins [46;47]. Recent theoretical studies indeed demonstrate the occurrence of key sites with high allosteric potential [48•;49] or strategically placed hot spot residues[30], and highlight pathways of allosteric signal propagation [50] consistent with experimental data [51]. These pathways predominantly involve conserved residues, as also deduced from sequence-based analysis [52;53], as illustrated in a recent MD study[54•]. Recent work also shows how communication pathways relate to collective motions [55]. Notably, binding sites are reported to be located at regions that strongly affect the network of interactions, and such properties have been suggested to result from evolutionary pressure [56•].
The spread of signals/perturbations via coupled motions, including in particular the most cooperative (lowest frequency) modes of motions, and conserved residues, thus emerges as a plausible mechanism in allostery. This also suggests hinge sites in these modes to serve as ‘messengers’ for mediating allosteric communication, a conjecture to be tested by further studies.
Conclusion
Increasingly larger body of experimental and theoretical studies concurs on the ability of enzymes and/or allosteric proteins to sample, even in their inactive or substrate-free forms, conformations that approximate those required for biological function. This ability is structure-encoded.
Based on recent observations, can we reconcile the MWC and KNF models? The accumulating data appear to support the MWC model, in general. On the other hand, there exist examples where sequences of events or intermediate states are reported [45;47;57;58]. While the intrinsic dynamics pre-dispose the protein to bind its substrate, the final conformation is usually stabilized upon local rearrangements induced after binding [12••;16;21•;24–26;39•;59], suggestive of the mechanism schematically described in Figure 2, and similar changes in energy landscape are entailed by allostery[60]. Therein a pre-existing equilibrium followed by the selection of the optimal conformation by the substrate and an induced fit to stabilize the final bound form is anticipated. Also, evidence has been presented for networks of coupled motions that facilitate enzyme catalysis, energy propagation or signal transmission [1•;30–32;35;36;42;50;51;55;56], which may elicit a sequence of events, reminiscent of the KNF model. Clearly in some systems the sequential events may be too fast and subtle to be detectable by experiments resulting in an apparent all-or-none process dominated by a highly cooperative single mode of motion.
Figure 2. Combination of a pre-existing equilibrium and post-binding rearrangement.

The protein (E) originally samples an ensemble of conformations, accessible via fluctuations in the global energy well (top panel), occasionally involving passages between substates within the well. The well is approximated by a harmonic potential (dashed curve) in coarse-grained NMA. Two conformations/substates are schematically shown, which are in a dynamic equilibrium prior to substrate (S) binding, as shown on the left box (bottom). The substrate (S) selects the conformer(s) that allows for optimal interaction (conformational selection). The stabilization of the final complex is subject to further rearrangement (induced fit) to stabilize the final bound form. The energy profile/landscape of the protein E in the presence of the substrate differs from its unbound form (top right diagram) inducing a shift in the population of the substates in favor of the conformer that binds the substrate.
Irrespective of the kinetics of conformational change, the intrinsic ability of the protein structures to undergo conformational changes along directions that enable their function unequivocally supports the mapping structure→dynamics→function. Structures are usually accepted to be designable when they meet the criterion of being thermodynamically stable (the lowest energy conformer) for a diversity of sequences [61]. With the emerging functionality and robustness (insensitivity to structural and energetic details) of proteins intrinsic dynamics, the pre-disposition to perform functional changes in conformation may probably be viewed as another criterion for designing structures. Structures may have evolved to ‘move’ in the right directions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- • 1.Hammes-Schiffer S, Benkovic SJ. Relating protein motion to catalysis. Annu Rev Biochem. 2006;75:519–541. doi: 10.1146/annurev.biochem.75.103004.142800. This is an extensive review of experimental and theoretical evidence supporting the interplay between conformational dynamics of enzymes and their catalytic activities, with focus on two enzymes, dehydrofolate reductase and liver alcohol dehydrogenase. A network of coupled motions involving both fast thermal vibrations and low frequency modes is pointed out to modulate the interactions between enzyme, substrate and cofactor at the active site, facilitating the hydride transfer reaction. [DOI] [PubMed] [Google Scholar]
- •• 2.Changeux JP, Edelstein SJ. Allosteric mechanisms of signal transduction. Science. 2005;308:1424–1428. doi: 10.1126/science.1108595. This is an insightful review of the progress made in the last forty years in understanding the allosteric mechanisms of signal tranduction. The basic concepts underlying the MWC model are summarized, such as the pre-disposition of symmetric oligomeric structures to undergo cooperative changes and the occurrence of reversible transitions, or spontaneous ‘switches’ between discrete conformations, accessible in the absence of ligand. The importance of binding ligands at strategic locations such as interfaces between subunits or along symmetry axes is emphasized. [DOI] [PubMed] [Google Scholar]
- 3.Bahar I, Rader AJ. Coarse-grained normal mode analysis in structural biology. Curr Opin Struct Biol. 2005;15:586–592. doi: 10.1016/j.sbi.2005.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- • 4.Tama F, Brooks CL. Symmetry, form, and shape: guiding principles for robustness in macromolecular machines. Annu Rev Biophys Biomol Struct. 2006;35:115–133. doi: 10.1146/annurev.biophys.35.040405.102010. An excellent review where the authors draw attention to the utility of multi-resolution methods based on elastic network models for understanding the functional dynamics of biological systems. They highlight the role of shape and form in the defining robust modes of motions. [DOI] [PubMed] [Google Scholar]
- 5.Koshland DE. Application of a theory of enzyme specificity to protein synthesis. Proc Natl Acad Sci. 1958;44:98–104. doi: 10.1073/pnas.44.2.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ma B, Shatsky M, Wolfson HJ, Nussinov R. Multiple diverse ligands binding at a single protein site: A matter of pre-existing populations. Protein Sci. 2002;11:184–197. doi: 10.1110/ps.21302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weber G. Ligand binding and internal equilibrium in proteins. Biochemistry. 1972;11:864–868. doi: 10.1021/bi00755a028. [DOI] [PubMed] [Google Scholar]
- 8.Monod J, Wyman J, Changeux JP. On the nature of allosteric transitions: A plausible model. J Mol Biol. 1965;12:88–118. doi: 10.1016/s0022-2836(65)80285-6. [DOI] [PubMed] [Google Scholar]
- 9.Koshland DE, Jr, Neméthy G, Filmer D. Comparison of experimental binding data and theoretical models in proteins containing subunits. Biochemistry. 1966;5:365–385. doi: 10.1021/bi00865a047. [DOI] [PubMed] [Google Scholar]
- 10.Frauenfelder H, McMahon BH, Austin RH, Chu K, Groves JT. The role of structure, energy landscape, dynamics, and allostery in the enzymatic function of myoglobin. Proc Natl Acad Sci USA. 2001;98:2370–2374. doi: 10.1073/pnas.041614298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- • 11.James LC, Roversi P, Tawfik DS. Antibody multispecificity mediated by conformational diversity. Science. 2003;299:1362–1367. doi: 10.1126/science.1079731. [DOI] [PubMed] [Google Scholar]
- •• 12.James LC, Tawfik DS. Structure and kinetics of a transient antibody binding intermediate reveal a kinetic discrimination mechanism in antigen recognition. Proc Natl Acad Sci USA. 2005;102:12730–12735. doi: 10.1073/pnas.0500909102. Binding of ligands to the antibody SPE7 was analyzed using kinetic measurements and X-ray crystallography to show that small ligands bind using the SPE7 pre-existing equilibrium followed by an induced fit that produces a high affinity complex. Nonspecific ligands are also able to bind the antibody. However, they are unable to induce the conformational changes for optimizing their interaction with SPE7, and consequently dissociate from the antibody. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kern D, Zuiderweg ER. The role of dynamics in allosteric regulation. Curr Opin Struct Biol. 2003;13:748–757. doi: 10.1016/j.sbi.2003.10.008. [DOI] [PubMed] [Google Scholar]
- • 14.Eisenmesser EZ, Millet O, Labeikovsky W, Korzhnev DM, Wolf-Watz M, Bosco DA, Skalicky JJ, Kay LE, Kern D. Intrinsic dynamics of an enzyme underlies catalysis. Nature. 2005;438:117–121. doi: 10.1038/nature04105. Cyclophilin motions observed during catalysis are detected by NMR relaxation to be already present in the unbound (free) form of the enzyme. This suggests that the motions necessary for catalysis are an intrinsic property of the enzymes. [DOI] [PubMed] [Google Scholar]
- 15.Labeikovsky W, Eisenmesser EZ, Bosco DA, Kern D. Structure and dynamics of pin1 during catalysis by NMR. J Mol Biol. 2007;367:1370–1381. doi: 10.1016/j.jmb.2007.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fetler L, Kantrowitz ER, Vachette P. Direct observation in solution of a preexisting structural equilibrium for a mutant of the allosteric aspartate transcarbamoylase. Proc Natl Acad Sci USA. 2007;104:495–500. doi: 10.1073/pnas.0607641104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eyal E, Yang LW, Bahar I. Anisotropic network model: systematic evaluation and a new web interface. Bioinformatics. 2006;22:2619–2627. doi: 10.1093/bioinformatics/btl448. [DOI] [PubMed] [Google Scholar]
- 18.Temiz NA, Meirovitch E, Bahar I. Escherichia coli adenylate kinase dynamics: comparison of elastic network model modes with mode-coupling (15)N-NMR relaxation data. Proteins. 2004;57:468–480. doi: 10.1002/prot.20226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xu C, Tobi D, Bahar I. Allosteric changes in protein structure computed by a simple mechanical model: hemoglobin T<-->R2 transition. J Mol Biol. 2003;333:153–168. doi: 10.1016/j.jmb.2003.08.027. [DOI] [PubMed] [Google Scholar]
- 20.Tama F, Sanejouand YH. Conformational change of proteins arising from normal mode calculations. Protein Eng. 2001;14:1–6. doi: 10.1093/protein/14.1.1. [DOI] [PubMed] [Google Scholar]
- • 21.Tobi D, Bahar I. Structural changes involved in protein binding correlate with intrinsic motions of proteins in the unbound state. Proc Natl Acad Sci USA. 2005;102:18908–18913. doi: 10.1073/pnas.0507603102. In this application of ANM to three different enzyme-substrate systems, the authors show that the motions of enzymes along the global modes facilitate ligand binding, while final stabilization is achieved by post-binding induced fit. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cui Q, Bahar IE. Normal Mode Analysis. In: Qiang C, Bahar I, editors. Theory and Applications to Biological and Chemical Systems. Taylor & Francis Group; 2006. [Google Scholar]
- • 23.Lou H, Cukier RI. Molecular dynamics of apo-adenylate kinase: a distance replica exchange method for the free energy of conformational fluctuations. J Phys Chem B. 2006;110:24121–24137. doi: 10.1021/jp064303c. The potential of mean force for the reaction coordinate between the open and closed forms of AK is demonstrated to have a rather flat region between the open and relatively closed forms. These results imply that apo AK can fluctuate between the open and closed conformations in the absence of its substrate. [DOI] [PubMed] [Google Scholar]
- 24.Hornak V, Okur A, Rizzo RC, Simmerling C. HIV-1 protease flaps spontaneously close to the correct structure in simulations following manual placement of an inhibitor into the open state. J Am Chem Soc. 2006;128:2812–2813. doi: 10.1021/ja058211x. MD simulations of HIV-1 protease demonstrate that the protein exists in a dynamic equilibrium between its close (bound), semi-open and open (unbound) conformations in the absence of the ligand. [DOI] [PMC free article] [PubMed] [Google Scholar]
- • 25.Grünberg R, Leckner J, Nilges M. Complementarity of structure ensembles in protein-protein binding. Structure. 2004;12:2125–2136. doi: 10.1016/j.str.2004.09.014. Substates populated by unbound receptor and ligand structures were generated by MD simulations for 17 protein complexes, which were subjected to combinatorial docking computations. Shape complementarity between substates emerges as a major determinant of the selection of particular pairs to form complexes, some of which are further rearranged to adopt stable structures. The authors proposed a 3-step mechanism for binding: diffusion, free conformer selection and refolding. [DOI] [PubMed] [Google Scholar]
- 26.Formaneck MS, Ma L, Cui Q. Reconciling the “old” and “new” views of protein allostery: a molecular simulation study of chemotaxis Y protein (CheY) Proteins. 2006;63:846–867. doi: 10.1002/prot.20893. [DOI] [PubMed] [Google Scholar]
- 27.Okazaki K, Koga N, Takada S, Onuchic JN, Wolynes PG. Multiple-basin energy landscapes for large-amplitude conformational motions of proteins: Structure-based molecular dynamics simulations. Proc Natl Acad Sci USA. 2006;103:11844–11849. doi: 10.1073/pnas.0604375103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kong Y, Karplus M. The signaling pathway of rhodopsin. Structure. 2007;15:611–623. doi: 10.1016/j.str.2007.04.002. [DOI] [PubMed] [Google Scholar]
- 29.Trylska J, Tozzini V, Chang CE, McCammon JA. HIV-1 protease substrate binding and product release pathways explored with coarse-grained molecular dynamics. Biophys J. 2007;92:4179–4187. doi: 10.1529/biophysj.106.100560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yu H, Ma L, Yang Y, Cui Q. Mechanochemical coupling in the myosin motor domain. II Analysis of critical residues. PLoS Comput Biol. 2007;3:e23. doi: 10.1371/journal.pcbi.0030023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yu H, Ma L, Yang Y, Cui Q. Mechanochemical coupling in the myosin motor domain. I Insights from equilibrium active-site simulations. PLoS Comput Biol. 2007;3:e21. doi: 10.1371/journal.pcbi.0030021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Agarwal PK. Role of protein dynamics in reaction rate enhancement by enzymes. J Am Chem Soc. 2005;127:15248–15256. doi: 10.1021/ja055251s. [DOI] [PubMed] [Google Scholar]
- 33.Yang LW, Bahar I. Coupling between catalytic site and collective dynamics: a requirement for mechanochemical activity of enzymes. Structure. 2005;13:893–904. doi: 10.1016/j.str.2005.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kurkcuoglu O, Jernigan RL, Doruker P. Loop motions of triosephosphate isomerase observed with elastic networks. Biochemistry. 2006;45:1173–1182. doi: 10.1021/bi0518085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Agarwal PK, Billeter SR, Rajagopalan PT, Benkovic SJ, Hammes-Schiffer S. Network of coupled promoting motions in enzyme catalysis. Proc Natl Acad Sci USA. 2002;99:2794–2799. doi: 10.1073/pnas.052005999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Benkovic SJ, Hammes-Schiffer S. A perspective on enzyme catalysis. Science. 2003;301:1196–1202. doi: 10.1126/science.1085515. [DOI] [PubMed] [Google Scholar]
- 37.Olsson MH, Parson WW, Warshel A. Dynamical contributions to enzyme catalysis: critical tests of a popular hypothesis. Chem Rev. 2006;106:1737–1756. doi: 10.1021/cr040427e. [DOI] [PubMed] [Google Scholar]
- 38.Liu H, Warshel A. The catalytic effect of dihydrofolate reductase and its mutants is determined by reorganization energies. Biochemistry. 2007;46:6011–6025. doi: 10.1021/bi700201w. [DOI] [PubMed] [Google Scholar]
- • 39.Gunasekaran K, Ma B, Nussinov R. Is allostery an intrinsic property of all dynamic proteins? Proteins. 2004;57:433–443. doi: 10.1002/prot.20232. In this review, allostery is proposed to derive from redistribution in population following ligand binding and as a consequence all proteins are pointed out to be potentially allosteric. [DOI] [PubMed] [Google Scholar]
- 40.Taly A, Delarue M, Grutter T, Nilges M, Le NN, Corringer PJ, Changeux JP. Normal mode analysis suggests a quaternary twist model for the nicotinic receptor gating mechanism. Biophys J. 2005;88:3954–3965. doi: 10.1529/biophysj.104.050229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shrivastava IH, Bahar I. Common mechanism of pore opening shared by five different potassium channels. Biophys J. 2006;90:3929–3940. doi: 10.1529/biophysj.105.080093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu T, Whitten ST, Hilser VJ. Ensemble-based signatures of energy propagation in proteins: a new view of an old phenomenon. Proteins. 2006;62:728–738. doi: 10.1002/prot.20749. [DOI] [PubMed] [Google Scholar]
- 43.Hawkins RJ, McLeish TC. Coupling of global and local vibrational modes in dynamic allostery of proteins. Biophys J. 2006;91:2055–2062. doi: 10.1529/biophysj.106.082180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- • 44.Ma L, Cui Q. The activation mechanism of a signaling protein at atomic resolution from advanced computations. J Am Chem Soc. 2007 doi: 10.1021/ja073059f. in press. To clarify the activation mechanism of CheY as a prototypical signaling molecule, the authors generate and analyze 160 unbiased activation trajectories using transition path sampling and free energy simulations. The rotational isomerization of Tyr106 (functional residue at substrate binding site) is shown to be a low-energy-barrier transition readily sampled during simulations, irrespective of the phosphorylation of Thr87. This supports a population shift mechanism in line with MWC model, as opposed to the traditional Y-T coupling model which would require the Thr87 phosphorylation to elicit the isomerization of Tyr106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dyer CM, Dahlquist FW. Switched or not? The structure of unphosphorylated CheY bound to the N terminus of FliM. J Bacteriol. 2006;188:7354–7363. doi: 10.1128/JB.00637-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rousseau F, Schymkowitz J. A systems biology perspective on protein structural dynamics and signal transduction. Curr Opin Struct Biol. 2005;15:23–30. doi: 10.1016/j.sbi.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 47.Stock AM, Guhaniyogi J. A new perspective on response regulator activation. J Bacteriol. 2006;188:7328–7330. doi: 10.1128/JB.01268-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- • 48.Ming D, Wall ME. Allostery in a coarse-grained model of protein dynamics. Phys Rev Lett. 2005;95:198103. doi: 10.1103/PhysRevLett.95.198103. This study provides a quantitative measure based on KL divergence for the change in conformational dynamics induced by ligand binding. [DOI] [PubMed] [Google Scholar]
- 49.Ming D, Wall ME. Interactions in native binding sites cause a large change in protein dynamics. J Mol Biol. 2006;358:213–223. doi: 10.1016/j.jmb.2006.01.097. [DOI] [PubMed] [Google Scholar]
- 50.Chennubhotla C, Bahar I. Markov propagation of allosteric effects in biomolecular systems: application to GroEL-GroES. Mol Syst Biol. 2006;2:36. doi: 10.1038/msb4100075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ranson NA, Clare DK, Farr GW, Houldershaw D, Horwich AL, Saibil HR. Allosteric signaling of ATP hydrolysis in GroEL-GroES complexes. Nat Struct Mol Biol. 2006;13:147–152. doi: 10.1038/nsmb1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lockless SW, Ranganathan R. Evolutionarily conserved pathways of energetic connectivity in protein families. Science. 1999;286:295–299. doi: 10.1126/science.286.5438.295. [DOI] [PubMed] [Google Scholar]
- 53.Russ WP, Lowery DM, Mishra P, Yaffe MB, Ranganathan R. Natural-like function in artificial WW domains. Nature. 2005;437:579–583. doi: 10.1038/nature03990. [DOI] [PubMed] [Google Scholar]
- • 54.Ota N, Agard DA. Intramolecular signaling pathways revealed by modeling anisotropic thermal diffusion. J Mol Biol. 2005;351:345–354. doi: 10.1016/j.jmb.2005.05.043. A novel MD simulation method, anisotropic thermal diffusion, is introduced and used to examine the long-range propagation of conformational changes that mediate allosteric communication. Successful application to a member of the PDZ domain is presented. [DOI] [PubMed] [Google Scholar]
- 55.Chennubhotla C, Bahar I. Markov Propagation of Signals in Proteins and its Relation to Equilibrium Fluctuations. PLoS Comput Biol. 2007 doi: 10.1371/journal.pcbi.0030172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- • 56.Liu T, Whitten ST, Hilser VJ. Functional residues serve a dominant role in mediating the cooperativity of the protein ensemble. Proc Natl Acad Sci USA. 2007;104:4347–4352. doi: 10.1073/pnas.0607132104. An ensemble-based description of proteins is utilized to evaluate the extent to which perturbations at different sites propagate to the entire structure. Application to a database indicates that perturbations at binding sites affect the cooperativity between all residues. The authors suggest that binding residues are inherently located at sites poised to effectively propagate signals. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ragona L, Catalano M, Luppi M, Cicero D, Eliseo T, Foote J, Fogolari F, Zetta L, Molinari H. NMR dynamic studies suggest that allosteric activation regulates ligand binding in chicken liver bile acid-binding protein. J Biol Chem. 2006;281:9697–9709. doi: 10.1074/jbc.M513003200. [DOI] [PubMed] [Google Scholar]
- 58.Popovych N, Sun S, Ebright RH, Kalodimos CG. Dynamically driven protein allostery. Nat Struct Mol Biol. 2006;13:831–838. doi: 10.1038/nsmb1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rajamani D, Thiel S, Vajda S, Camacho CJ. Anchor residues in protein-protein interactions. Proc Natl Acad Sci USA. 2004;101:11287–11292. doi: 10.1073/pnas.0401942101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Swain JF, Gierasch LM. The changing landscape of protein allostery. Curr Opin Struct Biol. 2006;16:102–108. doi: 10.1016/j.sbi.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 61.Li H, Helling R, Tang C, Wingreen N. Emergence of preferred structures in a simple model of protein folding. Science. 1996;273:666–669. doi: 10.1126/science.273.5275.666. [DOI] [PubMed] [Google Scholar]
- 62.Wang Y, Rader AJ, Bahar I, Jernigan RL. Global ribosome motions revealed with elastic network model. J Struct Biol. 2004;147:302–314. doi: 10.1016/j.jsb.2004.01.005. [DOI] [PubMed] [Google Scholar]
- 63.Tama F, Valle M, Frank J, Brooks CL., III Dynamic reorganization of the functionally active ribosome explored by normal mode analysis and cryo-electron microscopy. Proc Natl Acad Sci USA. 2003;100:9319–9323. doi: 10.1073/pnas.1632476100. [DOI] [PMC free article] [PubMed] [Google Scholar]