

Abstract
Most of the effects of the signaling molecule nitric oxide (NO) are mediated by cGMP, which is synthesized by soluble guanylyl cyclase and degraded by phosphodiesterases. Here we show that in platelets and aortic tissue, NO led to a biphasic response characterized by a tremendous increase in cGMP (up to 100-fold) in less than 30 s and a rapid decline, reflecting the tightly controlled balance of guanylyl cyclase and phosphodiesterase activities. Inverse to the reported increase in sensitivity caused by NO shortage, concentrating NO attenuated the cGMP response in a concentration-dependent manner. We found that guanylyl cyclase remained fully activated during the entire course of the cGMP response; thus, desensitization was not due to a switched off guanylyl cyclase. However, when intact platelets were incubated with NO and then lysed, enhanced activity of phosphodiesterase type 5 was detected in the cytosol. Furthermore, this increase in cGMP degradation is paralleled by the phosphorylation of phosphodiesterase type 5 at Ser-92. Thus, our data suggest that NO-induced desensitization of the cGMP response is caused by the phosphorylation and subsequent activity increase of phosphodiesterase type 5.
Keywords: phosphodiesterase; cGMP; NO-sensitive guanylyl cyclase; platelets; desensitization
Introduction
Nitric oxide (NO)* has been identified as an important signaling molecule playing a crucial role in the cardiovascular and nervous systems (Waldman and Murad, 1987; Moncada et al., 1991; Garthwaite and Boulton, 1995; Ignarro et al., 1999). Most of NO's effects are mediated via the stimulation of soluble guanylyl cyclase (sGC), which leads to enhanced conversion of GTP to cGMP. sGC, a heterodimer consisting of an α and β subunit each (Koesling and Friebe, 1999), contains a prosthetic heme group that is required for NO stimulation. The cellular effects of cGMP are mediated by the activation of different cGMP effector proteins such as the cGMP-dependent protein kinases (cGKs; Hofmann et al., 2000) and the cGMP-regulated phosphodiesterases (PDEs; Juilfs et al., 1999), and the cGMP-gated ion channels (Biel et al., 1999). The NO-induced increase in the cGMP concentration is counteracted by the activation of PDEs, which are responsible for the degradation of cGMP (Francis et al., 2001).
During the last few years, several reports indicated a possible modulatory influence of NO on the cGMP response in addition to its sGC stimulatory action. In fact, a lack of NO caused by either endothelium removal, gene disruption, or inhibition of the endothelial NO synthase has been reported to lead to increased NO sensitivities in the aortic vessels; which was paralleled by an augmentation of NO-induced cGMP levels (Moncada et al., 1991; Brandes et al., 2000). Decreased sensitivities of the NO–cGMP system have been demonstrated under various conditions after treatment with NO-releasing agents (Waldman et al., 1986; Schroder et al., 1988; Ujiie et al., 1994; Filippov et al., 1997). In a recent report, Bellamy et al. (2000) showed a rapid desensitization of sGC within seconds.
It was the aim of the present study to investigate the short-term NO-dependent desensitization of the cGMP response. As the properties of the NO–cGMP system in cultured cells change very rapidly during cell cultivation (Wyatt et al., 1998; unpublished data), we chose platelets and aortic smooth muscle as model systems. Here, we show that short-term preincubation of human platelets and rat aortic strips with submaximally activating S-nitrosoglutathione (GSNO) concentrations blunted the NO-induced cGMP response. In contrast to a recent report (Bellamy et al., 2000), the decrease in the cGMP response was not based on sGC desensitization, as in our experiments the enzyme remained fully activated. Our data show that within a few seconds of the NO incubation of intact platelets, an increase in PDE5 activity can be detected in the cytosolic fractions; a result that is paralleled by an increase in phosphorylation of PDE5 at Ser-92. Therefore, we postulate that activation of PDE5, not desensitization of sGC, is responsible for the NO-induced desensitization of the cGMP response.
Results
One of the important functions of NO in the cardiovascular system is the inhibition of platelet aggregation. In these cells, NO stimulation of sGC and the resulting cGMP increase leads to the activation of the cGMP-dependent protein kinase type I (cGKI), which subsequently phosphorylates several substrate proteins including vasodilator-stimulated phosphoprotein (VASP). The rapid GSNO-induced cGMP response in human platelets is shown in Fig. 1 A. This biphasic response is characterized by a fast increase in cGMP reaching a maximum after 5–10 s, and a subsequent decrease leading to a plateau close to basal levels after 40–60 s. At the highest concentration, GSNO elicited a 30-fold increase in intraplatelet cGMP levels compared with basal level (300 ± 40 and 10 ± 3 pmol/109 platelets, respectively). As expected, at lower GSNO concentrations, peak cGMP levels reached after 5–10 s were reduced, even though the biphasic profile of the cGMP response remained identical.
Figure 1.

Time course of cGMP levels and VASP phosphorylation in human platelets after stimulation with GSNO. (A) Washed human platelets were stimulated with increasing concentrations of GSNO. After stopping the reaction at the indicated time points, cGMP was extracted and measured by RIA. Shown is a representative experiment out of a total of five experiments performed in duplicates. (B) After stimulation with 300 μM GSNO for the time indicated, platelets were lysed and cytosolic proteins were separated by SDS-PAGE and blotted. VASP phosphorylation was detected with a phosphospecific monoclonal antibody. The shift of the protein band from the apparent mass of 46 to 50 kD was caused by further phosphorylation of VASP at a second residue. Shown is a representative experiment out of three performed with similar results.
To assess the physiological relevance of the rapid increase in the intracellular cGMP concentration, the activity of the downstream cGMP effector, cGKI, was studied in human platelets. Activity of cGKI was determined by monitoring the phosphorylation of VASP, a well-characterized substrate of cGKI (Smolenski et al., 1998). As shown in Fig. 1 B, a marked immunoreactive signal with the appropriate molecular mass of 46 kD was detected as soon as 3 s after addition of GSNO, indicating a rapid transduction of the cGMP signal to cGKI and further onto VASP. The observed shift of the protein band from the apparent mass of 46 to 50 kD was caused by further phosphorylation of VASP at a second residue (Smolenski et al., 1998).
Next, to investigate the possible modulation of the cGMP response by NO, we performed preincubation experiments. First, we applied submaximally effective GSNO concentrations (3, 10, and 30 μM) for 3 min (Fig. 2), a period after which the intraplatelet cGMP levels had declined to almost basal levels (Fig. 1 A). After this 3-min preincubation, platelets were stimulated with the maximally effective concentration of GSNO (300 μM; Fig. 1), and cGMP levels were determined over time. As expected, under control conditions (0 μM GSNO during preincubation), the cGMP response to 300 μM GSNO resembled that seen in Fig. 1. However, GSNO preincubation attenuated the cGMP responses in a concentration-dependent manner, when induced by maximally effective GSNO. The use of 30 μM GSNO during preincubation almost totally abrogated the NO-induced cGMP response in platelets. These data indicate a rapidly induced desensitization of the cGMP response. Furthermore, the degree of NO sensitivity of the cGMP system appears to be inversely related to the amount of NO present during preincubation of the platelets.
Figure 2.

NO-induced desensitization of the cGMP response in human platelets. Platelets preincubated for 3 min with the indicated concentrations of GSNO were stimulated with a maximally effective GSNO concentration (300 μM; indicated by the arrow). cGMP accumulation at the indicated time points was determined by RIA. Shown is a representative experiment out of a total of three experiments performed in duplicates.
To see whether the observed NO-induced decrease in the cGMP response was a special feature of platelets, we chose rat aortic strips as another system; here, the cGMP system is known to play an important role in the regulation of smooth muscle tone. To prevent endogenously produced NO from affecting the sensitivity state of the cGMP system, the experiments with aortic strips were performed in the presence of the NO synthase inhibitor N-nitro-l-arginine methyl ester (200 μM). Application of GSNO (300 μM) led to a biphasic cGMP response similar to that seen in human platelets (Fig. 3 A), consisting of a fast and enormous increase in cGMP levels (150-fold; maximum after 20–30 s) followed by a rapid and pronounced decrease. Although the amount of cGMP measured in the aortic preparations from different animals varied considerably (0.5–1.5 and 100–250 pmol/mg protein under basal and stimulated conditions, respectively), the biphasic shapes of the responses were qualitatively similar.
Figure 3.

Time course of NO-induced cGMP levels and NO-induced desensitization of the cGMP response in rat aortic strips. (A) Aortic strips were stimulated with 300 μM GSNO for the times indicated. After shock freezing and homogenization of the tissue, the extracted cGMP was measured by RIA. As the amount of cGMP measured in the aortic preparations from different animals varied considerably, data are expressed as the percentage of maximal stimulation. (B) Aortic strips preincubated for 3 min with the indicated concentrations of GSNO were stimulated with 300 μM GSNO, and cGMP levels were measured after 30 s. Data represent means ± SEM of nine independent experiments performed in duplicates (A) or means ± SEM of triplicate determinations in one representative experiment out of a total of three similar experiments (B).
Next, we tested whether NO-induced desensitization of the cGMP system also occurred in aortic tissue, using a similar experimental setup as described for the platelets in Fig. 2. Aortic strips were preincubated for 3 min with submaximally effective GSNO concentrations and then stimulated with 300 μM GSNO. cGMP levels were determined at one single time point, i.e., 30 s after NO stimulation (Fig. 3 B). Preincubation with NO attenuated the NO-induced cGMP response in a concentration-dependent manner. As seen in platelets, these data show the desensitizing effect of NO on the cGMP response in vascular smooth muscle.
Our next experiment investigated the underlying mechanism of the observed desensitization in the platelets. Intracellular cGMP levels reflect the state of activity of the cGMP-forming sGC and cGMP-degrading PDEs. Therefore, decreased synthesis or enhanced degradation should account for the desensitization of the cGMP response. To find out whether desensitization occurred on the level of sGC, NO-induced cGMP formation was determined in the presence of PDE inhibitors. As PDE5 and PDE2 are the major enzymes responsible for cGMP degradation in platelets, we chose the respective, specific inhibitors sildenafil and erythro-9-(2-hydroxy-3-nonyl)-adenine (EHNA). Under a blockade of the two PDEs, cGMP levels did not decrease after the initial rise in cGMP but rather came to an enormously elevated plateau within 60–120 s (Fig. 4 A). On this plateau, cGMP levels were ∼10-fold higher than the maximal levels seen in the absence of PDE inhibitors (3,000 vs. 300 pmol/109 platelets; Fig. 4 A). Fig. 4 B shows NO preincubation experiments in the presence of PDE inhibitors performed similar to those in Fig. 2. GSNO preincubation (3, 10, and 30 μM for 3 min) in the presence of PDE inhibitors led to augmented cGMP levels at time point zero, since the cGMP formed during preincubation was not degraded. Although the initial rate of cGMP formation in the control sample appeared greater than the rates of the NO-preincubated samples, cGMP levels reached a similar plateau in all cases. Calculation revealed an extremely high concentration, ∼600 μM, of intraplatelet cGMP (see Discussion). Therefore, reduction of the substrate GTP concentration could not be ruled out.
Figure 4.

Time course of NO-induced cGMP accumulation in human platelets in the presence of PDE inhibitors. (A) Platelets preincubated for 20 min with inhibitors specific for PDE5 and PDE2, sildenafil and EHNA, respectively, (100 μM each; filled circles) were stimulated with 300 μM GSNO. Then, platelets were lysed at the indicated time points, and cGMP was determined by RIA. For comparison, the cGMP response in the absence of PDE inhibitors is shown (open circles). (B) Platelets were preincubated for 20 min with sildenafil and EHNA (100 μM each). After 17 min, GSNO was added at the indicated concentrations (i.e., 3-min preincubation). At time point zero, a maximally effective GSNO concentration (300 μM; indicated by the arrow) was administered. Platelets were lysed at the indicated time points, and cGMP accumulation was determined by RIA. For comparison, cGMP accumulation without NO preincubation is also shown (filled circles with dotted line). Data represent means ± SEM from three independent experiments performed in duplicates.
To find out whether NO-induced cGMP formation in the presence of PDE inhibitors leads to a significant reduction of the substrate GTP, we measured intraplatelet GTP concentrations using HPLC. Original traces of the GTP peaks of NO-stimulated platelets in the absence (control; dotted line) and presence of both PDE inhibitors (100 μM each; solid line) are shown in Fig. 5 A. Clearly, the inhibition of PDEs in NO-stimulated platelets led to a 50% reduction of the intracellular GTP concentration, as shown in the statistical analysis in Fig. 5 B. Because of this massive loss of available GTP in the stimulated platelets in the presence of PDE inhibitors, kinetic analysis of cGMP synthesis is not appropriate.
Figure 5.


HPLC analysis of intraplatelet GTP levels under NO-stimulated conditions in the absence and presence of PDE inhibitors. After preincubation for 10 min with DMSO (control) or the combination of sildenafil and EHNA (100 μM each), platelets were stimulated with 300 μM GSNO for 60 s. Subsequently, reactions were stopped by the addition of HClO4 (final concentration 0.7 M). After precipitation of proteins and adjustment of the pH, nucleotides were separated on a Mono-Q column as described under Materials and methods. (A) Original trace data shows elution of GTP at 13.3% buffer B (see Materials and methods). (B) Bars show the quantitative analysis as means ± SEM from four independent experiments.
Under substrate-depleting conditions, the actual activity of sGC cannot be determined. Therefore, we tried to bypass this problem by choosing a different experimental approach that would not generate extraordinarily elevated cGMP levels. Platelets were stimulated with GSNO and PDE inhibitors were added at different stages of the cGMP response, as indicated in the inset of Fig. 6 (simultaneous to NO, 0 s; near maximum, 15 s; within the cGMP reduction phase, 30 s; and during plateau phase, 60 s). After addition of PDE inhibitors, aliquots of the platelet suspensions were taken every 3 s (series 1: 0, 3, 6 s; series 2: 18, 21, 24 s; series 3: 33, 36, 39 s; series 4: 63, 66, 69 s after NO stimulation), and cGMP accumulation was measured. The increases in cGMP measured in the four series are shown in Fig. 6. Because of the short-term application of PDE inhibitors, depletion of substrate did not occur, and cGMP synthesis, i.e., sGC activity, was unmasked within the cGMP response. Within this time range cGMP increased linearly, and in the different series, the rates of cGMP synthesis were similar (series 1: 109 ± 23, series 2: 102 ± 7, series 3: 108 ± 6, series 4: 110 ± 20 pmol cGMP × s−1/109 platelets), indicating that the apparent NO-stimulated sGC activities were similar, regardless of the stage of the cGMP response. We conclude that sGC is not responsible for desensitizing the cGMP response.
Figure 6.

sGC does not desensitize during the time course of the NO-stimulated cGMP response in human platelets. Platelets were stimulated with 300 μM GSNO at time point zero. PDE inhibitors sildenafil and EHNA (100 μM each) were administered at the time points indicated by the arrows in the inset corresponding to different states of the cGMP response (see text). After addition of PDE inhibitors, aliquots of the platelet suspension were removed into ice cold ethanol every 3 s. Extracted cGMP was then determined by RIA. For the very first data point at 0 s, basal cGMP levels were subtracted; for better comparison, the cGMP levels 3 s after addition of PDE inhibitors (i.e., 18, 33 or 63 s) were subtracted from the following cGMP levels. Thus, the y axis shows the increase in cGMP values within every 3 s. Values represent means ± SEM of hexaplicate determinations in one representative experiment out of three similar experiments.
The rapid desensitization of the cGMP response (Fig. 2) appears not to be based on a decrease in cGMP synthesis, which leaves cGMP degradation as the mechanism most likely to account for this phenomenon. Therefore, we focused on a possible modulation of PDE activity. At the different stages of the NO-stimulated cGMP response (0, 15, 30, and 60 s; Fig. 6, inset), platelets were lysed by sonification, and PDE activity was then determined in the cytosolic fraction. As shown in Fig. 7 A, cGMP degradation measured in the presence of 1 μM cGMP showed 1.6-, 2.1-, and 2.4-fold increases in enzyme activity after 15, 30, and 60 s of NO stimulation of intact platelets, respectively. Thus, activation of PDE began simultaneously with NO exposure in intact cells. A changed activity due to translocation of PDEs can be ruled out, as PDE activity in the cytosol was ∼95% of that in the homogenate samples and was not altered by NO stimulation (control: 94% ± 12%; NO-stimulated: 96% ± 12%). To confirm that sGC does not desensitize under these conditions, NO-stimulated cGMP formation was measured in cytosolic fractions. As shown in Fig. 7 B , sGC activity remained unchanged.
Figure 7.
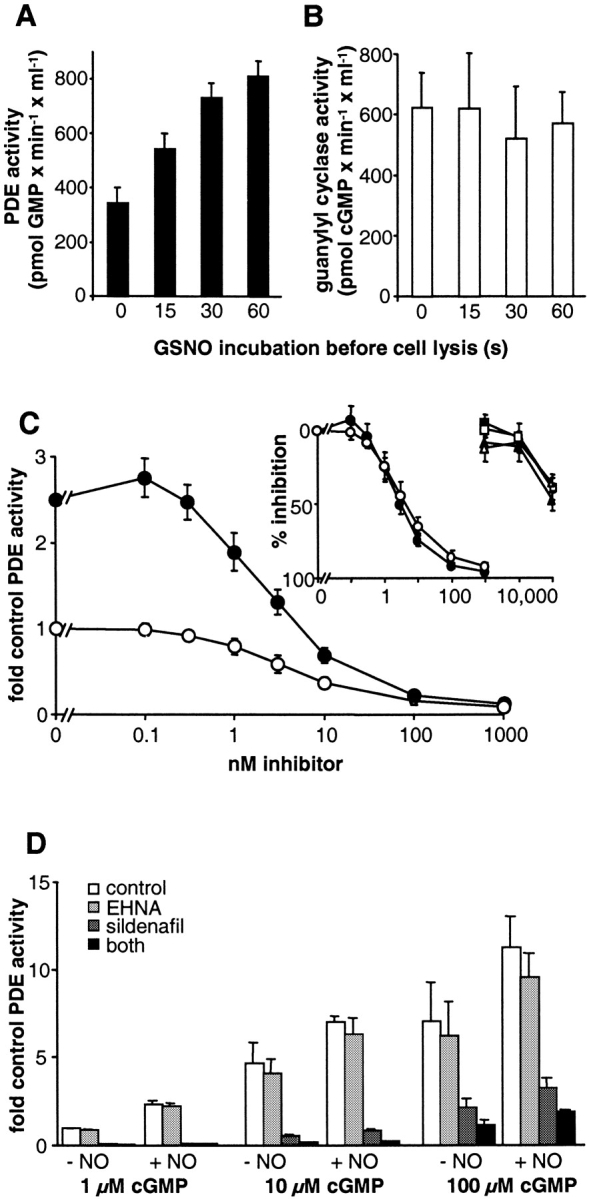
Cytosolic PDE and sGC activities after NO incubation of intact platelets. Platelets were stimulated with 300 μM GSNO and lysed after 15, 30, or 60 s. (A) PDE (1 μM cGMP as substrate) and (B) NO-stimulated sGC activities were determined in the supernatant fraction as described in Materials and methods. Values represent means ± SEM of quadruplicate determinations in one representative experiment out of three similar experiments. (C) PDE activities in the cytosol of control platelets (open symbols) and NO-incubated platelets (60 s; closed symbols) were measured in the presence of isoform-specific inhibitors (sildenafil, circles; EHNA, squares; milrinone, triangles). The activities were normalized relative to that of the control cytosol (no NO incubation, absence of sildenafil). Data represent means ± SEM of two independent experiments performed in duplicates. (D) Cytosolic PDE activities were measured with 1, 10, and 100 μM cGMP in the presence of the indicated inhibitors (20 μM EHNA, 1 μM sildenafil). The activities were normalized relative to that of the control cytosol (no NO incubation, 1 μM cGMP). Data represent means ± SEM of two independent experiments performed in duplicates.
To identify the isoform of PDE responsible for the enhanced degradation, cGMP breakdown was measured in the presence of PDE inhibitors specific for PDE2 (EHNA), PDE3 (milrinone), and PDE5 (sildenafil). Fig. 7 C shows that cytosolic cGMP degradation was inhibited in a concentration-dependent manner by sildenafil (IC50 of ∼3 nM), whereas EHNA and milrinone only prevented cGMP breakdown at very high concentrations, at which an effect on PDE5 cannot be ruled out. Even at higher cGMP concentrations (10 and 100 μM), sildenafil inhibited the majority of PDE activity (Fig. 7 D), whereas the addition of EHNA (20 μM) caused only up to 10% inhibition of PDE activity. The increase in PDE activity after NO incubation was still detectable under PDE2-inhibiting conditions but almost abrogated under PDE5 inhibition. Therefore, the increase in PDE activity can be attributed to PDE5.
The fact that enhanced PDE activity was evident after breaking up the cells suggested a covalent modification as the mechanism underlying desensitization. Phosphorylation of PDE5 at Ser-92 has been shown to be associated with an increase in enzyme activity (Wyatt et al., 1998; Corbin et al., 2000). Therefore, we studied this possible phosphorylation by using a novel phospho-specific antibody against PDE5, raised against the phosphorylated Ser-92–containing peptide. Fig. 8 A shows that the antibody only reacts with the phosphorylated and not with the unphosphorylated form of PDE5. Platelets were stimulated with GSNO, and at the different stages of the NO-stimulated cGMP response (Fig. 6, inset) aliquots of the platelet suspension were subjected to SDS-PAGE and blotted onto nitrocellulose (Fig. 8 B). Under nonstimulated conditions, PDE5 was not recognized. Immunoreactive signals appeared already 15 s after NO stimulation and paralleled the increased enzyme activity within a time range of 60 s (Fig. 7 A). Phosphorylation of PDE5 was detected with concentrations of GSNO as low as 3 μM (unpublished data). Thus, our data show, for the first time, the concomitant phosphorylation and activity increase of PDE5 over the course of the NO-stimulated cGMP response. In conclusion, phosphorylation of PDE5 by a yet unidentified kinase appears to be responsible for the NO-induced desensitization of the cGMP response.
Figure 8.
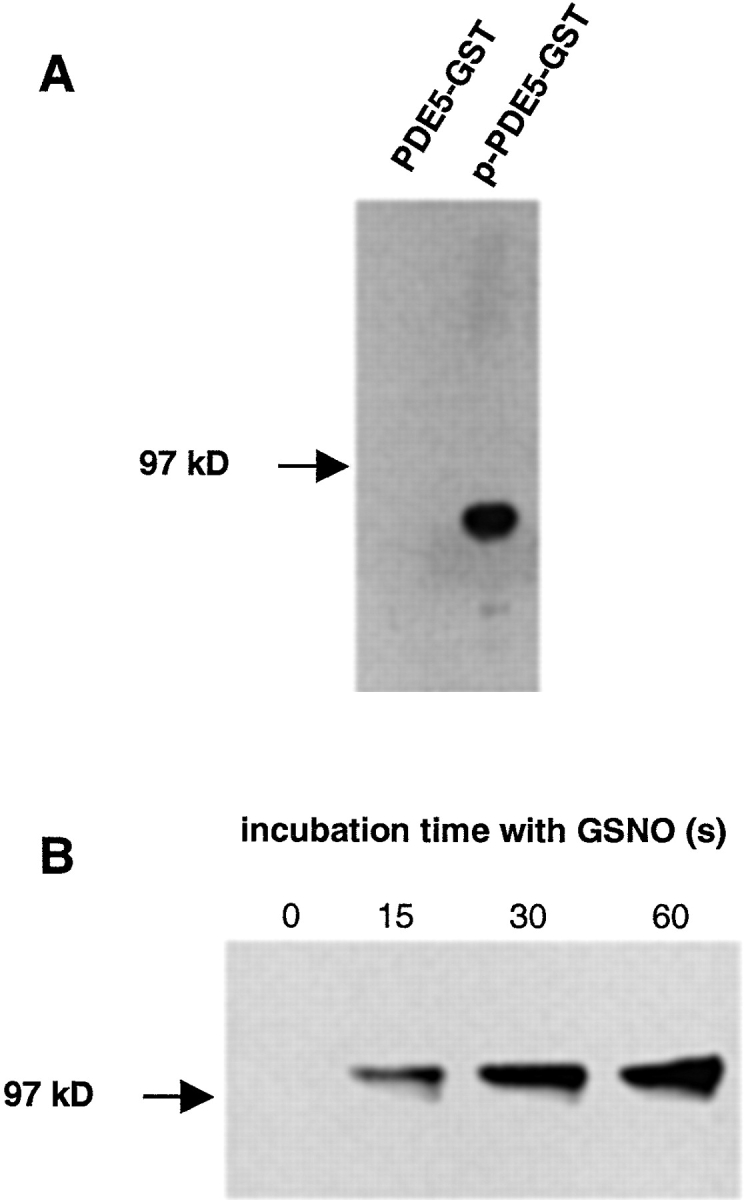
Detection of PDE5 phosphorylation. (A) Purified unphosphorylated and phosphorylated GST–PDE5 fusion proteins (∼85 kD; 200 ng, each) were subjected to SDS-PAGE and transferred to nitrocellulose. Membranes were incubated with the antiphospho-PDE5 antibody in a dilution of 1:10,000. (B) Platelets were stimulated with 300 μM GSNO. At the indicated time points, aliquots (3.6 × 107 platelets) were removed and proteins were separated by SDS-PAGE and transferred to nitrocellulose. Membranes were incubated with phospho-PDE5–specific antibody in a dilution of 1:50,000.
Discussion
In this article, we show that NO not only induces a rapid cGMP response in platelets and in aortic strips but also serves to alter the responsiveness of the cGMP cascade. In both models, the NO-induced cGMP response is biphasic and characterized by a very fast increase in cGMP, which amounts to a calculated peak concentration of ∼60 μM in platelets (see below). Subsequently, the concentration of cGMP declines rapidly, and it can be assumed that PDE activity has outcompeted cGMP synthesis. Thus, the biphasic cGMP accumulation profiles are indicative of a complex, thus far poorly understood interplay of cGMP-forming and -degrading activities.
The rapid desensitizing effect of NO is demonstrated by preincubating platelets or aortic strips, which reveals that the extent of the cGMP response is inversely related to the amount of NO present during the preincubation (Figs. 2 and 3 B). At high NO concentrations, the cGMP system becomes desensitized almost completely, whereas at low tissue concentrations of NO, the system retains a higher sensitivity state. Physiologically, modulation of the sensitivity of the cGMP response reflects the ability of the NO–cGMP system to adapt to acute changes in NO exposition. Thus, information is not only transduced within the NO–cGMP system but is also processed for adaptation to the amount of NO available.
The reduced sensitivity of the cGMP response after NO preincubation can be explained by either reduced cGMP synthesis and/or enhanced cGMP degradation. To solely monitor cGMP formation (i.e., the actual sGC activity), PDE inhibitors can be used to prevent GMP breakdown. Based mainly on this approach, i.e., measurements of cGMP accumulation in the presence of PDE inhibitors, a recent report suggested a rapid NO-induced desensitization of platelet sGC (Bellamy et al., 2000).
These results and our data (Fig. 4 A, inset) are in perfect accordance with the 10-fold elevation of NO-induced cGMP levels caused by the addition of PDE inhibitors (3,000 pmol/109 platelets equaling 1,500 pmol/mg protein, assuming a protein content of 2 mg/109 platelets; Eigenthaler et al., 1992). The plateau of the cGMP response, reached after 60 s in the presence of PDE inhibitors, indicates low cGMP-forming activity, which could be attributed to a switched-off sGC. Peculiarly, NO-preincubated platelets reached a similar plateau (Fig. 4 B) suggesting that this intracellular cGMP concentration may represent an “ultimate” level for cGMP accumulation. However, the decrease in sGC activity could also be explained by a reduction in substrate GTP. Assuming a single platelet volume of 5.2 fl (Corash et al., 1977), these peak cGMP levels correspond to an intraplatelet concentration of ∼600 μM (intracellular GTP levels between 400 and 800 μM have been published; Traut, 1994). Under normal conditions, sGC will cyclize GTP to cGMP, which is then hydrolyzed to GMP by PDEs. GMP will be readily phosphorylated to GDP and GTP by nucleoside mono- and diphosphate kinases, respectively. The only enzymes able to convert cGMP to GMP and hence back into the GMP/GDP/GTP metabolism are the PDEs. Production of cGMP in the presence of PDE inhibitors can be viewed as a “dead-end” for guanosine phosphates; accordingly, we see a dramatic accumulation of cGMP and a 50% reduction of intraplatelet GTP levels. Taking compartmentalization as well as protein-bound GTP into account, it is very likely that this decrease in GTP leads to a massive reduction in accessible substrate for sGC.
Realizing that substrate depletion could account for the reduction in cGMP forming activity, our next experiment was designed to avoid GTP depletion, and sGC was revealed to be fully active during the entire cGMP response. Here, sGC activities obtained at various time points during the NO-induced cGMP response were practically identical to the initial velocity measured directly after NO/sildenafil/EHNA coadministration (Fig. 6).
If sGC is not responsible, a change in PDE activity has to account for the desensitization of the cGMP response. In fact, we were able to show increased PDE activity in the cytosol of NO-incubated, intact platelets, in which sGC activity remained unaffected. Although the NO-induced increases in PDE activity were modest, they may well be sufficient to counteract cGMP synthesis. Rough calculation reveals a PDE activity in platelet cytosol of ∼40 pmol/s per 109 platelets at 1 μM cGMP (Fig. 7 A; 1 ml of cytosolic fraction contains 1.37 × 108 platelets). NO-stimulated cGMP formation by guanylyl cyclase ranges between 100 and 110 pmol cGMP/s per 109 platelets (Fig. 6). Thus, a 2.4-fold increase in cGMP degradation, after 60 s (Fig. 7 A), is sufficient to increase PDE activity to the level of sGC activity. The resulting net cGMP accumulation of zero explains the “plateau” phase, after 30 s, in which cGMP levels are low although guanylyl cyclase remains active (Fig. 6). In combination with the NO-induced activation, substrate-linked activation of PDE (Fig. 7 D) leads to PDE activity exceeding that of sGC, which is required for net cGMP reduction. The observed increase in PDE activity can be attributed to PDE5, as shown by sildenafil inhibition. Furthermore, the detection of enhanced PDE5 activity after disruption of cell integrity indicates a covalent modification as the mechanism underlying desensitization.
In fact, Wyatt et al. (1998) have shown that ANP-induced cGMP accumulation leads to phosphorylation of PDE5 in primary vascular smooth muscle cells concomitant with a two- to fourfold increase in the catalytic activity of the immunoprecipitated enzyme. In vitro, phosphorylation of this PDE isozyme was shown to result in a 50–70% increase in catalytic activity (Corbin et al., 2000). Our experiments (Fig. 8) with a novel antibody show that phosphorylation of PDE5 occurs within the same time range as the observed increase in PDE activity (Fig. 7 A). Furthermore, phosphorylation of PDE5 was detected in a sample with a GSNO concentration as small as 3 μM. Interestingly, this was the lowest concentration that led to increased cGMP formation and desensitization of the cGMP response (Figs. 1 A and 2). In sum, our data suggest that phosphorylation of PDE5 is the underlying mechanism of the NO-induced desensitization of the cGMP response. Although it is tempting to speculate on cGKI, the kinase responsible for PDE5 phosphorylation remains to be identified. The presented data emphasize the complexity of the tightly regulated concert of cGMP synthesis and degradation in the intact cell, and underline the therapeutic importance of PDEs as pharmacological targets.
Materials and methods
Time course of intraplatelet cGMP accumulation
Venous blood from healthy volunteers was drawn into ACD (85 mM trisodium citrate, 65 mM citric acid, 100 mM glucose) and platelets were prepared as described previously (Friebe et al., 1998). Aliquots (900 μl) of the platelet suspension containing 1.5 × 108 platelets/ml were equilibrated at 37°C for 10 min, followed by addition of GSNO at the indicated concentrations. At the indicated time points, an aliquot of the suspension corresponding to 1.35 × 107 platelets was removed into ice cold ethanol (final concentration 66%). Vials were put on ice for 30 min and centrifuged at 4°C and 20,000 g for 15 min. Supernatants were dried at 95°C and the cGMP content was determined using a radioimmunoassay (RIA) as described before (Friebe et al., 1998). Assays were performed in duplicates, unless otherwise indicated.
Phosphorylation of VASP
Platelets (5 × 108/ml) were stimulated with 300 μM GSNO; at the indicated time points, an aliquot (4.5 × 107 platelets) was removed into Laemmli buffer and heated to 95°C for 5 min. Detection of VASP phosphorylation was performed as described previously (Friebe et al., 1998).
Determination of cGMP levels in intact aortic strips
Aortas from male Wistar-Kyoto rats were cleaned of connective tissue and cut into strips of 2–5 mg wet weight. Before stimulation with GSNO, strips were allowed to equilibrate in the presence of 200 μM N-nitro-l-arginine methyl ester for 1 h (37°C) in Krebs-Henseleit solution (118 mM NaCl, 4.7 mM KCl, 2.5 mM CaCl2, 1.2 mM KH2PO4, 1.2 mM MgSO4, 25 mM NaHCO3, 7.5 mM glucose), pH 7.4, gassed with 95% O2 and 5% CO2. In the case of preincubation, samples were treated with the indicated GSNO concentrations for 3 min and then washed twice. Stimulation with the maximally effective GSNO concentration was performed 2 min after the second wash. To terminate the reactions, tissue was shock frozen using metal forceps precooled in liquid nitrogen. Isolation and measurement of cGMP was performed as described (Rothermund et al., 2000). Protein content was determined using the bicinchoninic acid method.
HPLC detection of GTP
After GSNO stimulation of platelets in the absence or presence of the PDE inhibitors sildenafil and EHNA (100 μM, respectively), the reactions were stopped by the addition of 0.8 M HClO4. After centrifugation (15 min at 20,000 g and 4°C), supernatants were adjusted to pH 12.0 with KOH and frozen at –80°C. After a second centrifugation step, supernatants were diluted into running buffer A (see below), and pH was adjusted to that of the running buffer. Samples were loaded onto a Mono-Q HR5/5 column (Amersham Pharmacia Biotech) and eluted with a linear gradient (buffer A: 20 mM K2HPO4, pH 8.0; buffer B: 1 mM NaCl, 20 mM K2HPO4, pH 8.0; 0–20% B, 240 min; flow rate 0.5 ml/min). Elution of nucleotides was monitored at 254 nm; GTP was identified by co-chromatography of 32P–GTP.
Determination of cGMP synthesis in platelets
NO-stimulated cGMP synthesis in platelets was assessed by adding the indicated PDE inhibitors (100 μM sildenafil and EHNA) either simultaneously with GSNO or 15, 30, or 60 s after addition of GSNO. Subsequently, aliquots of the platelet suspension were removed every 3 s for cGMP determination. Experiments were performed in triplicates or hexaplicates.
Measurement of PDE and sGC activities in the cytosolic fraction of platelets
Platelet suspensions were adjusted to 3 × 108 platelets/ml. Aliquots of 450 μl were equilibrated at 37°C for 10 min and stimulated with 50 μl GSNO yielding a final concentration of 300 μM. After the indicated incubation time, 500 μl of an ice cold protease inhibitor cocktail (2 μM pepstatin A, 0.4 μM benzamidine, 0.5 mM PMSF, 2 mM sodium vanadate, 1 mg/ml BSA, 4 mM DTT) was added, and the suspension was briefly sonicated (one pulse of 5 s) on ice using a Branson Sonifier B-12. After centrifugation (15 min, 4°C, 20,000 g), PDE activity in the supernatant was measured by the conversion of 32P-cGMP (synthesized from [α-32P]GTP using purified sGC) to guanosine and 32P-phosphate in the presence of alkaline phosphatase at 37°C for 10 min. Reaction mixtures contained 1 μl of the supernatants, 32P-cGMP (10,000–50,000 cpm), 1 μM cGMP, 12 mM MgCl2, 3 mM DTT, 0.5 mg/ml BSA, 1 U of alkaline phosphatase, and 50 mM triethanolamine/HCl, pH 7.4, in a total volume of 0.1 ml. Reactions were stopped by the addition of 900 μl ice cold charcoal suspension (20% activated charcoal in 50 mM KH2PO4, pH 2.3). After pelleting the charcoal by centrifugation, 32P-phosphate was measured in the supernatant. For the determination of sGC activity, 10 μl of the supernatant was measured in the presence of [α-32P]GTP (500,000 cpm), 300 μM GTP, 3 mM MgCl2, 3 mM DTT, 1 mM cGMP, 0.5 mg/ml BSA, 300 μM GSNO, 1 mM IBMX and a GTP-regenerating system (0.025 mg creatine kinase, 5 mM creatine phosphate), and 50 mM triethanolamine/HCl, pH 7.4, in a total volume of 0.1 ml as described previously (Friebe et al., 1996).
Generation of antiphospho-Ser92-PDE5 antibody and GST-PDE5
Antiphospho-Ser92-PDE5 antibody was raised by immunizing rabbit with keyhole limpet hemocyanin conjugate of synthetic phospho-Ser92 peptide (CTRKIS-PO3-ASEFDR). The immune serum was purified over two sequential affinity columns, CTRKISASEFDR peptide immunosorbent and the CTRKIS-PO3-ASEFDR peptide immunosorbent. Preparation of the recombinant GST-PDE5 was as described (Liu et al., 2001).
Detection of in vitro–phosphorylated PDE5
200 ng of the purified nonphosphorylated and phosphorylated GST–PDE5 fusion proteins, respectively, were subjected to SDS-PAGE and transferred to nitrocellulose. Membranes were incubated with the antiphospho-PDE5 antibody in a dilution of 1:10,000 followed by a peroxidase-coupled anti–rabbit antibody (Sigma-Aldrich). Detection was performed using an ECL kit (Amersham Pharmacia Biotech).
Detection of phosphorylated PDE5 in platelets
Platelet suspensions (4 × 108 platelets/ml) were stimulated with GSNO. At the indicated time points, an aliquot (3.6 × 107 platelets) was removed into Laemmli buffer and boiled for 10 min. Proteins were separated by SDS-PAGE and transferred to nitrocellulose. Membranes were incubated with phospho-PDE5–specific antibody in a dilution of 1:50,000. Detection was performed as described above.
Materials
Sildenafil was a gift from Pfizer (Sandwich, UK). GSNO and monosuccinyl-tyrosyl-cGMP were obtained from Sigma-Aldrich. Anti-VASP antibody was from Alexis Biochemicals Corp. Alkaline phosphatase was purchased from Boehringer, EHNA was from Tocris Cookson. Activated charcoal was from Riedel-de Haën. [α-32P]GTP (800 Ci/mmol) was purchased from DuPont, Na125I was from Amersham Pharmacia Biotech. The bicinchoninic acid protein determination kit was from Pierce Chemical Co. Chemicals used for HPLC detection of GTP were of HPLC grade.
Acknowledgments
We thank Friedrich Eichhorst, Katja Rezny, Gaby Scheibel, and Arkadius Pacha for excellent technical assistance. The critical reading of the manuscript by Dr. Günter Schultz is gratefully acknowledged.
This work was supported by the Deutsche Forschungsgemeinschaft.
Footnotes
Abbreviations used in this paper: cGKI, cGMP-dependent protein kinases type I; EHNA, erythro-9-(2-hydroxy-3-nonyl)-adenine; GSNO, S-nitrosoglutathione; NO, nitric oxide; PDE, phosphodiesterase; RIA, radioimmunoassay; sGC, soluble guanylyl cyclase; VASP, vasodilator-stimulated phosphoprotein.
References
- Bellamy, T.C., J. Wood, D.A. Goodwin, and J. Garthwaite. 2000. Rapid desensitization of the nitric oxide receptor, soluble guanylyl cyclase, underlies diversity of cellular cGMP responses. Proc. Natl. Acad. Sci. USA. 97:2928–2933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biel, M., X. Zong, A. Ludwig, A. Sautter, and F. Hofmann. 1999. Structure and function of cyclic nucleotide-gated channels. Rev. Physiol. Biochem. Pharmacol. 135:151–171. [DOI] [PubMed] [Google Scholar]
- Brandes, R.P., D. Kim, F.H. Schmitz-Winnenthal, M. Amidi, A. Godecke, A. Mulsch, and R. Busse. 2000. Increased nitrovasodilator sensitivity in endothelial nitric oxide synthase knockout mice: role of soluble guanylyl cyclase. Hypertension. 35:231–236. [DOI] [PubMed] [Google Scholar]
- Corash, L., H. Tan, and H.R. Gralnick. 1977. Heterogeneity of human whole blood platelet subpopulations. I. Relationship between buoyant density, cell volume, and ultrastructure. Blood. 49:71–87. [PubMed] [Google Scholar]
- Corbin, J.D., I.V. Turko, A. Beasley, and S.H. Francis. 2000. Phosphorylation of phosphodiesterase-5 by cyclic nucleotide-dependent protein kinase alters its catalytic and allosteric cGMP-binding activities. Eur. J. Biochem. 267:2760–2767. [DOI] [PubMed] [Google Scholar]
- Eigenthaler, M., C. Nolte, M. Halbrugge, and U. Walter. 1992. Concentration and regulation of cyclic nucleotides, cyclic-nucleotide-dependent protein kinases and one of their major substrates in human platelets. Estimating the rate of cAMP-regulated and cGMP-regulated protein phosphorylation in intact cells. Eur. J. Biochem. 205:471–481. [DOI] [PubMed] [Google Scholar]
- Filippov, G., D.B. Bloch, and K.D. Bloch. 1997. Nitric oxide decreases stability of mRNAs encoding soluble guanylate cyclase subunits in rat pulmonary artery smooth muscle cells. J. Clin. Invest. 100:942–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis, S.H., I.V. Turko, and J.D. Corbin. 2001. Cyclic nucleotide phosphodiesterases: relating structure and function. Prog. Nucleic Acid. Res. Mol. Biol. 65:1–52. [DOI] [PubMed] [Google Scholar]
- Friebe, A., G. Schultz, and D. Koesling. 1996. Sensitizing soluble guanylyl cyclase to become a highly CO-sensitive enzyme. EMBO J. 15:6863–6868. [PMC free article] [PubMed] [Google Scholar]
- Friebe, A., F. Mullershausen, A. Smolenski, U. Walter, G. Schultz, and D. Koesling. 1998. YC-1 potentiates nitric oxide- and carbon monoxide-induced cyclic GMP effects in human platelets. Mol. Pharmacol. 54:962–967. [DOI] [PubMed] [Google Scholar]
- Garthwaite, J., and C.L. Boulton. 1995. Nitric oxide signaling in the central nervous system. Annu. Rev. Physiol. 57:683–706. [DOI] [PubMed] [Google Scholar]
- Hofmann, F., A. Ammendola, and J. Schlossmann. 2000. Rising behind NO: cGMP-dependent protein kinases. J. Cell Sci. 113:1671–1676. [DOI] [PubMed] [Google Scholar]
- Ignarro, L.J., G. Cirino, A. Casini, and C. Napoli. 1999. Nitric oxide as a signaling molecule in the vascular system: an overview. J. Cardiovasc. Pharmacol. 34:879–886. [DOI] [PubMed] [Google Scholar]
- Juilfs, D.M., S. Soderling, F. Burns, and J.A. Beavo. 1999. Cyclic GMP as substrate and regulator of cyclic nucleotide phosphodiesterases (PDEs). Rev. Physiol. Biochem. Pharmacol. 135:67–104. [DOI] [PubMed] [Google Scholar]
- Koesling, D., and A. Friebe. 1999. Soluble guanylyl cyclase: structure and regulation. Rev. Physiol. Biochem. Pharmacol. 135:41–65. [DOI] [PubMed] [Google Scholar]
- Liu, L., T. Underwood, H. Li, R. Pamukcu, and W.J. Thompson. 2001. Specific cGMP binding by the cGMP binding domains of cGMP-binding cGMP specific phosphodiesterase. Cell. Signal. In press. [DOI] [PubMed] [Google Scholar]
- Moncada, S., D.D. Rees, R. Schulz, and R.M. Palmer. 1991. Development and mechanism of a specific supersensitivity to nitrovasodilators after inhibition of vascular nitric oxide synthesis in vivo. Proc. Natl. Acad. Sci. USA. 88:2166–2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothermund, L., A. Friebe, M. Paul, D. Koesling, and R. Kreutz. 2000. Acute blood pressure effects of YC-1-induced activation of soluble guanylyl cyclase in normotensive and hypertensive rats. Br. J. Pharmacol. 130:205–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder, H., D.C. Leitman, B.M. Bennett, S.A. Waldman, and F. Murad. 1988. Glyceryl trinitrate-induced desensitization of guanylate cyclase in cultured rat lung fibroblasts. J. Pharmacol. Exp. Ther. 245:413–418. [PubMed] [Google Scholar]
- Smolenski, A., C. Bachmann, K. Reinhard, P. Honig-Liedl, T. Jarchau, H. Hoschuetzky, and U. Walter. 1998. Analysis and regulation of vasodilator-stimulated phosphoprotein serine 239 phosphorylation in vitro and in intact cells using a phosphospecific monoclonal antibody. J. Biol. Chem. 273:20029–20035. [DOI] [PubMed] [Google Scholar]
- Traut, T.W. 1994. Physiological concentrations of purines and pyrimidines. Mol. Cell. Biochem. 140:1–22. [DOI] [PubMed] [Google Scholar]
- Ujiie, K., L. Hogarth, R. Danziger, J.G. Drewett, P.S. Yuen, I.H. Pang, and R.A. Star. 1994. Homologous and heterologous desensitization of a guanylyl cyclase-linked nitric oxide receptor in cultured rat medullary interstitial cells. J. Pharmacol. Exp. Ther. 270:761–767. [PubMed] [Google Scholar]
- Waldman, S.A., and F. Murad. 1987. Cyclic GMP synthesis and function. Pharmacol. Rev. 39:163–196. [PubMed] [Google Scholar]
- Waldman, S.A., R.M. Rapoport, R. Ginsburg, and F. Murad. 1986. Desensitization to nitroglycerin in vascular smooth muscle from rat and human. Biochem. Pharmacol. 35:3525–3531. [DOI] [PubMed] [Google Scholar]
- Wyatt, T.A., A.J. Naftilan, S.H. Francis, and J.D. Corbin. 1998. ANF elicits phosphorylation of the cGMP phosphodiesterase in vascular smooth muscle cells. Am. J. Physiol. 274:H448–455. [DOI] [PubMed] [Google Scholar]