

Abstract
The cargo that the molecular motor kinesin moves along microtubules has been elusive. We searched for binding partners of the COOH terminus of kinesin light chain, which contains tetratricopeptide repeat (TPR) motifs. Three proteins were found, the c-jun NH2-terminal kinase (JNK)–interacting proteins (JIPs) JIP-1, JIP-2, and JIP-3, which are scaffolding proteins for the JNK signaling pathway. Concentration of JIPs in nerve terminals requires kinesin, as evident from the analysis of JIP COOH-terminal mutants and dominant negative kinesin constructs. Coprecipitation experiments suggest that kinesin carries the JIP scaffolds preloaded with cytoplasmic (dual leucine zipper–bearing kinase) and transmembrane signaling molecules (the Reelin receptor, ApoER2). These results demonstrate a direct interaction between conventional kinesin and a cargo, indicate that motor proteins are linked to their membranous cargo via scaffolding proteins, and support a role for motor proteins in spatial regulation of signal transduction pathways.
Keywords: kinesin, molecular motors, scaffolding protein, JNK signaling, Reelin
Introduction
Kinesins are a family of motor proteins that use the energy of ATP hydrolysis to move cargoes along microtubules (MTs). The founding member of the family, conventional kinesin, was identified in squid axoplasm over 15 years ago as a motor that could drive fast axonal transport. A large body of biochemical and localization experiments have implicated kinesin in a variety of processes in many cell types (Brendza et al. 2000a,Brendza et al. 2000b; Goldstein and Gunawardena 2000; Goldstein and Yang 2000; Kamal and Goldstein 2000; Rogers and Gelfand 2000; Terada et al. 2000; Yabe et al. 2000). However, evidence linking kinesin directly to any of the proposed cellular roles is lacking; that is, a molecular relationship between kinesin and a cargo has yet to be established.
Genetic studies in Caenorhabditis elegans, Drosophila melanogaster, and mice have failed to identify the primary cargo of conventional kinesin although, they have demonstrated that kinesin is required for axonal transport in neuronal cells (Saxton et al. 1991; Gho et al. 1992; Patel et al. 1993; Hurd and Saxton 1996; Hurd et al. 1996; Gindhart et al. 1998; Tanaka et al. 1998; Rahman et al. 1999). Mammalian conventional kinesin (kinesin family member [KIF]5B) associates with the actin-based motor MyoVA, but neither motor is envisioned as the cargo of the other; rather, this interaction is likely to be important for coordinating the transition of vesicle transport from MTs to actin filaments (Brown 1999; Huang et al. 1999; Goode et al. 2000). Attempts to identify the kinesin–cargo linker molecule have also met with limited success. The protein kinectin was identified biochemically as a potential kinesin–cargo linker; however, its role has been controversial. No homologue for kinectin exists in the Drosophila genome, arguing against its role in kinesin-driven transport (Goldstein and Gunawardena 2000).
Conventional kinesin is a heterotetramer of two kinesin heavy chains (KHCs) and two kinesin light chains (KLCs). KHC contains three domains: an NH2-terminal motor domain, a central coiled coil stalk domain for dimerization, and a COOH-terminal globular domain. KLC contains an NH2-terminal α-helical domain that associates with the KHC stalk and six tetratricopeptide repeat (TPR) motifs (Vale and Fletterick 1997; Diefenbach et al. 1998; Verhey et al. 1998). The roles of most of the domains of kinesin have been elucidated, and, in particular, many of the molecular details of how the motor domain of kinesin walks processively along an MT have been clarified (for reviews see Vale and Fletterick 1997; Manning and Snyder 2000; Vale and Milligan 2000; Wade and Kozielski 2000; Woehlke and Schliwa 2000).
Given the processive nature of the kinesin motor in vitro, it must be tightly regulated in vivo to prevent its accumulation at the cell periphery. Indeed, cargoless kinesin is inhibited from movement along MTs due to a folded conformation that allows the KHC COOH-terminal domain to associate physically with the NH2-terminal motor domain and thus inhibit motor activity (Verhey et al. 1998; Coy et al. 1999; Friedman and Vale 1999; Stock et al. 1999; Manning and Snyder 2000). KLC stabilizes the inhibitory interaction of the KHC COOH-terminal and motor domains (Verhey et al. 1998). How the motor is activated is unclear. In the simplest scenario, cargo binding alone would activate kinesin, but it is possible that additional activation, such as posttranslational modification (Hollenbeck 1993; Lee and Hollenbeck 1995; De Vos et al. 2000), local changes in the cellular environment (Verhey et al. 1998), or chaperone binding (Tsai et al. 2000), is required.
The tail of the kinesin molecule, encompassing the KHC COOH terminus and KLC, is the most likely region to be involved in cargo binding, although it is not established whether regions of both KHC and KLC are required. Several studies point to a role for the KHC COOH terminus in cargo binding, as this region binds membranes (Skoufias et al. 1994; Bi et al. 1997). In addition, several studies in the filamentous fungus Neurospora crassa have implied that KHC alone binds to cargo (Steinberg and Schliwa 1995; Kirchner et al. 1999a; Seiler et al. 2000). However, the results of disruption of KLC in Drosophila and mice suggest an essential role for KLC in kinesin function (Gindhart et al. 1998). Our previous work led us to suggest that the TPR motifs of KLC are involved in cargo binding, as all other notable domains of KHC and KLC could be assigned a function either in motor activity or in the interaction between the two chains (Verhey et al. 1998). The KLC TPR motifs are highly conserved across species, and TPR motifs are known to be involved in protein–protein interactions. A role for the KLC TPRs in cargo binding is supported by experiments in which an antibody to this region, when injected into squid axoplasm, dissociates organelles from MTs (Stenoien and Brady 1997).
TPR motifs consist of degenerate 34–amino acid repeats, often arranged as multiple copies in tandem, and are present in proteins involved in diverse cellular processes such as phosphate transfer, cell cycle control, protein folding, and mitochondrial and peroxisomal import (for reviews see Blatch and Lassle 1999; Groves and Barford 1999). In the case of two TPR-containing proteins, the Hsp70/Hsp90 organizing protein Hop and the peroxisomal import receptor PAS8, three or more TPR motifs form a TPR domain that recognizes short stretches of primary amino acid sequence at the COOH termini of their target proteins, −EEVD in Hsp70 and Hsp90 and −SKL in peroxisomal proteins, respectively (Terlecky et al. 1995; Scheufler et al. 2000).
To identify proteins that interact with kinesin, we undertook a yeast two-hybrid screen using the TPR motifs of KLC as a bait. Three proteins were identified, c-jun NH2-terminal kinase (JNK)–interacting protein (JIP)-1, JIP-2, and JIP-3. All three have been proposed to act as scaffolding proteins for the JNK kinase signaling pathway. Here, we report that disruption of the kinesin–JIP interaction in neuronal cells results in mislocalization of the JIP-1 scaffolding protein, suggesting that JIPs are a cargo for kinesin. Coprecipitation of kinesin and JIP-1 with the upstream kinase dual leucine zipper kinase (DLK) and the Reelin receptor, ApoER2, suggests that kinesin carries the JIP scaffolding proteins as part of a multiprotein complex. Our results provide the first demonstration of a direct interaction between conventional kinesin and a cargo. In addition, these results suggest a general model in which motor proteins are linked to their membranous cargo via scaffolding proteins. Finally, we discuss a model in which motor proteins localize signal transduction pathways and may in turn be regulated by them.
Materials and Methods
Plasmids
Plasmids encoding Myc-tagged rat KHC and the truncation Myc-KHC-891, as well as HA (hemagglutinin)-tagged rat KLC and the truncation HA-KLC-176, have been described previously (Verhey et al. 1998). To make a construct containing the six TPR motifs of rat KLC in pcDNA3 expression vector, the plasmid pcDNA3-HA-KLC-488 (Verhey et al. 1998) was digested with XhoI, which cuts immediately after the HA tag, and NotI, which cuts in the rat KLC gene preceding the TPR motifs. The linear piece was religated with a linker designed to conserve the reading frame and introduce an NheI site between the HA tag and the TPR motifs. For the two-hybrid system, sequences encoding the TPR motifs of KLC were subcloned from pcDNA3-HA-KLC-TPR6 into vector pGBDU-C1 (James et al. 1996), whereas sequences encoding the TPR motifs of protein phosphatase 5 (PP5) (cDNA gift of D. Barford, University of Oxford, Oxford, UK) were obtained by PCR with primers containing the appropriate restriction sites and were subcloned into pGBDU-C1 (James et al. 1996).
Flag-tagged JIP-1, JIP-2, and JIP-3 in mammalian expression vectors were a gift of Dr. Roger Davis (University of Massachusetts Medical School, Worcester, MA) and have been described previously (Whitmarsh et al. 1998; Yasuda et al. 1999; Kelkar et al. 2000). Myc-tagged JIP-1 (307–711) was described previously (Meyer et al. 1999). Myc-tagged full-length JIP-1 was generated by annealing overlapping oligonucleotides encoding human JIP-1 amino acids 1–120. This sequence was amplified by PCR and then cloned into RK5 Myc–JIP-1 plasmid containing the rest of the human JIP-1 sequence to generate the full-length gene. Myc JIP-1 (307–700) was generated by mutating tyrosine 701 to a stop codon, and Myc JIP-1 (P704A) and (Y709A) were generated by making the appropriate point mutations within the Myc JIP-1 (307–711) plasmid using the QuickChange site-directed mutagenesis kit (Stratagene).
Two-Hybrid Screen
The yeast two-hybrid screen was carried out using a MATCHMAKER GAL4 two-hybrid system 3 (CLONTECH Laboratories, Inc.), and all methods were done according to the manufacturer's protocols. In brief, PJ69-4A cells (James et al. 1996) were transformed with pGBDU-KLC TPRs and selected for growth on media lacking uracil. PJ69-4A KLC TPR yeast were subsequently transformed with a mouse brain MATCHMAKER cDNA library in plasmid pACT2 (CLONTECH Laboratories, Inc.). Positive interacting clones were selected for growth on plates lacking uracil, leucine, and histidine and were supplemented with 10 mM 3-aminotriazole. Positive clones were subsequently tested for growth on plates lacking uracil, leucine, and adenine and for β-galactosidase activity. To confirm the specificity of the interacting plasmids, PJ69-4A yeast expressing only the library plasmid were mated to Y187 yeast expressing the KLC TPRs, the PP5 TPRs, or the bait plasmid pGBDU alone.
Cells, Transfection, Immunofluorescence, and Immunoprecipitation
African green monkey kidney cells (COS) were grown as described (Verhey et al. 1998), except that FuGene 6 (Roche) was used for transfections according to the manufacturer's instructions. Human embryonic kidney HEK293 cells were grown and transfected as described (Meyer et al. 1999). For immunoprecipitation, COS or 293 cells were washed, lysed, and processed for immunoprecipitation with mAbs to the HA, Myc, and Flag tags (12CA5, 9E10, and M2 [Sigma-Aldrich]) as described (Verhey et al. 1998; Meyer et al. 1999). Immunoprecipitates were detected by immunoblotting with polyclonal antibodies to the Myc and HA tags (Santa Cruz Biotechnology, Inc.). A polyclonal antibody to the Flag tag was generated in rabbits against the Flag peptide (DYKDDDDKSHC) and affinity-purified as described (Verhey et al. 1998).
The mouse central nervous system catecholaminergic cell line CAD (Qi et al. 1997) was grown in a 1:1 mixture of F12–DME (Bio-Whittaker) plus 10% FBS (HyClone). Cells were transfected using FuGene 6 (Roche) in Opti-MEM (GIBCO BRL) according to the manufacturer's instructions. The mouse neuroblastoma cell line NIE 115 was maintained as described previously (Meyer et al. 1999) and transfected using Effectene (QIAGEN) according to the manufacturer's instructions. 12 h after transfection, cells were replated on coverslips and switched to differentiation media (F12–DMEM without serum or DME without serum). 48–72 h later, the cells were fixed and processed for indirect immunofluorescence as described (Verhey et al. 1998; Meyer et al. 1999). Localization of endogenous JIP-1 protein was detected using an affinity-purified polyclonal antibody that is specific for JIP-1 (Meyer et al. 1999). Microtubules and neurofilaments were detected with mAbs to tubulin (DM1α; Sigma-Aldrich) and neurofilaments H + M (Zymed Laboratories), whereas mitochondria were labeled with MitoTracker (Molecular Probes). Goat anti–mouse fluorescein isothiocyanate–conjugated and goat anti–rabbit rhodamine-conjugated secondary antibodies were used (Molecular Probes).
Immunoprecipitation and MT Binding from Rat Brain
Rat brains were homogenized in buffer A (50 mM Hepes-KOH, pH 7.4, 50 mM potassium acetate, 6 mM magnesium acetate, 1 mM EDTA, 250 mM sucrose, 1 mM DTT, 1 mM PMSF plus protease inhibitors [10 μg/ml leupeptin, 5 μg/ml chymostatin, 3 μg/ml elastatinal, 1 μg/ml pepstatin]). The homogenate was subjected to a low speed spin (10,000 g, 10 min, 4°C), followed by a high speed spin (>100,000 g, 45 min, 4°C). The resultant supernatant was removed, and the pellet was resuspended in buffer C (50 mM Hepes-KOH, 250 mM sucrose, 1 mM PMSF, protease inhibitors). Aliquots were frozen in liquid nitrogen and stored at −80°C. 1,000–1,500 μg of high speed supernatant was diluted with an equal volume of buffer B (20 mM Hepes-KOH, pH 7.4, 115 mM potassium acetate, 5 mM sodium acetate, 2 mM MgCl2, 1 mM EGTA, 10% glycerol, 25 mM β-glycerol phosphate, 10 mM NaF, 1 mM sodium orthovanadate, 1 mM PMSF, protease inhibitors) containing no detergent, 1% Triton X-100, 2% digitonin, or 1% octyl glucoside. After solubilizing by rotation for 20 min at 4°C, the insoluble material was removed by spinning at 14,000 rpm, 15 min, 4°C. The supernatant was used for immunoprecipitation and MT binding experiments.
For immunoprecipitation, 100 μl of mouse ascites (H2) against KHC (gift of G. Bloom, University of Texas Southwestern Medical Center, Dallas, TX) was added to the supernatant, and immunoprecipitates were isolated by the addition of protein G (Amersham Pharmacia Biotech). The immunoprecipitates were washed three times with buffer B containing no detergent. Associated proteins were detected by immunoblotting the precipitates with rabbit polyclonal antibodies to KHC (peptide khc13; Verhey et al. 1998), to JIP-1/IB1 (number 176; Meyer et al. 1999), to DLK (gift of Lawrence Holzman, University of Michigan Medical School, Ann Arbor, MI; Holzman et al. 1994), to ApoER2 (anti-23, gift of Johannes Nimpf, University of Vienna, Vienna, Austria; Stockinger et al. 2000), to mitogen-activated kinase (MAP) kinase kinase (MKK) 3 (Santa Cruz Biotechnology, Inc.), to TrkA (Upstate Biotechnology), and to the type I sodium channel (Upstate Biotechnology).
MT binding experiments were carried out essentially as described (Verhey et al. 1998). In brief, 2.5 mM ATP or AMP-PNP, 20 μM taxol, and 2.5 mg/ml taxol-stabilized MTs were added to Triton X-100–solubilized supernatant. After incubation at room temperature, the MTs and associated proteins were isolated by pelleting through a cushion of 10% sucrose in buffer B, with no detergent and 20 μM taxol. The presence of the polypeptides in the MT pellet was detected by immunoblotting with the indicated antibodies.
Results
Direct Interaction of KLC and the JIP Proteins
To identify proteins that interact directly with kinesin, we screened a mouse brain cDNA library using the yeast two-hybrid procedure with the TPR motifs of KLC as a bait. Nine of the clones isolated correspond to overlapping fragments of three different cDNAs encoding JIP-1, JIP-2, and JIP-3 (Fig. 1 B). No interaction of these clones was seen with either of two control bait proteins, the GAL4 DNA binding domain alone, or the TPR motifs of PP5 (data not shown).
Figure 1.

Schematic of protein constructs used in this study. (A) Schematic illustration of the domain structure of KHC, KLC, and their deletion mutants. (B) Schematic illustration of the domain structure of the JIP proteins. The independent overlapping clones isolated in the two-hybrid screen are indicated in red under each protein. JBD, JNK binding domain; coil, predicated coiled coil; L, leucine zipper; SH3, src homology domain 3; PTB, phosphotyrosine binding domain.
JIP-1 (also known as IB1) and JIP-2 are ∼50% identical, have a similar domain structure, and can form homodimeric and heterodimeric complexes (Yasuda et al. 1999). JIP-3 (also known as JSAP) can form oligomeric complexes with itself and JIP-2 but has no sequence relationship to JIP-1 or JIP-2 (Ito et al. 1999; Kelkar et al. 2000). The JIP proteins have all been proposed to be scaffolding proteins for the JNK family of MAP kinases (Dickens et al. 1997; Whitmarsh et al. 1998; Ito et al. 1999; Yasuda et al. 1999; Kelkar et al. 2000). They bind simultaneously to the three components of the JNK signaling pathway, the MAP kinase kinase kinase (DLK, MLK3, and others), the MAP kinase kinase (MKK4 and MKK7), and the MAP kinase JNK (for reviews see Davis 2000; Mielke and Herdegen 2000). JIP-1 and JIP-2 have additionally been found to interact with rhoGEF (Meyer et al. 1999) and with the cell surface receptor for Reelin, ApoER2 (Stockinger et al. 2000). Scaffolding proteins play a principal role in linking the cytoplasmic domains of transmembrane receptors or ion channels to cytoplasmic signaling molecules thereby maximizing the efficiency of signal propagation and maintaining the specificity of the pathway (for reviews see Garrington and Johnson 1999; Schillace and Scott 1999; Burack and Shaw 2000). An additional function of the JIP scaffolds may be to localize the JNK signaling pathway to a specific area of the cell. The JIPs display a highly polarized subcellular localization at the tips of neuronal cell processes and in extended projections from the surface of insulinoma cells (Meyer et al. 1999; Yasuda et al. 1999; Kelkar et al. 2000). JIPs are also highly enriched at synaptic junctions in adult and developing brains (Pellet et al. 2000). In each case, the JIPs localize to the region of the cell where the plus ends of MTs are found.
Given the two-hybrid interaction between KLC and the JIPs and the requisite transport of the JIPs to the plus ends of MTs, we considered the possibility that the JIPs are a cargo of kinesin. We confirmed the interaction between KLC and the JIP proteins by coimmunoprecipitation of HA-tagged KLC and Flag-tagged JIPs expressed together in COS cells. Using anti-Flag antibodies, precipitation of JIP-1, JIP-2, or JIP-3 resulted in coprecipitation of HA-tagged full-length KLC (Fig. 2, lanes 2, 5, and 8). Conversely, anti-HA antibodies precipitated KLC and coprecipitated the Flag-tagged JIP-1, JIP-2, and JIP-3 proteins (Fig. 2, lanes 3, 6, and 9). Identical results were obtained using the HA-tagged TPR motifs of KLC instead of the full-length protein (data not shown). These data confirm a direct interaction between the TPR motifs of KLC and the JIP proteins.
Figure 2.

Coimmunoprecipitation of KLC and the JIP proteins. Lysates of COS cells expressing Flag-tagged JIP-1, JIP-2, or JIP-3 together with HA-tagged KLC were immunoprecipitated (IP) with no primary antibody (−), with an anti-Flag mAb (F), or with an anti-HA mAb (H). Precipitates were immunoblotted to detect the expressed proteins using polyclonal antibodies to both epitope tags.
The JIP-1 COOH-terminal Residues Are Required for Binding to the KLC TPR Motifs
The TPR motifs of several proteins recognize short stretches of primary amino acid sequence at the COOH termini of their binding partners (Terlecky et al. 1995; Scheufler et al. 2000). Interestingly, the extreme COOH-terminal sequences of JIP-1 and JIP-2 (PTEDIYLE) are identical and are completely conserved across species (Fig. 3 A). To determine whether these sequences are responsible for binding to the TPR motifs of KLC, we constructed deletion mutants of JIP-1 (Fig. 3 B). We used an NH2-terminal deletion, JIP-1 (307–711), as the starting point for three additional mutants: deletion of the COOH-terminal 11 amino acids, JIP-1 (307–700), and mutation of P704 or Y709 within those 11 amino acids, JIP-1 (P704A) and JIP-1 (Y709A), respectively. These mutations do not interfere with folding of the COOH-terminal PTB domain as assayed by their ability to bind rhoGEF (data not shown).
Figure 3.
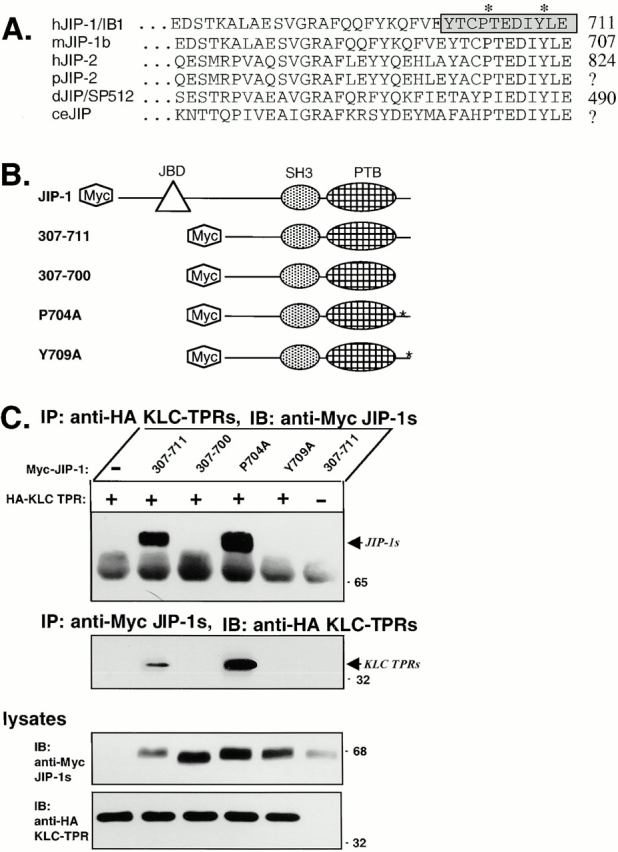
The COOH-terminal residues of JIP-1 are required for interaction with the KLC TPRs. (A) Sequence alignment of the COOH-terminal residues of JIP-1 and JIP-2 across species. The 11 residues deleted in the mutant JIP-1 (307–700) are indicated (boxed and shaded) as are the residues mutated in JIP-1 (P704A) and JIP-1 (Y709A) (*). hJIP-1/IB1, human JIP-1; mJIP-1b, mouse JIP-1b; hJIP-2, human JIP-2; pJIP-2, pig JIP-2 derived from a partial EST (accession AW312953); dJIP/SP512, Drosophila JIP-1/2; ceJIP, C. elegans JIP sequence derived from cosmid C13A10. (B) Schematic illustration of the Myc-tagged JIP-1 constructs used in this study. (C) Lysates of 293 cells expressing Myc-tagged JIP-1 (307–711), JIP-1 (307–700), JIP-1 (P704A), or JIP-1 (Y709A) together with HA-tagged KLC TPRs were immunoprecipitated (IP) for the KLC TPRs using an anti-HA mAb (top) or for the JIP-1 variants using an anti-Myc mAb (middle). Precipitates were immunoblotted (IB) for associated proteins using anti-Myc (top) or anti-HA polyclonal antibodies (middle). The ∼65-kD band in the top panel is the heavy chain of immunoglobulin. Lysates were immunoblotted to detect the expressed proteins (bottom).
The interaction between KLC and the JIP-1 mutant proteins was investigated by coimmunoprecipitation of the Myc-tagged JIP-1 variants and HA-tagged KLC TPRs expressed together in 293 cells. Immunoprecipitation of the HA-tagged TPR motifs of KLC coprecipitated the NH2-terminal truncation JIP-1 (307–711), which contains the wild-type tail sequences (Fig. 3 C); this is consistent with the results of the two-hybrid screen where all of the positive JIP-1 and JIP-2 clones encode the extreme COOH termini of the proteins (Fig. 1 B). Immunoprecipitation of the HA-tagged TPR motifs of KLC also coprecipitated the proline to alanine point mutant, JIP-1 (P704A) (Fig. 3 C). In contrast, no coprecipitation of the JIP-1 construct missing the COOH-terminal 11 residues, JIP-1 (307–700), or the tyrosine to alanine mutant, JIP-1 (Y709A), was observed (Fig. 3 C). Conversely, immunoprecipitation of Myc-tagged JIP-1 (307–711) or the JIP-1 (P704A) point mutant, but not the JIP-1 (Y709A) point mutant or the JIP-1 (307–700) deletion mutant, resulted in coprecipitation of the TPR motifs of KLC (Fig. 3 C). Identical results were obtained using full-length HA-tagged KLC instead of the TPR motifs (data not shown). These data demonstrate that the interaction between KLC and JIP-1 requires the last 11 amino acids of JIP-1 and, in particular, the tyrosine residue at position −3 from the end. Thus, the mechanism of interaction between JIP-1 and the TPR motifs of KLC is similar to that of other TPR domain–containing proteins (Terlecky et al. 1995; Scheufler et al. 2000).
JIP-1 Is a Cargo for Kinesin
The demonstration of a direct interaction between kinesin and the JIP proteins, together with the highly polarized subcellular localization of the JIP proteins, raised the question whether JIP localization is dependent on its interaction with kinesin. To address this question, we undertook two approaches.
In the first approach, we asked whether the COOH-terminal residues of JIP-1 that are required for binding to KLC are also important for localization of JIP-1 to the tips of neuronal processes. Myc-tagged full-length JIP-1, the NH2-terminal truncation JIP-1 (307–711), and the three mutant constructs JIP-1 (307–700), JIP-1 (P704A), and JIP-1 (Y709A) were localized in differentiated N1E 115 cells by indirect immunofluorescence using antibodies to the Myc tag. Untransfected control cells showed nonspecific background staining of the cell body (Fig. 4 A). Correct localization of Myc–JIP-1 fusion proteins was scored as specific staining of the neurite tips compared with the background staining of the cell body. As seen in Fig. 4B and Fig. C, full-length JIP-1 and the NH2-terminal truncation JIP-1 (307–711) localize to the tips of the neurites (arrowheads indicate tip staining) identical to the staining seen for endogenous JIP-1 (Fig. 5; Meyer et al. 1999; Yasuda et al. 1999; Kelkar et al. 2000; Pellet et al. 2000). Likewise, mutation of the proline residue, JIP-1 (P704A), had no effect on JIP-1 localization (Fig. 4 E). In contrast, JIP-1 with a deletion of the 11 COOH-terminal residues, JIP-1 (307–700), or mutation of the tyrosine residue, JIP-1 (Y709A), exhibited localization throughout the cell with no concentration at the neurite tips (Fig. 4D and Fig. F, arrows indicate staining of the cell body). These data demonstrate that the residues of JIP-1 that are critical for binding to the TPR motifs of KLC are also required for the proper subcellular localization of JIP-1 and support the model that JIP proteins are a cargo for kinesin.
Figure 4.

The COOH-terminal residues of JIP-1 are required for proper subcellular localization. NIE 115 cells were transiently transfected with the parental plasmid (control) or with plasmids encoding the indicated JIP-1 variants, differentiated, and the expressed proteins were detected by indirect immunofluorescence microscopy using an anti-Myc mAb. Nonspecific background staining is visible in the control cells and is enhanced in A′–F′ to aid in visualization of the cells. Myc–JIP-1 variants were scored as positive for correct cellular localization (JIP-1, JIP-1 [307–711], and JIP-1 [P704A]) if fluorescence was more pronounced at the neurite tips (arrowheads), whereas transfected proteins were considered negative for localization (JIP-1 [307–700] and JIP-1 [Y709A]) if fluorescence was observed to be more prominent in the cell body (arrows).
Figure 5.
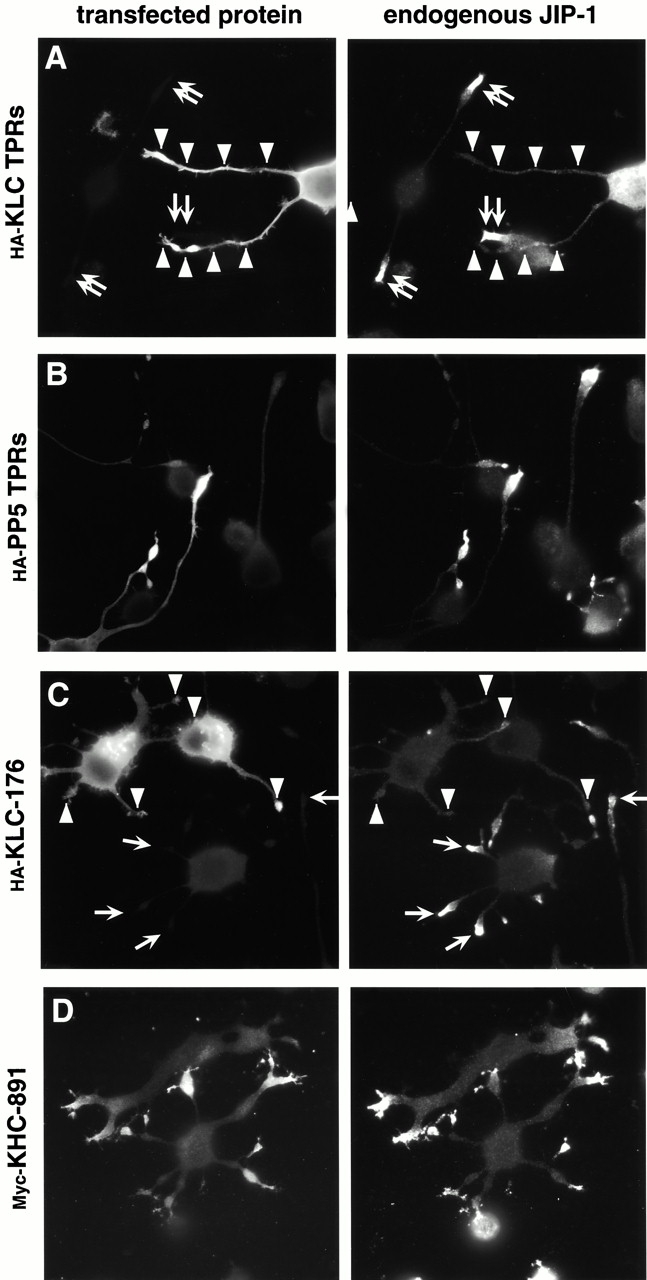
Expression of kinesin dominant negative constructs causes mislocalization of endogenous JIP-1 protein. CAD cells were transiently transfected with plasmids encoding the HA-tagged KLC TPRs (A), HA-tagged PP5 TPRs (B), HA-tagged KLC truncation KLC-176 (C), or Myc-tagged KHC truncation KHC-891 (D). After differentiation, the expressed proteins were detected by indirect immunofluorescence microscopy using mAbs to the epitope tags (left). Endogenous JIP-1 protein was detected with an affinity-purified polyclonal antibody (right). Note that the background fluorescence has been enhanced to show the entire neuronal cell. Arrowheads denote tips of transfected cells; arrows denote tips of untransfected cells.
In the second approach, we asked whether the proper subcellular localization of JIP-1 in cultured neuronal cells could be disrupted by the expression of dominant negative kinesin constructs. One region of kinesin that could act as a dominant negative is that containing the TPR motifs of KLC; this region should bind to kinesin's cargo and prevent cargo transport to the correct location due to the lack of an attached motor domain. Indeed, expression of the HA-tagged TPR motifs of KLC in differentiated CAD cells caused the localization of endogenous JIP-1 protein to shift from the tips of the neuronal processes (Fig. 5 A, arrows indicate untransfected cells) to throughout the entire cell (Fig. 5 A, arrowheads indicate transfected cells). In contrast, localization of endogenous JIP-1 is not disturbed in differentiated CAD cells expressing a control protein, the HA-tagged TPR motifs of PP5 (Fig. 5 B). Identical results were observed upon expression of these constructs in differentiated N1E 115 cells (data not shown).
Another region of kinesin that should act as a dominant negative are the heptad repeats of KLC, which are responsible for linking KLC to KHC (Gauger and Goldstein 1993; Diefenbach et al. 1998; Verhey et al. 1998). This domain should interact with endogenous KHC and prevent cargo binding due to the absence of the TPR motifs. Indeed, expression of the HA-tagged heptad repeats of KLC (KLC-176; Fig. 1 A) mislocalized endogenous JIP-1 protein in differentiated CAD cells (Fig. 5 C, arrowheads indicate transfected cells). In contrast, expression of a KHC construct lacking its COOH-terminal 64 amino acids (KHC-891; Fig. 1 A) has no effect on the localization of endogenous JIP-1 (Fig. 5 D), presumably because this construct is lacking only the autoinhibitory domain and still contains the regions necessary for motor function and cargo binding. Indeed, the truncated KHC protein was largely found at the tips of cellular processes (Fig. 5 D), consistent with its uninhibited migration towards the plus end of MTs and in agreement with similar results in COS cells (Verhey et al. 1998) and Neurospora (Seiler et al. 2000).
Together, these data demonstrate that a direct interaction with kinesin is required for the correct cellular localization of JIP-1. It should be noted that the dominant negative kinesin mutants had no effect on the rate of axonal outgrowth (data not shown), suggesting that kinesin may not have a crucial role in the differentiation of neuronal cells. We have not tested the effects of expression of the kinesin dominant negatives on the localization of endogenous JIP-2 or JIP-3 due to a lack of appropriate reagents. However, when expressed at low levels in differentiated CAD cells, Flag-tagged JIP-2 and JIP-3 localize to the tips of the neurites (Fig. 6A and Fig. B, arrows indicate untransfected cells), and this localization is abolished by coexpression of the kinesin dominant negative construct (Fig. 6A and Fig. B, arrowheads indicate transfected cells). These data support the assertion that JIP-2 and JIP-3 are also cargoes for kinesin and provide strong evidence that the JIP scaffolding proteins act not only as motor–cargo linkers but are integral components of the cargo as well. Our results argue against the alternative possibility that the JIPs are upstream of kinesin and that their only function is to regulate kinesin's transport of another cargo.
Figure 6.
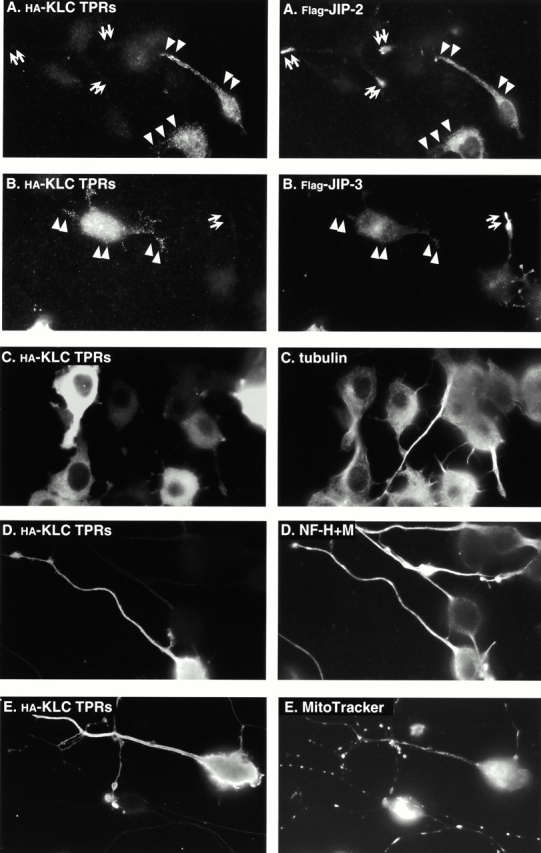
A kinesin dominant negative construct causes mislocalization of expressed JIP-2 and JIP-3 proteins but not endogenous mitochondria, neurofilaments, or MTs. (A and B) CAD cells were transiently transfected with plasmids encoding the HA-tagged KLC TPRs together with Flag-tagged JIP-2 (A) or Flag-tagged JIP-3 (B). After differentiation, the expressed proteins were detected by indirect immunofluorescence microscopy using a mAb to the HA tag (left) and a polyclonal antibody to the Flag tag (right). Arrowheads denote tips of transfected cells; arrows denote tips of untransfected cells. (C–E) CAD cells were transiently transfected with a plasmid encoding the HA-tagged KLC TPRs. After differentiation, the expressed protein was detected by indirect immunofluorescence microscopy using a polyclonal antibody to the HA tag (left). Endogenous MTs, neurofilaments (NF), and mitochondria were detected with mAbs to tubulin (C, right) or neurofilaments H + M (D, right), or with MitoTracker (E, right). Note that in C, the cells were only allowed to differentiate for 12 h, so that MT organization could be assessed before the cell bodies rounded up.
Importantly, in control experiments, expression of the dominant negative HA-tagged KLC TPRs had no effect on the localization of MTs in CAD cells (Fig. 6 C) or in a variety of other cell lines (data not shown), demonstrating that the effects on JIP localization are not due to a global effect on MT organization. In addition, expression of the HA-tagged KLC TPRs had no effect on the localization of neurofilaments (Fig. 6 D) or mitochondria (Fig. 6 E) in CAD cells, or on the localization of the endoplasmic reticulum, the Golgi complex, endosomes, lysosomes, actin, vimentin, or synaptophysin in a variety of cultured cell lines (data not shown).
Kinesin Associates with Proteins Assembled on the JIP Scaffold
The identification of the JIP proteins as a cargo for kinesin led us to consider whether kinesin carries the JIP proteins alone or together with proteins assembled on the JIP scaffold. To test this, we looked for an interaction in vivo between kinesin, the JIPs, and signaling molecules known to interact with JIPs. A high speed supernatant fraction from rat brain was used for immunoprecipitation experiments with an mAb to KHC (H2, gift of G. Bloom). As an additional method of precipitating kinesin and associated proteins, an MT binding assay was performed. The presence of associated proteins was detected by immunoblotting the immunoprecipitates and MT pellets.
We first looked for an association of kinesin with JIP-1 and with a neuronal-specific upstream kinase of the JNK signal transduction pathway, the DLK (Fan et al. 1996). As seen in Fig. 7 A, immunoprecipitation of kinesin resulted in the coprecipitation of JIP-1 and DLK. A complex of these proteins was also detected in the presence of the detergents digitonin (Fig. 7 A), Triton X-100, or octyl glucoside (data not shown). As seen in Fig. 7 B, precipitation of kinesins by binding to MTs in the presence of the nonhydrolyzable ATP analogue AMP-PNP also resulted in the coprecipitation of JIP-1 and DLK. Importantly, the upstream kinase MKK3 of another MAP kinase pathway, the p38 pathway, was not coprecipitated (Fig. 7 A).
Figure 7.
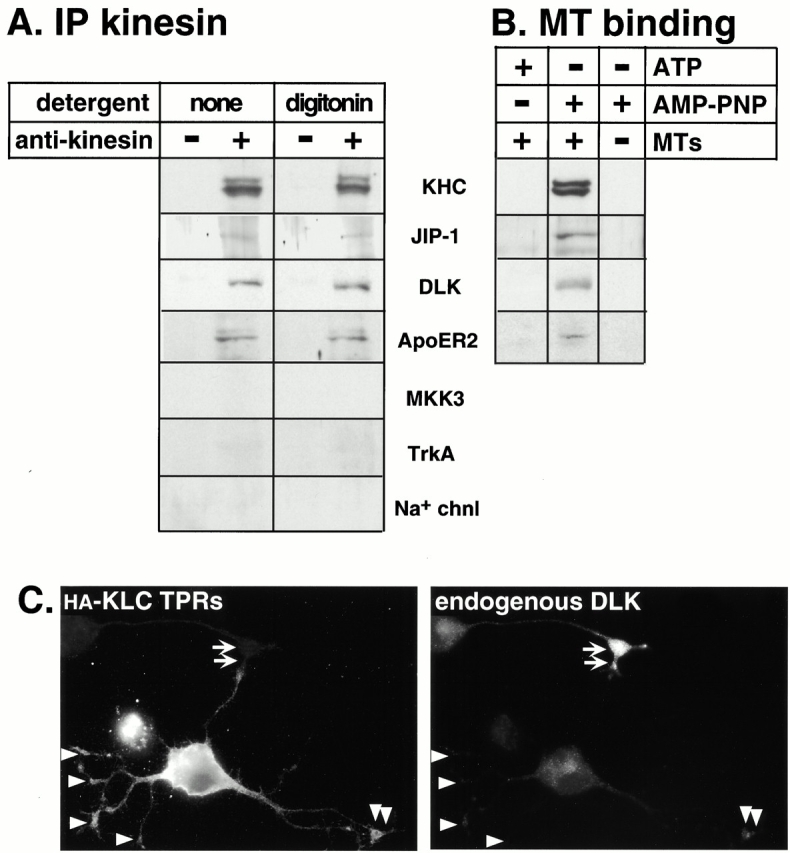
Kinesin and JIP-1 are associated with the upstream kinase DLK and the transmembrane receptor ApoER2. (A) An mAb to KHC (H2) was used to immunoprecipitate (IP) kinesin and associated proteins from a rat brain high speed supernatant in the presence of digitonin or no detergent. The presence of associated polypeptides was detected by immunoblotting the precipitates with polyclonal antibodies to the indicated proteins. (B) Rat brain high speed supernatant was subjected to an MT binding assay in the presence of Triton X-100 by adding ATP, AMP-PNP, and/or MTs as indicated. MTs and bound proteins were sedimented through a sucrose cushion, and the presence of the indicated proteins in the MT pellets was detected by immunoblotting. (C) CAD cells were transiently transfected with a plasmid encoding the HA-tagged KLC TPRs. After differentiation, the expressed protein was detected by indirect immunofluorescence microscopy using a mAb to the HA tag (left). Endogenous DLK kinase was detected with a polyclonal antibody (right). Arrowheads denote tips of transfected cells; arrows denote the tip of an untransfected cell.
Next, we looked for a link between kinesin and a vesicle by probing for the coprecipitation of the transmembrane protein ApoER2, the receptor for the Reelin ligand, which has been shown to interact directly with JIP-1 and JIP-2 (Gotthardt et al. 2000; Stockinger et al. 2000). Although most of the ApoER2 protein is found in a rapidly sedimenting fraction of rat brain homogenates, a small fraction of ApoER2 remains in the high speed supernatant fraction, presumably associated with small vesicles that do not pellet under these conditions (data not shown). As seen in Fig. 7 A, immunoprecipitation of kinesin resulted in the coprecipitation of ApoER2, and this interaction was not disturbed upon solubilization with digitonin (Fig. 7 A), Triton X-100, or octyl glucoside (data not shown). As seen in Fig. 7 B, precipitation of kinesins by binding to MTs in the presence of AMP-PNP also resulted in the coprecipitation of ApoER2. This is a small but significant portion of the ApoER2 found in the high speed supernatant fraction. Importantly, the transmembrane proteins TrkA, a receptor for nerve growth factor, and the type I sodium channel were not coprecipitated (Fig. 7 A), although they were present in the high speed supernatant fraction.
As an additional way of assessing whether kinesin is required for localization of proteins assembled on the JIP scaffold, we looked for an effect of the dominant negative HA-tagged KLC TPRs on the localization of DLK (the antibodies to ApoER2 were not suitable for these experiments). Indeed, localization of endogenous DLK to the tips of the neurites (Fig. 7 C, arrows indicate the tip of an untransfected cell) is abolished by coexpression of the kinesin dominant negative construct (Fig. 7 C, arrowheads indicate transfected cell). Together, these results suggest that the JIP scaffolding complex, which includes DLK and ApoER2, is preassembled before being transported by kinesin, rather than formed by diffusion of its individual components at the site of action.
Discussion
JIPs and Their Associated Proteins Are a Cargo for Kinesin
Our results demonstrate a direct interaction between conventional kinesin and a cargo, the JIP scaffolding proteins. Although kinesin has always been assumed to be an organelle and/or vesicle motor, these results suggest that the underlying assumption that kinesin links directly to its cargo via a membrane protein may not be true. Instead, our results demonstrate that the kinesin–cargo linker is a soluble scaffolding protein. We postulate that the scaffold assembles other proteins, some of which provide the direct link to a vesicle (Fig. 8). In addition to the proteins known to associate with the JIP scaffolding proteins, there are likely to be others that assemble in this macromolecular complex.
Figure 8.

Model for the transport of cargo by kinesins. JIP proteins form a scaffold, on which cytoplasmic as well as plasma membrane proteins are assembled. The entire complex is transported down an axonal process by conventional kinesin. Note that the JIP proteins are known to form homodimers and heterodimers, although only one polypeptide is drawn for clarity. Similarly, LIN-2, -7, and -10 form a scaffold, on which cytoplasmic and transmembrane proteins assemble. The entire complex is transported down a dendritic process by the kinesin superfamily member KIF17.
Kinesin and JIP-1 are connected via a direct interaction between the KLC TPR motifs and the JIP-1 COOH-terminal residues. In analogy to other TPR domain–containing proteins (Terlecky et al. 1995; Das et al. 1998; Groves and Barford 1999; Scheufler et al. 2000), it is likely that three or more TPR motifs of KLC generate an extended surface groove that provides the docking site for the linear JIP-1 (and presumably JIP-2) peptide. The six TPR motifs of KLC could thus bind two JIP-1 and/or JIP-2 molecules simultaneously. Interestingly, the TPR motifs of KLC must bind to internal residues in JIP-3 as the COOH terminus of JIP-3 bears no sequence relation to that of JIP-1 and JIP-2, and the JIP-3 clones isolated in our two-hybrid screen contain only the NH2-terminal portion of the protein (Fig. 1 B). These results are in agreement with a recent report that demonstrates a direct interaction between the NH2-terminal half of JIP-3 (Sunday Driver) and the TPR motifs of KLC (Bowman et al. 2000). Our work extends this study by demonstrating that all JIP scaffolding proteins interact with KLC and that this interaction is essential for proper localization of the JIPs. Bowman et al. 2000 claim that JIP-3 is a transmembrane protein that provides a direct link between kinesin and the membrane. However, this is not supported by the sequence of JIP-3, the cytosolic localization of Flag-tagged JIP-3 expressed in CAD cells (Fig. 6 B) or in COS cells (data not shown), or the localization of endogenous JIP-3 in neuronal cells (Kelkar et al. 2000).
Although our results demonstrate that the TPR motifs of KLC interact with a cargo, the JIP scaffolding proteins, they do not exclude that there are regions in kinesin that interact with other cargoes. Additional cargo binding sites could be the variable COOH termini of KLC, which arise from alternative splicing (Khodjakov et al. 1998; Liao and Gundersen 1998; Gyoeva et al. 2000), or the region of the KHC stalk domain that is highly conserved from fungi to humans (Kirchner et al. 1999a,Kirchner et al. 1999b; Seiler et al. 2000). The possibility of multiple cargo interactions may explain at least in part the apparently conflicting reports that kinesin transports a large variety of organelles (Goldstein and Philp 1999; Goldstein and Yang 2000; Kamal and Goldstein 2000; Rogers and Gelfand 2000). Alternatively, kinesin's sole cargo may be the JIPs; it may be via the JNK signaling pathway that kinesin exerts its various effects on cells, as known JNK effectors include tau, neurofilaments, and the p150-Spir actin–regulating protein (Mielke and Herdegen 2000; Otto et al. 2000).
Role of Kinesin and JNK Signaling in Neurogenesis
The identification of the JIP scaffold proteins as a cargo for kinesin raises intriguing questions on the role of kinesin and the JNK signaling pathway in nerve cell development. Kinesin and JIPs are not necessary for axonal growth because mislocalization of JIP-1 by dominant negative kinesin constructs does not prevent neurite outgrowth (Fig. 5); rather, kinesin and JIPs may be involved in neuronal migration, pathfinding, and synaptogenesis. This is supported by the fact that, in KHC or KLC mutants in Drosophila, the morphology of the larval neuromuscular system remains intact but there are defects in the number of synaptic boutons in action potential propagation and in neurotransmitter release (Gho et al. 1992; Hurd and Saxton 1996; Hurd et al. 1996; Gindhart et al. 1998). Consistent with the postulated connection between kinesin, JNK signaling, and neurogenesis, mice lacking the ubiquitously expressed conventional kinesin gene kif5b and mice lacking the jnk1 and jnk2 genes exhibit embryonic lethality with severe defects in early brain development (Kuan et al. 1999). A role for kinesin and JNK signaling in synaptogenesis is also suggested by the developmental expression profile of JIP-1 message and protein, which coincide with the transition from axonal and dendritic outgrowth to a phase in which synaptic contacts are stabilized (Pellet et al. 2000). A possible mechanism may be through the interaction of JIP-1 and/or JIP-2 with ApoER2, the receptor for Reelin. The Reelin protein, which is defective in the naturally occurring reeler strain of mouse, has been suggested to function in the maturation of neuronal processes, axonal branching, and synaptogenesis (for reviews see D'Arcangelo and Curran 1998; Willnow et al. 1999; Bothwell and Giniger 2000). Together, these results support a model in which localization of kinesin, JIPs, and JNK signaling pathways at the nerve terminal is necessary for neurogenesis.
KIFs and Localization of Signal Transduction Pathways
Kinesins may play a general role in localizing signal transduction cascades. The neuronal-specific KIF17 binds to the first of two PDZ domains in the scaffold protein mLin-10 (Mint1/X11) and thereby transports an associated transmembrane protein, the NMDA receptor, to the ends of dendrites (Fig. 7 A) (Setou et al. 2000). PDZ domain–containing proteins are scaffolding proteins that play a fundamental role in the synaptic localization of neurotransmitter receptors and ion channels and in the assembly of synaptic junctions (for reviews see Fanning and Anderson 1999; Garner et al. 2000; Sheng and Pak 2000). Although PDZ domains have no structural relationship to TPR domains, they also bind to short peptide sequences located on the COOH-terminal tails of their target proteins as well as to internal sequences in their binding partners (for reviews see Saras and Heldin 1996; Cowburn 1997; Fanning and Anderson 1999; Oschkinat 1999). A link between kinesins and signaling pathways was also suggested by the identification of KIF3X, a member of the kinesin II subfamily, in a yeast two-hybrid screen for proteins that interact with MLK2, an upstream kinase that activates the extracellular signal-regulated kinase, JNK, and p38 MAP kinases (Nagata et al. 1998).
Together, these results suggest a general model for kinesins in which the motor binds to a scaffolding protein that then links it to a vesicle (Fig. 7 A). Thus, in addition to their known role in assembling and stabilizing protein complexes, scaffolding proteins can now be seen as a link between motors and membrane trafficking. This linkage would serve to localize not just membrane proteins, but also cytoplasmic signaling molecules, to the appropriate cellular domain and would provide the targeting mechanism required for spatial regulation of signaling pathways. Indeed, one of the long-standing questions in signal transduction is how cells generate specific responses out of generic signaling pathways (Jordan et al. 2000; Schlessinger 2000). Spatial concentration of signaling molecules is one mechanism, in addition to regulation of signal strength and timing, by which cells could accomplish this.
A provocative implication raised by our results, and those recently reported for KIF17, is that scaffolding proteins assemble multiprotein complexes at great distances from their final functional locations. Particularly in the case of neuronal cells but applicable to all cells, it seems most advantageous for a cell to assemble the components of a signaling complex in the cell body where synthesis of cytoplasmic and membrane proteins takes place, load it on a motor protein, and thereby carry the entire complex to the tips of the neuronal processes. In addition, since one of the components is a transmembrane receptor, it seems likely that the cargo is not simply soluble signaling molecules but a module of signaling components attached to a vesicle. Further studies are required to address whether these components, and presumably others, are actually associated together on vesicles that move along MTs.
Signal Transduction Pathways May Regulate the Activity of Their Motors
Another provocative implication raised by the model in Fig. 7 is that scaffolds and their associated proteins are required not only for localized signal transduction but also for regulating the motor that delivers them. Regulation of motor activity is required in two cellular contexts: activation of the motor at the site of assembly and/or loading and inactivation of the motor upon delivery of the cargo to its destination. In the case of conventional kinesin, the binding of JIPs to the TPR motifs in the tail of kinesin may unfold the native kinesin protein and activate it for MT binding. In this case, the KLCs would play a role both in directing cargo binding and in regulating motor activity (Verhey et al. 1998). The bound cargo could then stabilize the active state of the motor complex, ensuring that only cargo-bound kinesins are capable of movement along MTs. When kinesin and its cargo reach their destination, the fusion of the transported vesicle with the plasma membrane would expose the receptor, e.g., ApoER2, to its extracellular ligand, e.g., Reelin. This may result in the activation of the JNK signaling cascade, which may in turn inactivate kinesin so it can be recycled back to the cell body or be degraded. This model is consistent with previous reports pointing to a role for phosphorylation of KHC and KLC in regulating kinesin's activity (Sato-Yoshitake et al. 1992; Hollenbeck 1993; Matthies et al. 1993; McIlvain et al. 1994; Lee and Hollenbeck 1995; Lindesmith et al. 1997; De Vos et al. 2000). Other potential mechanisms that may play a role in regulating kinesin's activity include local changes in cellular pH (Verhey et al. 1998) and chaperone proteins such as hsc70 (Tsai et al. 2000). Further experiments are required to determine whether JIP binding is necessary and sufficient for activation of the kinesin motor and whether any of the known signal transduction pathways are involved in regulating kinesin activity.
Acknowledgments
For helpful advice and discussions, we thank Seth Sadis, Eric Olsen, Kathy Buckley, Joachim Herz, and the members of the Margolis and Rapoport laboratories, particularly Pascal Stein, Melissa Rolls, and Will Prinz. We are grateful to Drs. Kathy Buckley, Martin Myers, Diane Fingar, Bill Saxton, and Virgil Muresan for critical reading of the manuscript.
The work in the laboratory of T.A. Rapoport by K.J. Verhey and R.M. Deehan was supported by the Howard Hughes Medical Institute. The work in the laboratory of B. Margolis by D. Meyer was supported by the Howard Hughes Medical Institute. B.J. Schnapp and J. Blenis are supported by the National Institutes of Health.
Footnotes
K.J. Verhey and D. Meyer contributed equally to this work.
B. Schnapp, Department of Cell Biology, Harvard Medical School, 240 Longwood Ave., Boston, MA 02115. Tel.: (617) 432-3818. E-mail:bschnapp@hms.harvard.edu
Abbreviations used in this paper: DLK, dual leucine zipper kinase; HA, hemagglutinin; JIP, JNK-interacting protein; JNK, c-jun NH2-terminal kinase; KHC, kinesin heavy chain; KIF, kinesin family member; KLC, kinesin light chain; MAP, mitogen-activated kinase; MKK, MAP kinase kinase; MT, microtubule; PP5, protein phosphatase 5; TPR, tetratricopeptide repeat.
References
- Bi G.Q., Morris R.L., Liao G., Alderton J.M., Scholey J.M., Steinhardt R.A. Kinesin- and myosin-driven steps of vesicle recruitment for Ca2+-regulated exocytosis. J. Cell Biol. 1997;138:999–1008. doi: 10.1083/jcb.138.5.999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blatch G.L., Lassle M. The tetratricopeptide repeata structural motif mediating protein-protein interactions. Bioessays. 1999;21:932–939. doi: 10.1002/(SICI)1521-1878(199911)21:11<932::AID-BIES5>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Bothwell M., Giniger E. Alzheimer's diseaseneurodevelopment converges with neurodegeneration. Cell. 2000;102:271–273. doi: 10.1016/s0092-8674(00)00032-5. [DOI] [PubMed] [Google Scholar]
- Bowman A.B., Kamal A., Ritchings B.W., Philp A.V., McGrail M., Gindhart J.G., Goldstein L.S.B. Kinesin-dependent axonal transport is mediated by the sunday driver (SYD) protein. Cell. 2000;103:583–594. doi: 10.1016/s0092-8674(00)00162-8. [DOI] [PubMed] [Google Scholar]
- Brendza R.P., Serbus L.R., Duffy J.B., Saxton W.M. A function for kinesin I in the posterior transport of oskar mRNA and Staufen protein Science 289 2000. 2120 2122a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brendza R.P., Sheehan K.B., Turner F.R., Saxton W.M. Clonal tests of conventional kinesin function during cell proliferation and differentiation Mol. Biol. Cell 11 2000. 1329 1343b [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown S.S. Cooperation between microtubule- and actin-based motor proteins. Annu. Rev. Cell Dev. Biol. 1999;15:63–80. doi: 10.1146/annurev.cellbio.15.1.63. [DOI] [PubMed] [Google Scholar]
- Burack W.R., Shaw A.S. Signal transductionhanging on a scaffold. Curr. Opin. Cell Biol. 2000;12:211–216. doi: 10.1016/s0955-0674(99)00078-2. [DOI] [PubMed] [Google Scholar]
- Cowburn D. Peptide recognition by PTB and PDZ domains. Curr. Opin. Struct. Biol. 1997;7:835–838. doi: 10.1016/s0959-440x(97)80155-8. [DOI] [PubMed] [Google Scholar]
- Coy D.L., Hancock W.O., Wagenbach M., Howard J. Kinesin's tail domain is an inhibitory regulator of the motor domain. Nat. Cell Biol. 1999;1:288–292. doi: 10.1038/13001. [DOI] [PubMed] [Google Scholar]
- D'Arcangelo G., Curran T. Reelernew tales on an old mutant mouse. Bioessays. 1998;20:235–244. doi: 10.1002/(SICI)1521-1878(199803)20:3<235::AID-BIES7>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- Das A.K., Cohen P.T.W., Barford D. The structure of the tetratricopeptide repeats of protein phosphatase 5implications for TPR-mediated protein–protein interactions. EMBO (Eur. Mol. Biol. Organ.) J. 1998;17:1192–1199. doi: 10.1093/emboj/17.5.1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis R.J. Signal transduction by the JNK group of MAP kinases. Cell. 2000;103:239–252. doi: 10.1016/s0092-8674(00)00116-1. [DOI] [PubMed] [Google Scholar]
- De Vos K., Severin F., Van Herreweghe F., Vancompernolle K., Goossens V., Hyman A., Grooten J. Tumor necrosis factor induces hyperphosphorylation of kinesin light chain and inhibits kinesin-mediated transport of mitochondria. J. Cell Biol. 2000;149:1207–1214. doi: 10.1083/jcb.149.6.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickens M., Rogers J.S., Cavanagh J., Raitano A., Xia Z., Halpern J.R., Greenberg M.E., Sawyers C.L., Davis R.J. A cytoplasmic inhibitor of the JNK signal transduction pathway. Science. 1997;277:693–696. doi: 10.1126/science.277.5326.693. [DOI] [PubMed] [Google Scholar]
- Diefenbach R.J., Mackay J.P., Armati P.J., Cunningham A.L. The C-terminal region of the stalk domain of ubiquitous human kinesin heavy chain contains the binding site for kinesin light chain. Biochemistry. 1998;37:16663–16670. doi: 10.1021/bi981163r. [DOI] [PubMed] [Google Scholar]
- Fan G., Merritt S.E., Kortenjann M., Shaw P.E., Holzman L.B. Dual leucine zipper-bearing kinase (DLK) activates p46SAPK and p38mapk but not ERK2. J. Biol. Chem. 1996;271:24788–24793. doi: 10.1074/jbc.271.40.24788. [DOI] [PubMed] [Google Scholar]
- Fanning A.S., Anderson J.M. PDZ domainsfundamental building blocks in the organization of protein complexes at the plasma membrane. J. Clin. Invest. 1999;103:767–772. doi: 10.1172/JCI6509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman D.S., Vale R.D. Single-molecule analysis of kinesin motility reveals regulation by the cargo-binding tail domain. Nat. Cell Biol. 1999;1:293–297. doi: 10.1038/13008. [DOI] [PubMed] [Google Scholar]
- Garner C.C., Nash J., Huganir R.L. PDZ domains in synapse assembly and signalling. Trends Cell Biol. 2000;10:274–280. doi: 10.1016/s0962-8924(00)01783-9. [DOI] [PubMed] [Google Scholar]
- Garrington T.P., Johnson G.L. Organization and regulation of mitogen-activated protein kinase signaling pathways. Curr. Opin. Cell Biol. 1999;11:211–218. doi: 10.1016/s0955-0674(99)80028-3. [DOI] [PubMed] [Google Scholar]
- Gauger A.K., Goldstein L.S. The Drosophila kinesin light chain. Primary structure and interaction with kinesin heavy chain. J. Biol. Chem. 1993;268:13657–13666. [PubMed] [Google Scholar]
- Gho M., McDonald K., Ganetzky B., Saxton W.M. Effects of kinesin mutations on neuronal functions. Science. 1992;258:313–316. doi: 10.1126/science.1384131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gindhart J.G., Jr., Desai C.J., Beushausen S., Zinn K., Goldstein L.S. Kinesin light chains are essential for axonal transport in Drosophila . J. Cell Biol. 1998;141:443–454. doi: 10.1083/jcb.141.2.443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein L.S., Philp A.V. The road less traveledemerging principles of kinesin motor utilization. Annu. Rev. Cell Dev. Biol. 1999;15:141–183. doi: 10.1146/annurev.cellbio.15.1.141. [DOI] [PubMed] [Google Scholar]
- Goldstein L.S.B., Gunawardena S. Flying through the Drosophila cytoskeletal genome. J. Cell Biol. 2000;150:F63–F68. doi: 10.1083/jcb.150.2.f63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein L.S., Yang Z. Microtubule-based transport systems in neuronsthe roles of kinesins and dyneins. Annu. Rev. Neurosci. 2000;23:39–71. doi: 10.1146/annurev.neuro.23.1.39. [DOI] [PubMed] [Google Scholar]
- Goode B.L., Drubin D.G., Barnes G. Functional cooperation between the microtubule and actin cytoskeletons. Curr. Opin. Cell Biol. 2000;12:63–71. doi: 10.1016/s0955-0674(99)00058-7. [DOI] [PubMed] [Google Scholar]
- Gotthardt M., Trommsdorff M., Nevitt M.F., Shelton J., Richardson J.A., Stockinger W., Nimpf J., Herz J. Interactions of the low density lipoprotein receptor gene family with cytosolic adaptor and scaffold proteins suggest diverse biological functions in cellular communication and signal transduction. J. Biol. Chem. 2000;275:25616–25624. doi: 10.1074/jbc.M000955200. [DOI] [PubMed] [Google Scholar]
- Groves M.R., Barford D. Topological characteristics of helical repeat proteins. Curr. Opin. Struct. Biol. 1999;9:383–389. doi: 10.1016/s0959-440x(99)80052-9. [DOI] [PubMed] [Google Scholar]
- Gyoeva F.K., Bybikova E.M., Minin A.A. An isoform of kinesin light chain specific for the Golgi complex. J. Cell Sci. 2000;113:2047–2054. doi: 10.1242/jcs.113.11.2047. [DOI] [PubMed] [Google Scholar]
- Hollenbeck P.J. Phosphorylation of neuronal kinesin heavy and light chains in vivo. J. Neurochem. 1993;60:2265–2275. doi: 10.1111/j.1471-4159.1993.tb03513.x. [DOI] [PubMed] [Google Scholar]
- Holzman L.B., Merritt S.E., Fan G. Identification, molecular cloning, and characterization of dual leucine zipper bearing kinase. A novel serine/threonine protein kinase that defines a second subfamily of mixed lineage kinases. J. Biol. Chem. 1994;269:30808–30817. [PubMed] [Google Scholar]
- Huang J.D., Brady S.T., Richards B.W., Stenolen D., Resau J.H., Copeland N.G., Jenkins N.A. Direct interaction of microtubule- and actin-based transport motors. Nature. 1999;397:267–270. doi: 10.1038/16722. [DOI] [PubMed] [Google Scholar]
- Hurd D.D., Saxton W.M. Kinesin mutations cause motor neuron disease phenotypes by disrupting fast axonal transport in Drosophila . Genetics. 1996;144:1075–1085. doi: 10.1093/genetics/144.3.1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurd D.D., Stern M., Saxton W.M. Mutation of the axonal transport motor kinesin enhances paralytic and suppresses Shaker in Drosophila . Genetics. 1996;142:195–204. doi: 10.1093/genetics/142.1.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito M., Yoshioka K., Akechi M., Yamashita S., Takamatsu N., Sugiyama K., Hibi M., Nakabeppu Y., Shiba T., Yamamoto K.I. JSAP1, a novel jun N-terminal protein kinase (JNK)-binding protein that functions as a Scaffold factor in the JNK signaling pathway. Mol. Cell. Biol. 1999;19:7539–7548. doi: 10.1128/mcb.19.11.7539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James P., Halladay J., Craig E.A. Genomic libraries and a host strain designed for highly efficient two-hybrid selection in yeast. Genetics. 1996;144:1425–1436. doi: 10.1093/genetics/144.4.1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan J.D., Landau E.M., Iyengar R. Signaling networksthe origins of cellular multitasking. Cell. 2000;103:193–200. doi: 10.1016/s0092-8674(00)00112-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamal A., Goldstein L.S. Connecting vesicle transport to the cytoskeleton. Curr. Opin. Cell Biol. 2000;12:503–508. doi: 10.1016/s0955-0674(00)00123-x. [DOI] [PubMed] [Google Scholar]
- Kelkar N., Gupta S., Dickens M., Davis R.J. Interaction of a mitogen-activated protein kinase signaling module with the neuronal protein JIP3. Mol. Cell. Biol. 2000;20:1030–1043. doi: 10.1128/mcb.20.3.1030-1043.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khodjakov A., Lizunova E.M., Minin A.A., Koonce M.P., Gyoeva F.K. A specific light chain of kinesin associates with mitochondria in cultured cells. Mol. Biol. Cell. 1998;9:333–343. doi: 10.1091/mbc.9.2.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirchner J., Seiler S., Fuchs S., Schliwa M. Functional anatomy of the kinesin molecule in vivo EMBO (Eur. Mol. Biol. Organ.) J. 18 1999. 4404 4413a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirchner J., Woehlke G., Schliwa M. Universal and unique features of kinesin motorsinsights from a comparison of fungal and animal conventional kinesins Biol. Chem 380 1999. 915 921b [DOI] [PubMed] [Google Scholar]
- Kuan C.Y., Yang D.D., Samanta Roy D.R., Davis R.J., Rakic P., Flavell R.A. The Jnk1 and Jnk2 protein kinases are required for regional specific apoptosis during early brain development. Neuron. 1999;22:667–676. doi: 10.1016/s0896-6273(00)80727-8. [DOI] [PubMed] [Google Scholar]
- Lee K.D., Hollenbeck P.J. Phosphorylation of kinesin in vivo correlates with organelle association and neurite outgrowth. J. Biol. Chem. 1995;270:5600–5605. doi: 10.1074/jbc.270.10.5600. [DOI] [PubMed] [Google Scholar]
- Liao G., Gundersen G.G. Kinesin is a candidate for cross-bridging microtubules and intermediate filaments. Selective binding of kinesin to detyrosinated tubulin and vimentin. J. Biol. Chem. 1998;273:9797–9803. doi: 10.1074/jbc.273.16.9797. [DOI] [PubMed] [Google Scholar]
- Lindesmith L., McIlvain J.M., Jr., Argon Y., Sheetz M.P. Phosphotransferases associated with the regulation of kinesin motor activity. J. Biol. Chem. 1997;272:22929–22933. doi: 10.1074/jbc.272.36.22929. [DOI] [PubMed] [Google Scholar]
- Manning B.D., Snyder M. Drivers and passengers wanted! the role of kinesin-associated proteins. Trends Cell Biol. 2000;10:281–289. doi: 10.1016/s0962-8924(00)01774-8. [DOI] [PubMed] [Google Scholar]
- Matthies H.J., Miller R.J., Palfrey H.C. Calmodulin binding to and cAMP-dependent phosphorylation of kinesin light chains modulate kinesin ATPase activity. J. Biol. Chem. 1993;268:11176–11187. [PubMed] [Google Scholar]
- McIlvain J.M., Jr., Burkhardt J.K., Hamm-Alvarez S., Argon Y., Sheetz M.P. Regulation of kinesin activity by phosphorylation of kinesin-associated proteins. J. Biol. Chem. 1994;269:19176–19182. [PubMed] [Google Scholar]
- Meyer D., Liu A., Margolis B. Interaction of c-Jun amino-terminal kinase interacting protein-1 with p190 rhoGEF and its localization in differentiated neurons. J. Biol. Chem. 1999;274:35113–35118. doi: 10.1074/jbc.274.49.35113. [DOI] [PubMed] [Google Scholar]
- Mielke K., Herdegen T. JNK and p38 stress kinases—degenerative effectors of signal-transduction-cascades in the nervous system. Prog. Neurobiol. 2000;61:45–60. doi: 10.1016/s0301-0082(99)00042-8. [DOI] [PubMed] [Google Scholar]
- Nagata K., Puls A., Futter C., Aspenstrom P., Schaefer E., Nakata T., Hirokawa N., Hall A. The MAP kinase kinase kinase MLK2 co-localizes with activated JNK along microtubules and associates with kinesin superfamily motor KIF3. EMBO (Eur. Mol. Biol. Organ.) J. 1998;17:149–158. doi: 10.1093/emboj/17.1.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oschkinat H. A new type of PDZ domain recognition. Nat. Struct. Biol. 1999;6:408–410. doi: 10.1038/8203. [DOI] [PubMed] [Google Scholar]
- Otto I.M., Raabe T., Rennefahrt U.E.E., Bork P., Rapp U.R., Kerkhoff E. The p150-Spir protein provides a link between c-Jun N-terminal kinase function and actin reorganization. Curr. Biol. 2000;10:345–348. doi: 10.1016/s0960-9822(00)00388-2. [DOI] [PubMed] [Google Scholar]
- Patel N., Thierry-Mieg D., Mancillas J.R. Cloning by insertional mutagenesis of a cDNA encoding Caenorhabditis elegans kinesin heavy chain. Proc. Natl. Acad. Sci. USA. 1993;90:9181–9185. doi: 10.1073/pnas.90.19.9181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellet J.B., Haefliger J.A., Staple J.K., Widmann C., Welker E., Hirling H., Bonny C., Nicod P., Catsicas S., Waeber G., Riederer B.M. Spatial, temporal and subcellular localization of islet-brain 1 (IB1), a homologue of JIP-1, in mouse brain. Eur. J. Neurosci. 2000;12:621–632. doi: 10.1046/j.1460-9568.2000.00945.x. [DOI] [PubMed] [Google Scholar]
- Qi Y., Wang J.K.T., McMillian M., Chikaraishi D.M. Characterization of a CNS cell line, CAD, in which morphological differentiation is initiated by serum deprivation. J. Neurosci. 1997;17:1217–1225. doi: 10.1523/JNEUROSCI.17-04-01217.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman A., Kamal A., Roberts E.A., Goldstein L.S. Defective kinesin heavy chain behavior in mouse kinesin light chain mutants. J. Cell Biol. 1999;146:1277–1288. doi: 10.1083/jcb.146.6.1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers S.L., Gelfand V.I. Membrane trafficking, organelle transport, and the cytoskeleton. Curr. Opin. Cell Biol. 2000;12:57–62. doi: 10.1016/s0955-0674(99)00057-5. [DOI] [PubMed] [Google Scholar]
- Saras J., Heldin C.H. PDZ domains bind carboxy-terminal sequences of target proteins. Trends Biochem. Sci. 1996;21:455–458. doi: 10.1016/s0968-0004(96)30044-3. [DOI] [PubMed] [Google Scholar]
- Sato-Yoshitake R., Yorifuji H., Inagaki M., Hirokawa N. The phosphorylation of kinesin regulates its binding to synaptic vesicles. J. Biol. Chem. 1992;267:23930–23936. [PubMed] [Google Scholar]
- Saxton W.M., Hicks J., Goldstein L.S., Raff E.C. Kinesin heavy chain is essential for viability and neuromuscular functions in Drosophila, but mutants show no defects in mitosis. Cell. 1991;64:1093–1102. doi: 10.1016/0092-8674(91)90264-y. [DOI] [PubMed] [Google Scholar]
- Scheufler C., Brinker A., Bourenkov G., Pegoraro S., Moroder L., Bartunik H., Hartl F.U., Moarefi I. Structure of TPR domain-peptide complexescritical elements in the assembly of the Hsp70-Hsp90 multichaperone machine. Cell. 2000;101:199–210. doi: 10.1016/S0092-8674(00)80830-2. [DOI] [PubMed] [Google Scholar]
- Schillace R.V., Scott J.D. Organization of kinases, phosphatases, and receptor signaling complexes. J. Clin. Invest. 1999;103:761–765. doi: 10.1172/JCI6491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2000;103:211–225. doi: 10.1016/s0092-8674(00)00114-8. [DOI] [PubMed] [Google Scholar]
- Seiler S., Kirchner J., Horn C., Kallipolitou A., Woehlke G., Schliwa M. Cargo binding and regulatory sites in the tail of fungal conventional kinesin. Nat. Cell Biol. 2000;2:333–338. doi: 10.1038/35014022. [DOI] [PubMed] [Google Scholar]
- Setou M., Nakagawa T., Seog D.H., Hirokawa N. Kinesin superfamily motor protein KIF17 and mLin-10 in NMDA receptor-containing vesicle transport. Science. 2000;288:1796–1802. doi: 10.1126/science.288.5472.1796. [DOI] [PubMed] [Google Scholar]
- Sheng M., Pak D.T. Ligand-gated ion channel interactions with cytoskeletal and signaling proteins. Annu. Rev. Physiol. 2000;62:755–778. doi: 10.1146/annurev.physiol.62.1.755. [DOI] [PubMed] [Google Scholar]
- Skoufias D.A., Cole D.G., Wedaman K.P., Scholey J.M. The carboxyl-terminal domain of kinesin heavy chain is important for membrane binding. J. Biol. Chem. 1994;269:1477–1485. [PubMed] [Google Scholar]
- Steinberg G., Schliwa M. The Neurospora organelle motora distant relative of conventional kinesin with unconventional properties. Mol. Biol. Cell. 1995;6:1605–1618. doi: 10.1091/mbc.6.11.1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenoien D.L., Brady S.T. Immunochemical analysis of kinesin light chain function. Mol. Biol. Cell. 1997;8:675–689. doi: 10.1091/mbc.8.4.675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stock M.F., Guerrero J., Cobb B., Eggers C.T., Huang T.G., Li X., Hackney D.D. Formation of the compact confomer of kinesin requires a COOH-terminal heavy chain domain and inhibits microtubule-stimulated ATPase activity. J. Biol. Chem. 1999;274:14617–14623. doi: 10.1074/jbc.274.21.14617. [DOI] [PubMed] [Google Scholar]
- Stockinger W., Brandes C., Fasching D., Hermann M., Gotthardt M., Herz J., Schneider W.J., Nimpf J. The reelin receptor ApoER2 recruits JNK-interacting proteins-1 and -2. J. Biol. Chem. 2000;275:25625–25632. doi: 10.1074/jbc.M004119200. [DOI] [PubMed] [Google Scholar]
- Tanaka Y., Kanai Y., Okada Y., Nonaka S., Takeda S., Harada A., Hirokawa N. Targeted disruption of mouse conventional kinesin heavy chain, kif5B, results in abnormal perinuclear clustering of mitochondria. Cell. 1998;93:1147–1158. doi: 10.1016/s0092-8674(00)81459-2. [DOI] [PubMed] [Google Scholar]
- Terada S., Kinjo M., Hirokawa N. Oligomeric tubulin in large transporting complex is transported via kinesin in squid giant axons. Cell. 2000;103:141–155. doi: 10.1016/s0092-8674(00)00094-5. [DOI] [PubMed] [Google Scholar]
- Terlecky S.R., Nuttley W.M., McCollum D., Sock E., Subramani S. The Pichia pastoris peroxisomal protein PAS8p is the receptor for the C-terminal tripeptide peroxisomal targeting signal. EMBO (Eur. Mol. Biol. Organ.) J. 1995;14:3627–3634. doi: 10.1002/j.1460-2075.1995.tb00032.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai M.Y., Morfini G., Szebenyi G., Brady S.T. Release of kinesin from vesicles by hsc70 and regulation of fast axonal transport. Mol. Biol. Cell. 2000;11:2161–2173. doi: 10.1091/mbc.11.6.2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vale R.D., Fletterick R.J. The design plan of kinesin motors. Annu. Rev. Cell Dev. Biol. 1997;13:745–777. doi: 10.1146/annurev.cellbio.13.1.745. [DOI] [PubMed] [Google Scholar]
- Vale R.D., Milligan R.A. The way things movelooking under the hood of molecular motor proteins. Science. 2000;288:88–95. doi: 10.1126/science.288.5463.88. [DOI] [PubMed] [Google Scholar]
- Verhey K.J., Lizotte D.L., Abramson T., Barenboim L., Schnapp B.J., Rapoport T.A. Light chain–dependent regulation of kinesin's interaction with microtubules. J. Cell Biol. 1998;143:1053–1066. doi: 10.1083/jcb.143.4.1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wade R.H., Kozielski F. Structural links to kinesin directionality and movement. Nat. Struct. Biol. 2000;7:456–460. doi: 10.1038/75850. [DOI] [PubMed] [Google Scholar]
- Whitmarsh A.J., Cavanagh J., Tournier C., Yasuda J., Davis R.J. A mammalian scaffold complex that selectively mediates MAP kinase activation. Science. 1998;281:1671–1674. doi: 10.1126/science.281.5383.1671. [DOI] [PubMed] [Google Scholar]
- Willnow T.E., Nykjaer A., Herz J. Lipoprotein receptorsnew roles for ancient proteins. Nat. Cell Biol. 1999;1:E157–E162. doi: 10.1038/14109. [DOI] [PubMed] [Google Scholar]
- Woehlke G., Schliwa M. Directional motility of kinesin motor proteins. Biochim. Biophys. Acta. 2000;1496:117–127. doi: 10.1016/s0167-4889(00)00013-6. [DOI] [PubMed] [Google Scholar]
- Yabe J.T., Jung C., Chan W.K., Shea T.B. Phospho-dependent association of neurofilament proteins with kinesin in situ. Cell Motil. Cytoskeleton. 2000;45:249–262. doi: 10.1002/(SICI)1097-0169(200004)45:4<249::AID-CM1>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- Yasuda J., Whitmarsh A.J., Cavanagh J., Sharma M., Davis R.J. The JIP group of mitogen-activated protein kinase scaffold proteins. Mol. Cell. Biol. 1999;19:7245–7254. doi: 10.1128/mcb.19.10.7245. [DOI] [PMC free article] [PubMed] [Google Scholar]