

A stable stoichiometric complex of archaeal DNA polymerase with proliferating cell nuclear antigen (PCNA) was formed using a PCNA monomer mutant and the complex was successfully crystallized.
Keywords: DNA polymerase, proliferating cell nuclear antigen
Abstract
Replicative DNA polymerase interacts with processivity factors, the β-subunit of DNA polymerase III or proliferating cell nuclear antigen (PCNA), in order to function with a long template DNA. The archaeal replicative DNA polymerase from Pyrococcus furiosus interacts with PCNA via its PCNA-interacting protein (PIP) motif at the C-terminus. The PCNA homotrimeric ring contains one PIP interacting site on each monomer and since the ring can accommodate up to three molecules simultaneously, formation of a stable stoichiometric complex of PCNA with its interacting protein has been difficult to control in vitro. A stable complex of the DNA polymerase with PCNA, using a PCNA monomer mutant, has been purified and crystallized. The best ordered crystal diffracted to 3.0 Å resolution using synchrotron radiation. The crystals belong to space group P21212, with unit-cell parameters a = 225.3, b = 123.3, c = 91.3 Å.
1. Introduction
DNA replication and repair processes play central roles in maintaining the integrity of genomic information. Extensive studies on the molecular mechanism of DNA replication revealed the existence of a protein that retains DNA polymerase on the template DNA strand for processive synthesis (reviewed in Waga & Stillman, 1998 ▶; Hingorani & O’Donnell, 2000 ▶). The DNA polymerase processivity factor, which is often called a sliding clamp, forms a dimer or trimer depending on the kingdom of life. Proliferating cell nuclear antigen (PCNA), which is an accessory protein essential for processive DNA synthesis in archaea and eukarya, forms a homotrimer. The central hole of the PCNA trimeric ring encircles double-stranded DNA so that DNA polymerases can operate for DNA synthesis with PCNA along a DNA template. The eukaryotic PCNA also anchors various proteins involved in DNA repair, cell-cycle control and apoptotic processes (reviewed in Kelman & Hurwitz, 1998 ▶; Tsurimoto, 1998 ▶).
Most of the PCNA-interacting proteins utilize the PIP (PCNA-interacting protein) motif, which mainly consists of seven hydrophobic residues (Warbrick et al., 1998 ▶) bound to the edge of the PCNA ring (Gulbis et al., 1996 ▶). A replicative DNA polymerase PolBI from Pyrococcus furiosus (Pfu) belongs to the family B DNA polymerases and is composed of a 3′-5′ exonuclease domain in its N-terminal half and the three polymerase domains in the remaining C-terminal region. This polymerase has a PIP motif at its C-terminal end. The DNA-synthesis activity of PolBI is stimulated by PfuPCNA (Cann et al., 1999 ▶). A mutant PolBI which lacked the PIP motif-like segment did not respond to PfuPCNA, suggesting that the PIP motif is essential for their interaction (Ishino et al., unpublished work).
The homotrimeric ring of PCNA possesses three PIP associating sites, belonging to a single subunit. Owing to difficulties in the in vitro formation of a stable stoichiometric complex between PCNA and the cognate proteins, only few successful structural analyses have been reported for such complexes. The recent structure determination of human FenI complexed with PCNA revealed an interesting feature in which all three interacting sites were occupied by FenI molecules in the crystal (Sakurai et al., 2005 ▶). In the case of the complex of Escherichia coli PolIV and the β-clamp, the two ‘little-finger’ domains of PolIV were bound to each of the two monomers in the β-clamp (Bunting et al., 2003 ▶). Both structures showed that the clamp-interacting proteins were located at the edge of the ring to maintain an inactive ‘locked-down’ orientation.
When we tried to produce a stoichiometric complex of PfuPolBI and PfuPCNA in the absence of DNA, three polymerase molecules were simultaneously loaded onto the PCNA ring in the resulting complex, as determined by a gel-filtration study including profile analyses on the components. In contrast, when we constructed the complex in the presence of various template DNA molecules, the stoichiometry of the protein components was 1:1. This complex was successfully crystallized, but the crystals diffracted to at most 6 Å resolution (Nishida et al., unpublished results). To overcome this poor diffraction, we used a PCNA monomer mutant (PCNAm, D143A/D147A) in which two aspartic acid residues were replaced by alanines in order to disrupt the ionic interactions between the monomers (Matsumiya et al., 2003 ▶). Thus, we succeeded in growing crystals of the complex which diffracted to 3 Å resolution. The crystals include whole molecules of PfuPolBI (775 amino acids, 90.1 kDa) and PfuPCNAm (249 amino acids, 28.0 kDa).
2. Expression and purification
The purification of PfuPolBI and PfuPCNAm was performed as described previously (Komori & Ishino, 2000 ▶; Matsumiya et al., 2001 ▶). Purified samples of each protein were mixed in a molar ratio of 1:1.2 (Pol:PCNAm). The mixture was dissolved in buffer A [50 mM Tris–HCl pH 8.0, 10%(v/v) glycerol, 0.1 mM EDTA, 0.5 mM DTT] with 0.5 M NaCl and was dialyzed against the same buffer containing 0.1 M NaCl. The equilibrated sample was concentrated and loaded onto a gel-filtration column (Superdex 200 HiLoad 26/60, Amersham Pharmacia) equilibrated with buffer A containing 0.1 M NaCl and was eluted with the same buffer. The fractions from a single peak corresponding to the complex of PfuPolBI (90.1 kDa) with PfuPCNAm (28.0 kDa) were collected and used for crystallization. Fig. 1 ▶ shows the gel-filtration profiles of the complex compared with those of purified PfuPolBI analyzed using the SMART system (Amersham Pharmacia) equipped with a Superdex 200 column. The molecular weight corresponding to the largest elution peak of the complex was equivalent to the value for a 1:1 PfuPolBI–PfuPCNAm complex.
Figure 1.

The elution profiles of PfuPolBI with PfuPCNAm gel-filtration chromatography measured at 260 and 280 nm are indicated by the elution volume; they are compared with those of purified PfuPolBI. The elution positions of the marker proteins are shown by arrowheads, with each molecular weight (in kDa) under the chromatographic profile. Each peak fraction was subjected to SDS–PAGE analysis as shown in Fig. 3 ▶.
3. Crystallization
Crystallization was initiated at 293 K using the hanging-drop vapour-diffusion method. For initial screening, 10, 20 and 40 mg ml−1 concentrations of the complex in storage buffer [10%(v/v) glycerol, 0.5 mM dithiothreitol, 0.1 M NaCl, 50 mM Tris–HCl pH 8] were used with 1 µl protein solution mixed with 0.5 µl precipitant solution. A cluster of crystals was obtained after 1–2 weeks from a mixture of protein solution [20 mg ml−1 protein, 10%(v/v) glycerol, 0.5 mM dithiothreitol, 0.1 M NaCl, 50 mM Tris–HCl pH 8] and precipitant solution [0.2 M ammonium sulfate, 30%(w/v) polyethyleneglycol monomethylether 5000, 0.1 M MES pH 6.5; Crystal Screen 2 No. 26, Hampton Research] (Fig. 2 ▶ a). The addition of 1%(v/v) 1,8-diaminooctane improved the crystal dimensions (1 × 0.5 × 0.05 mm) and helped to prevent cluster formation (Fig. 2 ▶ b). Fig. 3 ▶ shows the SDS–PAGE analysis of the obtained crystals in comparison with the peak samples from the gel-filtration analysis.
Figure 2.

(a) Crystals obtained from the precipitant solution of ammonium sulfate and polyethylene glycol monomethylether 5000. (b) The addition of 1%(v/v) 1,8-diaminooctane to the above solution dramatically improved the crystal size and inhibited the clustering.
Figure 3.

5 µl aliquots of the peaks from the gel-filtration analysis and a dissolved sample of the obtained crystals were analyzed by SDS–PAGE. The gel was stained with Coomassie blue dye. Lane 1, molecular-weight markers (sizes are shown in kDa on the left); lane 2, PfuPolBI; lane 3, PfuPolBI + PfuPCNAm, gel filtration; lane 4, PfuPolBI + PfuPCNAm, crystal. The crystals were washed in the precipitant solution and then dissolved in buffer A.
4. X-ray analysis
The crystals were cryoprotected by soaking briefly in mother liquor supplemented with 10%(v/v) ethylene glycol before flash-freezing in a boiled-off N2 stream. The crystals belong to space group P21212, with unit-cell parameters a = 225.3, b = 123.3, c = 91.3 Å. Assuming the presence of two 1:1 complexes in the asymmetric unit, the solvent content of the crystals is 54%, corresponding to a Matthews coefficient (Matthews, 1968 ▶) V M of 2.7 Å3 Da−1. The native data set was collected at beamline BL41XU (SPring-8, Japan) to a resolution of 3.2 Å (Fig. 4 ▶). Diffraction data were processed and scaled with HKL2000 (Otwinowski & Minor, 1997 ▶) and the data-collection statistics are summarized in Table 1 ▶.
Figure 4.

Diffraction from a crystal of the Pfu Pol–PfuPCNAm complex. The arrow indicates a diffraction spot at 3.1 Å resolution.
Table 1. Data-collection and processing statistics.
Values in parentheses are for the last shell.
| Space group | P21212 |
| Radiation | BL41XU, SPring-8 |
| Unit-cell parameters (Å) | a = 225.3, b = 123.3, c = 91.3 |
| Temperature (K) | 100 |
| Wavelength (Å) | 1.0 |
| Oscillation range (°) | 1.0 |
| Distance (mm) | 435 |
| No. of frames | 120 |
| Resolution limit (Å) | 50.0–3.2 (3.31–3.20) |
| Total reflections | 197722 (17898) |
| Independent reflections | 42642 (4073) |
| Average redundancy | 4.5 (4.4) |
| Average I/σ(I) | 5.5(3.3) |
| Completeness (%) | 99.4 (97.0) |
| Rmerge† | 0.112 (0.522) |
R
merge = 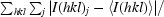
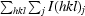 , where I(hkl)j is the jth measurement of the intensity of reflection hkl and 〈I(hkl)〉 is the mean intensity of reflection hkl.
, where I(hkl)j is the jth measurement of the intensity of reflection hkl and 〈I(hkl)〉 is the mean intensity of reflection hkl.
The stable stoichiometric complex of PfuPolBI and PCNA was easily obtained by making use of the PCNA monomer mutant D143A/D147A and the complex was successfully crystallized. We observed no stoichiometry other than 1:1, unlike the case when the wild-type PCNA homotrimeric ring was utilized for the basis of the complex. Although Asp mutations caused disruption of the PCNA ring, the crystal structure of this monomer mutant retained the same structure as found in the ring (Matsumiya et al., 2003 ▶) and the crystal structures of PCNA with its interacting protein (peptide) obtained to date have shown each interaction area to be restricted to within a single subunit in the PCNA ring and to be distant from the adjacent subunits (Gulbis et al., 1996 ▶; Matsumiya et al., 2001 ▶; Bunting et al., 2003 ▶; Sakurai et al., 2005 ▶). These observations suggest that our artificial 1:1 complex of PfuPolBI and the PfuPCNA monomer may represent a physiological interaction to some degree. We presume that the various complexes produced with different stoichiometries disturb the purification when wild-type PCNA is used for complex formation. A number of conformers are derived from the possible combinations between the PCNA monomer and the interacting proteins, as observed in the human PCNA–FenI complex. This heterogeneity in protein components could cause difficulties in crystallization.
Determination of this complex structure will provide crucial information regarding important aspects of the replication machinery, such as the possible reorganization of the exonuclease and polymerase domains of family B polymerases upon binding to the sliding clamp.
Acknowledgments
We are grateful to Dr T. Oyama for helpful discussions. We thank Dr S. Ishino for help with the purification of the PCNA monomer mutant. This work was partly supported by a research grant endorsed by the New Energy and Industrial Technology Development Organization (NEDO) and YI was supported by a grant from the Human Frontier Science Program (HFSP) and a Grant-in-Aid from the Ministry of Education, Science and Sports of Japan.
References
- Bunting, K. A., Roe, S. M. & Pearl, L. H. (2003). EMBO J.22, 5883–5892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cann, I. K. O., Ishino, S., Hayashi, I., Komori, K., Toh, H., Morikawa, K. & Ishino, Y. (1999). J. Bacteriol.181, 6591–6599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulbis, J. M., Kelman, Z., Hurwitz, J., O’Donnell, M. & Kuriyan, J. (1996). Cell, 87, 297–306. [DOI] [PubMed] [Google Scholar]
- Hingorani, M. M. & O’Donnell, M. (2000). Curr. Biol.10, R25–R29. [DOI] [PubMed] [Google Scholar]
- Kelman, Z. & Hurwitz, J. (1998). Trends Biochem. Sci.23, 236–238. [DOI] [PubMed] [Google Scholar]
- Komori, K. & Ishino, Y. (2000). Protein Eng.13, 41–47. [DOI] [PubMed] [Google Scholar]
- Matsumiya, S., Ishino, S., Ishino, Y. & Morikawa, K. (2003). Protein Sci.12, 823–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumiya, S., Ishino, Y. & Morikawa, K. (2001). Protein Sci.10, 17–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews, B. W. (1968). J. Mol. Biol.33, 491–497. [DOI] [PubMed] [Google Scholar]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Sakurai, S., Kitano, K., Yamaguchi, H., Hamada, K., Okada, K., Fukuda, K., Uchida, M., Ohtsuka, E., Morioka, H. & Hakoshima, T. (2005). EMBO J.24, 683–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsurimoto, T. (1998). Biochim. Biophys. Acta, 1443, 23–39. [DOI] [PubMed] [Google Scholar]
- Waga, S. & Stillman, B. (1998). Annu. Rev. Biochem.67, 721–751. [DOI] [PubMed] [Google Scholar]
- Warbrick, E., Heatherington, W., Lane, D. P. & Glover, D. M. (1998). Nucleic Acids Res.26, 3925–3932. [DOI] [PMC free article] [PubMed] [Google Scholar]