

Abstract
The Rho family GTPase Cdc42 is a key regulator of cell polarity and cytoskeletal organization in eukaryotic cells. In yeast, the role of Cdc42 in polarization of cell growth includes polarization of the actin cytoskeleton, which delivers secretory vesicles to growth sites at the plasma membrane. We now describe a novel temperature-sensitive mutant, cdc42-6, that reveals a role for Cdc42 in docking and fusion of secretory vesicles that is independent of its role in actin polarization. cdc42-6 mutants can polarize actin and deliver secretory vesicles to the bud, but fail to fuse those vesicles with the plasma membrane. This defect is manifested only during the early stages of bud formation when growth is most highly polarized, and appears to reflect a requirement for Cdc42 to maintain maximally active exocytic machinery at sites of high vesicle throughput. Extensive genetic interactions between cdc42-6 and mutations in exocytic components support this hypothesis, and indicate a functional overlap with Rho3, which also regulates both actin organization and exocytosis. Localization data suggest that the defect in cdc42-6 cells is not at the level of the localization of the exocytic apparatus. Rather, we suggest that Cdc42 acts as an allosteric regulator of the vesicle docking and fusion apparatus to provide maximal function at sites of polarized growth.
Keywords: Cdc42; Rho; GTPases; exocytosis; cell polarity
Introduction
Polarization is critical for the proper functioning of many, if not all, cells. For the budding yeast Saccharomyces cerevisiae, polarization of secretion is essential for successful bud formation and, hence, for cell proliferation. Because eukaryotic cells demonstrate a high degree of conservation in the molecules and mechanisms used to achieve cell polarity, S. cerevisiae has become an important model system to understand this process (Drubin and Nelson, 1996).
During the early stages of bud formation in yeast, membrane proteins, lipids, secretory, and cell wall proteins are delivered through the secretory pathway to a small region of the plasma membrane at the bud tip (for review, see Pruyne and Bretscher, 2000). This vectorial traffic requires a set of polarized actin cables that are thought to act as tracks along which the type V myosin, Myo2, delivers secretory vesicles. In turn, polarization of the actin cables requires the critical cell polarity regulator in yeast, Cdc42. Cdc42 is a very highly conserved Rho family GTPase which, like other members of the family, is presumed to act as a molecular switch that changes between a GTP-bound “on” and “off” state with the help of several regulatory proteins (Symons and Settleman, 2000). GTP-bound Cdc42 interacts with several effectors that are thought to contribute to the polarization of the actin cytoskeleton and, hence, to polarized growth (Pringle et al., 1995; Johnson, 1999).
Post-Golgi vesicles are assisted in targeting and docking at the plasma membrane by an evolutionarily conserved multisubunit complex known as the exocyst, containing, minimally, Sec3, Sec5, Sec6, Sec8, Sec10, Sec15, Exo70, and Exo84 (TerBush and Novick, 1995; Grindstaff et al., 1998; Guo et al., 1999a; Hazuka et al., 1999). The exocyst is concentrated at sites of active growth and is thought to act as a tether holding secretory vesicles close to the plasma membrane (TerBush and Novick, 1995; Guo et al., 1999b). Another important regulator of exocytosis in yeast is Rho3, as revealed by the phenotype of the rho3-V51 effector domain mutant that displays a pronounced defect in exocytosis without any detectable effect on the actin cytoskeleton (Imai et al., 1996; Adamo et al., 1999; Lehman et al., 1999). Identification of Exo70 as a candidate effector for Rho3 in a two-hybrid screen suggested that Rho3 might act through regulation of the exocyst complex (Robinson et al., 1999).
In addition to the exocyst, vesicle fusion with the plasma membrane requires SNARE family proteins located both on the secretory vesicle (v-SNAREs) and at the target membrane (t-SNAREs) (Rothman and Warren, 1994). Assembly of these SNAREs into a four-helix parallel bundle occurs just before the fusion event and is thought to bring the membranes into sufficiently close proximity for fusion to occur (Katz et al., 1998; McNew et al., 2000). SNARE complex formation is regulated by Rab family GTPases and SNARE accessory proteins, although the mechanism of this regulation is not fully understood (Brennwald et al., 1994; Lehman et al., 1999; Misura et al., 2000). Unlike the exocyst, the plasma membrane t-SNAREs Sec9 and Sso1/Sso2 are distributed throughout the plasma membrane (Brennwald et al., 1994).
Recent studies demonstrated that the Sec3 component of the exocyst is targeted to the bud tip even in the absence of polymerized actin or ongoing secretion (Finger and Novick, 1998; Guo et al., 2001). This result suggests that polarization of the secretory pathway entails specification of a fusion-competent patch of the plasma membrane, in addition to delivery of vesicles to that site along actin cables. Consistent with this idea, we now report that a novel temperature-sensitive allele of CDC42, cdc42-6, displays a specific defect in exocytosis during the early stages of bud formation even though actin cables remain competent to deliver secretory vesicles to the bud tip. Surprisingly, this secretory defect does not appear to be linked to defects in the localization of the exocyst, suggesting that Cdc42 acts to regulate the activity (rather than localization) of one or more of the components that mediate the exocytic fusion of secretory vesicles at sites of polarized growth.
Results
Isolation of the cdc42-6 allele
Cdc42 was identified through the temperature-sensitive cdc42-1 allele, isolated in a screen for mutants that show a uniform unbudded arrest (Adams et al., 1990). Subsequent studies employed site-directed mutagenesis to identify new alleles of CDC42, which in aggregate have demonstrated that Cdc42 carries out several genetically separable functions (Ziman et al., 1991; Richman et al., 1999; Kozminski et al., 2000; Moskow et al., 2000; Richman and Johnson, 2000; Gladfelter et al., 2001; Mosch et al., 2001). Seeking to gain a deeper understanding of the essential functions of Cdc42, we conducted an unbiased screen for new cdc42 ts− alleles and have begun to analyze their phenotypes in detail. New cdc42 mutants were isolated by a PCR mutagenesis and gap repair strategy (see Materials and methods), and temperature-sensitive alleles were selected that could support growth at 25 but not at 37°C. Of 78 mutants that fulfilled these criteria, the majority arrested as large round unbudded cells at 37°C. The remaining strains arrested heterogeneously containing lysed, elongated, and misshapen cells, as well as large round unbudded cells at 37°C. However, most of these already contained large numbers of misshapen cells even at 25°C. These unselected phenotypes confirm that a failure in bud emergence coupled with continued isotropic growth is by far the most frequent consequence of loss of Cdc42 function. Upon gene replacement to generate strains containing a single integrated copy of the mutant alleles as the only source of Cdc42, various strains displayed a range of minimal restrictive temperatures ranging from 28 to 33°C.
In contrast to the uniformity of the arrest phenotypes observed for the cdc42 mutants at 37°C, much greater heterogeneity was observed when some mutants were shifted to the minimal restrictive temperature. In this report, we focus on one mutant (cdc42-6) that displayed a mixture of budded and unbudded cells at the minimal restrictive temperature. cdc42-6 cells grew a little more slowly than isogenic wild-type cells at 25°C, and did not proliferate at all at 33°C (Fig. 1 A). Both budded and unbudded cells were present in the population even 7 h after a shift to 33°C (Fig. 1 B), and by this time there was a significant degree of cell lysis (unpublished data). It seemed possible that cdc42-6 cells that were unbudded at the time of shift might remain unbudded, whereas many cells that were budded at the time of shift might remain budded. To assess the behavior of individual cells in the population, we performed a microcolony assay in which cells from a log phase culture were sonicated to separate cell clumps, and spread on plates that were then incubated at permissive or restrictive temperature for 12 h. Individual cells varied greatly in their behavior in this assay: whereas many cells produced no new buds or one new bud, some cells went on to produce microcolonies of ten or even more cells (Fig. 1 C). Thus, the timing and cell cycle stage of death at the restrictive temperature were variable. Surprisingly, ∼40% of the cells failed to form microcolonies even at permissive temperature (Fig. 1 C), perhaps indicating that mutant cells are fragile and sensitive to sonication.
Figure 1.

Phenotype of cdc42-6 mutants. (A and B) CDC42 (open symbols) and cdc42-6 (closed symbols) cells were grown to a density of about 106 cells/ml in YPD at 25°C and split into flasks that were then incubated either at 25°C (circles) or 33°C (squares). Aliquots were fixed with 3.7% formaldehyde at hourly intervals and cell number (A) and the percentage of cells budded (B) were scored. (C) cdc42-6 cells were sonicated briefly and spread onto YPD plates. The number of cell bodies per microcolony was scored immediately after plating (T = 0) or after 12 h at 23 or 33°C, as indicated. An unbudded cell was counted as one cell body, and a budded cell was counted as two cell bodies.
We examined the organization of the actin and septin cytoskeleton, and again found heterogeneity within the population of cdc42-6 cells at the minimal restrictive temperature. Whereas many unbudded cells displayed a depolarized actin cytoskeleton, most of the budded cells had polarized actin and septins assembled at the neck (Fig. 2 A). The quantitation of overall actin polarity is shown in Fig. 2 B. After a shift to the restrictive temperature of 33°C, 86% of the small budded cdc42-6 cells had highly or somewhat polarized actin. A similar degree of polarization was observed in cdc42-6 cells that were kept at 25°C (unpublished data). CDC42 cells shifted to 33°C for 1 h showed 98% with highly or somewhat polarized actin, whereas cdc42-1 cells shifted to their restrictive temperature of 37°C for 1 h showed 0% of cells with highly polarized actin, and 80% had completely unpolarized actin, a defect consistent with previous reports (Ziman et al., 1991). Because polarization of the actin cytoskeleton was largely unaffected by the shift to 33°C, the basis for the death of this strain at the minimal restrictive temperature was not immediately apparent. Furthermore, the combination of phenotypes at this temperature is not reminiscent of that of any previously published cdc42 allele, although many of those mutants have not been studied in sufficient detail to allow direct comparison.
Figure 2.

Actin cytoskeleton is largely polarized in the cdc42-6 mutant at 33°C. (A) Wild-type (CDC42) and mutant (cdc42-6) cells were grown at 25°C and shifted to the indicated temperature for 4 h before fixation and processing for fluorescence microscopy. F-actin was visualized by staining with rhodamine-conjugated phalloidin and septin distribution was visualized using Cdc12-GFP. DIC images of the same cells are shown to the right. (B) Quantitation of the actin polarity phenotypes. Cells were fixed and then stained with rhodamine-phalloidin for analysis. Cells were considered highly polarized if they contained both polarized cortical actin patches and correctly positioned actin cables. Partially polarized cells met at least one of these two criteria while unpolarized cells met neither. (C) Localization of Myo2 and Tpm1. Immunofluorescence was performed to visualize Tpm1, a specific marker for actin cables (Liu and Bretscher, 1989), and Myo2, whose polarized localization is highly dependent on the presence of actin cables (Pruyne et al., 1998). Cells were shifted for 1 h shift to the restrictive temperature of 33°C before fixation. Bar, 5 μm.
A further verification of the proper polarity of the actin cytoskeleton was obtained by staining the cells for proteins that bind to actin or that are dependent on the actin cytoskeleton for their correct localization. Tpm1 is the major isoform of yeast tropomyosin, and binds specifically to actin cables but not cortical actin patches (Liu and Bretscher, 1989). Therefore, staining with Tpm1 antibodies can be used as specific probe to monitor the integrity and polarity of actin cables (Liu and Bretscher, 1989; Govindan et al., 1995; Pruyne et al., 1998). Myo2, a yeast unconventional type V myosin, is an essential gene that has been implicated in the targeting of secretory vesicles to the bud tip, and its polarized localization has been shown to be exquisitely sensitive to the presence of actin cables (Liu and Bretscher, 1989; Govindan et al., 1995; Pruyne et al., 1998). Therefore, we analyzed the localization of Myo2 and Tpm1 in both cdc42-6 and CDC42 cells at permissive (unpublished data) and restrictive temperatures using indirect immunofluorescence (Fig. 2 C). Consistent with our observations by phalloidin staining, we find that Tpm1, which binds exclusively to actin cables, gives similar staining patterns in cdc42-6 and wild-type cells after a shift to 33°C, thus demonstrating that the cdc42-6 mutant has properly polarized actin cables that are of the correct length and orientation. We also find that, as in wild-type cells, Myo2 localizes tightly to the bud tip and other sites of polarized growth in cdc42-6 after a 1-h shift to 33°C. Taken together, these data, along with the direct staining of the actin itself, clearly demonstrate that the cellular defect responsible for the temperature sensitivity of cdc42-6 cells at 33°C, is not due to a defect in the polarization of the actin cytoskeleton.
Analysis of the point mutations in cdc42-6
To determine the structural basis for the temperature-sensitive growth of the cdc42-6 mutant we subjected it to sequence analysis and mapping. The cdc42-6 allele was found to contain seven different point mutations, five of which resulted in changes in the amino acid sequence (Table I). Three of the mutations (P29L, D31V, Y32H) were clustered in the beginning of a region of Cdc42 known as the effector domain (Feltham et al., 1997) that is thought to be critical for the recognition of the GTP-bound state by downstream effectors. The cdc42-6 allele also contained two other mutations, V9A and K150E. The effector domain mutations are critical to the phenotype of this mutant, as cdc42-V9A, K150E displayed wild-type growth at all temperatures. Conversely, an allele containing the three effector mutations by themselves (cdc42-P29L, D31V, Y32H) displayed slow growth at elevated temperature. Interestingly, we found that the V9A mutation was additionally required in order to fully reconstruct the temperature sensitivity of the original cdc42-6 allele. The K150E mutation appeared to have no effect either by itself or in combination with the other mutations (Table I). Therefore, the cluster of three mutations in the effector domain is necessary, but not completely sufficient, for the temperature-sensitivity of this allele.
Table I. Mapping mutations responsible for temperature sensitivity of cdc42-6.
| Growth at | |||
|---|---|---|---|
| CDC42 allele | 25°C | 33°C | 37°C |
| CDC42 | +++ | +++ | +++ |
| cdc42-6 | +++ | − | − |
| cdc42-P29L, D31V, Y32H | +++ | ++ | + |
| cdc42-K150E | +++ | +++ | +++ |
| cdc42-P29L, D31V, Y32H, K150E | +++ | ++ | + |
| cdc42-V9A | +++ | +++ | +++ |
| cdc42-V9A, K150E | +++ | +++ | +++ |
| cdc42-V9A, P29L, D31V, Y32H | +++ | − | − |
| cdc42-V9A, P29L, D31V, Y32H, K150E | +++ | − | − |
The cdc42-6 allele contains seven mutations, two of which are silent and five of which result in amino acid substitutions. These five amino acid substitutions were separated into three groups of mutations: (a) the cluster of three effector domain mutations (P29L, D31V, Y32H); (b) V9A mutation; and (c) the K150E mutation. Each of the three sets of mutations and all combinations were generated by oligonucleotide mutagenesis and tested for their effect on growth of yeast cells as the only source of Cdc42 (see Materials and methods). The effector domain mutations are necessary, but are not sufficient for the full temperature sensitivity of the original allele that was found to require the V9A mutation to recapitulate the full thermosensitive phenotype. +++, strong growth; ++, intermediate growth; +, slight growth; −, no significant growth was seen.
Identification of RHO3 and components of the exocytic apparatus as suppressors of cdc42-6
To gain insight into the underlying defect(s) responsible for the death of cdc42-6 cells at the minimal restrictive temperature, we transformed a library of genomic DNA fragments in a high-copy (2 m) vector into the cdc42-6 mutant strain and selected colonies that could grow at 33°C. Of 233 colonies, 38 were able to grow at 37°C (several of these contained plasmids bearing wild-type CDC42), whereas the remaining 195 colonies grew at 33 but not at 37°C. The most frequent suppressor identified was RHO3 (found in 16 of the 40 colonies tested to date), and the next most common was KIN1 (found in 5 colonies tested). As CDC42 and KIN1 were identified independently in a screen for suppressors of rho3Δ and rho3-V51 strains (Adamo et al., 1999, and unpublished data), these findings suggested an intimate connection between Cdc42 and Rho3. Therefore, we tested whether any of the other genes that suppressed the cold sensitivity of rho3-V51 could also suppress the temperature sensitivity of cdc42-6. Strikingly, all of these genes suppressed cdc42-6. Most of these encode components of the exocytic apparatus, such as the t-SNAREs Sec9 and Sso2, the SNARE-binding protein Sro7, and the Rab GTPase Sec4. Even KIN1, which encodes a protein kinase, displays physical and genetic interactions with the exocytic apparatus (unpublished data). The relative effectiveness of these suppressors was quite similar in the two strains, with SEC9 consistently providing the strongest suppression (Table II). Combined with the findings that overexpression of wild-type CDC42 suppressed the rho3-V51 cold-sensitivity and that overexpression of wild-type RHO3 suppressed the cdc42-6 temperature sensitivity, these data argue strongly for a common defect in these two mutants.
Table II. Suppression of cdc42-6 .
| Suppression of | |||
|---|---|---|---|
| Suppressing plasmid | cdc42-6 ts− | rho3-V51 cs− | |
| 2 μm | CDC42 | ++++ | +++ |
| 2 μm | RHO3 | ++++ | ++++ |
| 2 μm | BEM1 | +++ | +++ |
| 2 μm | SEC9 | +++ | ++++ |
| CEN | SEC4 | ++ | ++ |
| 2 μm | SRO7 | ++ | ++ |
| 2 μm | KIN1 | ++ | ++ |
| 2 μm | SSO2 | + | + |
Both strains were transformed and grown at RT. Eight individual transformants for each multicopy or CEN plasmid were then picked, diluted, and replicated onto YPD or minimal media plates and tested for their ability to grow at their restrictive temperatures. cdc42-6 suppression is shown for 31°C, and the suppression of rho3-V51 was performed at 14°C. RHO3, BEM1, SEC9, and SEC4 also show strong suppression at 33°C. The temperature-sensitive suppression of cdc42-6 is indicated as follows: ++++, wild-type growth at 33°C; +++, strong growth at 33°C; ++, strong growth at 31°C and moderate growth at 32°C; and +, moderate growth at 31°C. The cold-sensitive suppression of rho3-V51 is indicated as follows: ++++, wild-type growth at 14°C; +++, strong growth at 14°C; ++, moderate growth at 14°C; and +, some growth at 14°C, but clearly better than the background of vector only.
The suppression of a rho3 mutant by high-copy CDC42 is consistent with previous observations of suppression of a rho3Δ mutant (Matsui and Toh-e, 1992). In contrast, previous studies reported that the temperature-sensitivity of the cdc42-1 mutant was exacerbated by high-copy RHO3 (Matsui and Toh-e, 1992). This suggested that the response to RHO3 was allele-specific, which we confirmed and extended by determining the ability of high-copy RHO3 and SEC9 to suppress cdc42-1 and eight other ts − alleles of cdc42 (generated in the random mutagenesis screen described earlier). The results demonstrated that only one other allele, cdc42-27, of the nine ts− alleles tested was detectably suppressed by both high-copy RHO3 and SEC9 (however, this mutant was not suppressed by KIN1 and was not further analyzed). In aggregate, these data strongly suggest that the cdc42-6 allele is specifically defective in a process that overlaps with the function of RHO3. Our previous demonstration of a direct role for RHO3 in exocytosis independent of its role in actin polarity (Adamo et al., 1999), as well as the observation that a number of components of the exocytic machinery act as potent suppressors, suggest the likelihood that the novel cellular defect associated with cdc42-6 is, in fact, a defect in exocytosis.
cdc42-6 is synthetically lethal with mutations in a subset of late-acting secretory genes
To examine the possible overlap in function between Cdc42 and Rho3, we sought to determine the phenotype of cells containing both cdc42-6 and rho3-V51 mutations. However, we found that we were unable to recover viable progeny containing both mutants. From the segregation patterns of the cross it was apparent that the progeny containing both mutations were inviable, even though the growth temperature was permissive for both single mutants. This behavior, known as synthetic lethality, was also found to be allele specific, as cdc42-1, rho3-V51 double mutants were viable (although slightly slower growing than either single mutant). Most of the late secretory (sec) mutants show extensive synthetic lethal interactions with other late-acting sec mutants, but not with temperature-sensitive mutants in other processes or even with sec mutants that are defective in earlier stages of the secretory pathway (Salminen and Novick, 1987). We found that mutations in eight of the ten of the late-acting sec genes were inviable in combination with cdc42-6 (Table III). This subset is similar (with sec3-2 as the only exception) to those that interact genetically with sec4-8 (Salminen and Novick, 1987) and with myo2-66 (Govindan et al., 1995). In contrast, combining cdc42-6 with mutations in early-acting sec genes did not cause synthetic lethality at 25°C, supporting the notion that the cdc42-6 defect is specific to a late secretory function. Similar crosses performed with the cdc42-1 allele did not cause synthetic lethality for any sec genes tested (Table III), consistent with another recent study (Finger and Novick, 2000). Thus, the synthetic lethal analysis strongly supports the suppression data indicating that the Cdc42 GTPase plays a role in exocytosis, which overlaps with that played by the Rho3 GTPase and which is specifically impaired in the cdc42-6 allele.
Table III. Viability of double mutants.
| sec/rho mutant | cdc42-6 | cdc42-1 |
|---|---|---|
| rho3-V51 | − | + |
| sec1-1 | + | ND |
| sec2-41 | − | + |
| sec3-2 | − | + |
| sec4-8 | − | + |
| sec5-24 | − | + |
| sec6-4 | + | |
| sec8-9 | − | + |
| sec9-4 | − | + |
| sec10-2 | − | + |
| sec15-1 | − | + |
| sec7-1 | + | ND |
| sec13-1 | + | ND |
| sec22-3 | 1 | ND |
cdc42-6 was crossed with each of the late secretory mutants. The presence of both mutations led to inviability for eight out of the ten late secretory mutants. In contrast, when the secretory mutants were tested with cdc42-1, we did not see this same inviability of the double mutants. Three mutants that cause blocks earlier in the secretory pathway were also tested.
Post-Golgi secretory defects in the cdc42-6 mutant
To directly determine whether cdc42-6 mutants had a secretory defect, we initially focused on two proteins secreted into the periplasmic space, the abundant exoglucanase Bgl2 and the sucrose cleaving enzyme invertase. These two enzymes were chosen because they represent well-characterized markers for each of the two major classes of post-Golgi vesicles that deliver proteins to the cell surface (Harsay and Bretscher, 1995). Surprisingly, secretion of invertase was not detectably perturbed in cdc42-6 cells even at restrictive temperature, in contrast to the control sec6-4 cells (Fig. 3 A). However, a very clear effect was apparent when we examined the secretion of Bgl2 (Fig. 3 B). After a 1-h shift to 33°C, there was a pronounced effect on the accumulation of internal Bgl2 in cdc42-6 cells, to a level even higher than that seen in the control sec6-4 cells. Unlike sec6-4 cells, cdc42-6 cells also have a detectable Bgl2 secretory defect at permissive temperature; however, the magnitude of the defect increased significantly following the 1-h shift to 33°C (Fig. 3 B). Consistent with the secretory defects observed for Bgl2, analysis of cdc42-6 cells by electron microscopy revealed that these cells accumulated 80–100-nm vesicles in a temperature-sensitive manner, with some accumulation of vesicles at permissive conditions but a large increase following a 1-h shift to 33°C (Fig. 3 D). In addition to an overall increase in numbers of vesicles per cell, the penetrance of the vesicle accumulation phenotype also increased following the temperature shift. This is discussed in greater detail below.
Figure 3.

Trafficking of Bgl2, invertase, and CPY in the cdc42-6 mutant. (A) The graph depicts the percentage of Bgl2 protein that remains internal in CDC42, cdc42-6, and sec6-4 strains. The percent distribution of Bgl2 was determined by immunoblot analysis using affinity-purified antibodies raised against the COOH terminus of Bgl2. Quantitation of the bands was done using ImageQuant software and the results are presented as a percentage of total Bgl2 that is found internally. (B) The graph depicts the percentage of invertase protein that remains internal in CDC42, cdc42-6, and sec6-4 strains. The percent of internal invertase was determined by enzyme assays and then shown in bar graph form. (C) Transport of the vacuolar protein CPY is not affected in cdc42-6, as it is found exclusively in the mature form, (mCPY, 61 kD). Wild-type CDC42 and the late secretory mutant, sec6-4, are also shown to process CPY fully to the mature form while sec18-1, an ER-to-Golgi mutant, accumulates the earlier forms. (D) Vesicle accumulation was quantitated for wild-type and cdc42-6 cells at both permissive and restrictive (33°C for 1 h) temperatures from randomly chosen fields of cells. Numbers shown were determined by dividing the total number of 80–100-nm vesicles observed per field of cells by the total number of cells present per field.
We also examined the maturation of the vacuolar protein carboxypeptidase Y (CPY).* We found that CPY was processed to its mature form with normal kinetics in cdc42-6 cells (Fig. 3 C). Thus, cdc42-6 cells can process vacuolar proteins and secrete invertase at near wild-type levels, but have pronounced defects in secreting Bgl2, consistent with a post-Golgi defect for this mutant.
Analysis of the vesicular cargo accumulated in cdc42-6 cells
Bgl2 does not exhibit a detectable change in electrophoretic mobility as it moves through the secretory pathway, presumably because of a limited level of outer chain mannose addition to the single glycosylation site (Mrsa et al., 1993). This precluded a simple gel mobility analysis of the step in the secretory pathway at which Bgl2 traffic was blocked. However, based on the genetic data described above, we suspected that Bgl2 would be retained in post-Golgi secretory vesicles. Mutations in all of the known factors involved in post-Golgi transport result in the accumulation of 80–100-nm vesicles that contain the v-SNAREs Snc1/2. We made use of velocity gradients to separate the cell's membrane components based on their size. Post-Golgi vesicles are quite uniform in size and run as a discrete peak when analyzed on these gradients. Cells were grown for 1 h at the restrictive temperature (37°C for sec6-4 and 33°C for CDC42 and cdc42-6), lysed, and spun through the sorbitol velocity gradient. Fractions were collected and subjected to SDS-PAGE and immunoblot analysis. As expected, gradients from the control sec6-4 mutant displayed a peak of vesicles containing the v-SNARE Snc1/2 and the Bgl2 cargo (Fig. 4). The accumulated Bgl2 in the cdc42-6 mutant was present in a peak of identical size, which also contained Snc1/2. As expected, these peaks of vesicles were absent in wild-type cells where post-Golgi vesicles are much more rare because of their rapid fusion with the plasma membrane. Therefore, the defect in secretion of Bgl2 found in the cdc42-6 mutant is due to a defect at the level of docking and/or fusion of post-Golgi secretory vesicles with the plasma membrane.
Figure 4.

Snc1, Bgl2, and Gas1 are all associated with vesicles that accumulate in cdc42-6 cells. Vesicle gradients of 20 to 40% sorbitol were prepared for each of CDC42, cdc42-6, and sec6-4. Normalized volumes of lysed cells (shifted for 1 h to 37°C for sec6-4 and 33°C for CDC42 and cdc42-6) for each strain were layered onto a gradient before a 1.5 h, 71,000 g spin. The gradients were then collected in 16 fractions and these fractions were subjected to SDS-PAGE analysis and blotted with the indicated antibodies.
As a third secretory marker, the glycosyl-phosphatidylinositol–anchored cell surface protein Gas1 was also examined. Gas1 also clearly accumulated in vesicle-sized intermediates in both the sec6-4 and cdc42-6 strains, in the same gradient fractions as Snc1/2 (Fig. 4). The vesicle peak for Gas1 was found to be somewhat broader in both sec6-4 and cdc42-6 cells than seen with either Bgl2 or Snc1/2, but the significance of this is unclear.
The cdc42-6 mutant accumulates 80–100-nm post-Golgi vesicles, specifically in small-budded cells
The accumulation of 80–100-nm secretory vesicles is a diagnostic feature of all post-Golgi secretory mutants (these vesicles are quite rare in wild-type cells, usually no more than 1–4 vesicles per cell per thin section). To examine the vesicle accumulation directly, we made use of thin-section electron microscopy. Cells with the cdc42-6 mutation were prepared for electron microscopy from cultures grown at both the permissive, and for 1 h, the restrictive temperature. Consistent with our other analyses suggesting a post-Golgi defect, we found that the cdc42-6 mutant accumulated numerous 80–100-nm vesicles in many, but not all, cells (Fig. 5 A). Fields of cells that were prepared from cultures grown at permissive temperature contained some cells that had accumulated a modest number of vesicles, whereas cells that had been shifted to restrictive temperature for 1 h before fixation contained more cells with significantly more vesicles per cell. In other words, the shift in temperature resulted in an increase in both the penetrance and severity of the vesicle accumulation phenotype.
Figure 5.
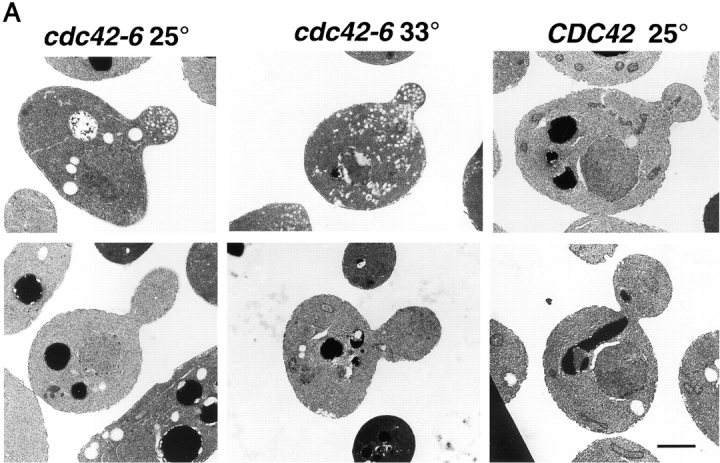

Vesicle accumulation in small-budded cdc42-6 cells. (A) CDC42 at 25°C and cdc42-6 at 25 and 33°C. An example of both small- (<1 μm) and large-budded (1 μm or greater) cells is shown. Preparation of cells is described in the Materials and methods. Cells were only counted as budded cells if the section clearly went through the neck region between the mother and the bud size is as described in the text. (B) Cells were scored for the number of accumulated vesicles and bar graphs show the percentage of small and large budded cells with vesicles. Approximately 30 cells were counted for cdc42-6 at each temperature. Bar, 1 μm.
We noted that even at the restrictive temperature, the penetrance of the vesicle accumulation phenotype was far from complete. Cells with large buds (1 μm in diameter or larger) rarely had many vesicles, whereas cells with small buds (<1 μm in diameter) often had many vesicles (Fig. 5 A shows representative profiles of large- and small-budded cells at each temperature). We quantitated the vesicle accumulation in cdc42-6 cells in a large number of budded cell profiles (Fig. 5 B), and found that the vesicle accumulation phenotype was highly dependent on the size of the bud. At both the permissive and the restrictive temperature, the majority of large-budded cdc42-6 cells (∼70%) showed no unusual accumulation of vesicles (0–5 vesicles/section), and very few cells accumulated >25 vesicles per cell. In contrast, small-budded cdc42-6 cells grown at the permissive temperature were heterogeneous with low, medium and high vesicle accumulation phenotypes each accounting for ∼1/3 of the total number of cells. After a shift to restrictive temperature, there was a dramatic increase in the vesicle numbers, with most small-budded cells accumulating more than 25 vesicles per cell. Consistent with the ability of cdc42-6 cells to polarize the actin cytoskeleton correctly at restrictive temperature, we also found that the accumulated vesicles were concentrated in the bud (Fig. 5 A) >90% of the time, indicating that these vesicles were not defective in myosin-mediated delivery, but rather in docking and fusion with the plasma membrane (Govindan et al., 1995; Novick and Botstein, 1985). Thus, similar to Rho3, Cdc42 has a role in post-Golgi vesicle docking and fusion that is independent of its role in regulation of actin polarity. Moreover, for Cdc42, we find that its function in this pathway is restricted to an early stage in bud formation.
Because our genetic analyses suggested that the role of Cdc42 in exocytosis overlaps with that of Rho3, we decided to reexamine our previous electron micrographs of the rho3-V51 mutant to determine if there was any detectable cell cycle dependence to the vesicle accumulation in this mutant that had been missed (Adamo et al., 1999). However, using precisely the same criteria as for the analysis cdc42-6, we found no difference in the vesicle accumulation phenotype of rho3-V51 cells as a function of bud size (Fig. 6). This suggests that whereas Cdc42 is especially important for exocytosis early during bud formation, Rho3 is required throughout the cell cycle.
Figure 6.

Vesicle accumulation in rho3-V51 mutants affects both large- and small-budded cells similarly. The rho3-V51 micrographs were reevaluated and vesicle accumulation was now counted with respect to bud size, using the same criteria as in Fig. 4.
Cell cycle dependence of Bgl2 secretion defect
We next investigated secretion of Bgl2 using populations of synchronized cells. Wild-type and cdc42-6 cells were arrested in G1 with the mating pheromone α-factor, and were released from arrest by washing and releasing them into fresh medium lacking α-factor. At various times between 5 and 90 min after release, aliquots of cells were transferred to restrictive temperature for a further 30 min. After processing the cells (as described in Materials and methods), the amount of internal and external Bgl2 protein was quantitated. The results, shown in Fig. 7, demonstrate that whereas wild-type cells have ∼25% of their Bgl2 accumulate internally throughout the time course of this assay, we find that cdc42-6 cells have a clear peak of internal Bgl2 accumulation (42%) after 15 min of release from α-factor arrest followed by a 30-min incubation at 33°C. This time point corresponds well to the first appearance of small buds on the cdc42-6 cells at the end of the assay (unpublished data). This strongly supports the data from electron microscopic analysis of asynchronously growing cultures, suggesting that the role of Cdc42 in exocytosis is important primarily during the emergence of small buds.
Figure 7.

Bgl2 secretion in synchronized cells traversing the cell cycle. CDC42 and cdc42-6 cells were synchronized using α-factor and then washed and released into fresh media for 5, 15, 30, 45, 60, or 90 min before undergoing a 30-min shift to restrictive temperature (33°C). Cells were then processed as in Fig. 3 for analysis of Bgl2 secretion.
Localization of Cdc42, Sec4, and exocyst components in cdc42-6 cells
Previous localization studies on Cdc42 using indirect immunofluorescence have revealed that Cdc42 is localized to the plasma membrane at sites of bud growth. This includes the presumptive bud site in unbudded cells, the bud tip in small budded cells, a more diffuse bud staining in larger budded cells and to the mother–daughter neck (Ziman et al., 1993; Richman et al., 1999). To examine the effects of the cdc42-6 mutation on Cdc42 localization, we generated a polyclonal antibody against Cdc42, affinity purified it, and verified that it reacted against a single band of the expected molecular weight that increased in abundance when Cdc42 dosage increased (unpublished data).
Immunoblot analysis indicated that there was a slight decrease in the amount of Cdc42-6 protein at 33°C and a slightly greater decrease at 37°C, but the change in proteins levels alone did not appear to account for the thermosensitivity of this allele (Fig. 8 A). This is especially clear when compared with the levels of protein present in a cdc42-1 mutant, which shows drastically reduced amounts of the protein at all temperatures, consistent with previous analyses, but which grows normally at permissive temperature. Indirect immunofluorescence analysis of CDC42, cdc42-1, and cdc42-6 strains after a 1-h shift to restrictive temperature (33°C) indicated that unlike Cdc42-1 which was only detectable in 15% of small-budded cells, Cdc42-6 was clearly polarized to the bud tip in 78% of small-budded cells, virtually the same as wild-type Cdc42 (Fig. 8 B). We conclude that the phenotype of the cdc42-6 cells is unlikely to be a result of mislocalization of the mutant Cdc42-6 protein, but rather an inability of the mutant protein to carry out some other function.
Figure 8.
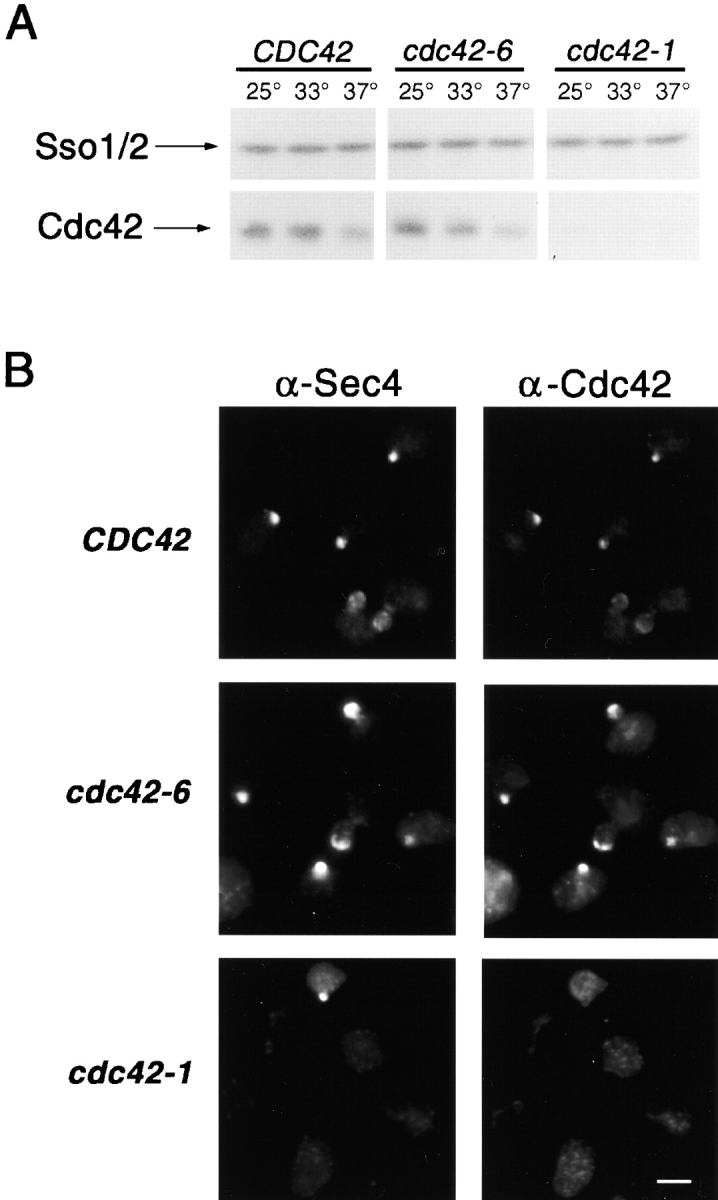
Levels of Cdc42 accumulation and localization of Cdc42 and Sec4 in CDC42 , cdc42-6 and cdc42-1 strains. (A) Whole cell lysates were prepared and subjected to SDS-PAGE analysis and then blotted with anti-Cdc42 or anti-Sso2 antibodies. (B) Immunofluorescence probing for Sec4 and Cdc42 localization was done after a shift to the restrictive temperature of 33°C for CDC42 (top), cdc42-6 (middle) and to 37°C for cdc42-1 (bottom). Bar, 5 μm.
We also examined the localization of Sec4 in the same cells using the α-Sec4 mAb1.2.3. First, we observed that Sec4 localization was indistinguishable in cdc42-6 and wild-type cells shifted to restrictive temperature, with 90% of small-budded cells showing a well-polarized patch of Sec4. In contrast, cdc42-1 cells processed in parallel show only ∼40% of cells with a polarized Sec4 staining (Fig. 8 B). Thus, localization of the Rab GTPase Sec4, like the secretory vesicles observed by electron microscopy, is largely unperturbed in the cdc42-6 mutant.
One plausible explanation for the secretory defect of cdc42-6 cells would be a failure to localize the exocyst complex to the sites of exocytosis. However, using functional Sec8-GFP and Exo70-GFP constructs, we found that in living cells these exocyst components are correctly localized in cdc42-6 cells at the restrictive temperature. Sec3-GFP constructs were also tested and showed similar results for wild-type and mutant strains at all temperatures, but are not shown here because a low signal associated with the particular constructs resulted in low percentage of cells (20–30%) with polarized staining. In a wild-type population of cells grown at either permissive or restrictive temperature, ∼70% of small-budded cells showed polarized Sec8-GFP, and 51% showed polarized Exo70-GFP (Fig. 9 A and B). In cdc42-6 cells grown for 1 h at the restrictive temperature, 65% of small-budded cells showed polarized Sec8-GFP and 56% showed polarized Exo70-GFP (Fig. 9, C and D). Therefore, we conclude that the cdc42-6 mutant is not defective for the localization of the exocyst complex.
Figure 9.

Localization of Exo70-GFP and Sec8-GFP in CDC42 and cdc42-6 strains. COOH terminally tagged GFP constructs were transformed into wild-type and cdc42-6 cells and examined by fluorescence microscopy. The results for wild-type (A and B) and cdc42-6 cells following a 1-h shift at 33°C (C and D) demonstrate that the polarization of the exocyst complex was unaffected in cdc42-6 cells. DIC and GFP fluorescence images were captured immediately following temperature shift. Similar were results were obtained by addition of formaldehyde directly to the media at the end of the temperature shift. Bar, 5 μm.
Discussion
Previous work had shown that Cdc42 was essential for the polarization of the actin cytoskeleton as well as a number of other events in yeast. Cells deprived of Cdc42 function continued to grow isotropically, which indicated that Cdc42 was not essential for the functioning of the secretory pathway. Thus, the requirement for Cdc42 in order for cells to perform the polarized secretion that underlies bud formation appeared to be entirely explicable as a consequence of Cdc42's role in actin polarization. We now report several lines of evidence that argue strongly against this view, and suggest that Cdc42 plays an additional important role in promoting the docking and fusion of secretory vesicles with the plasma membrane during polarized growth.
This evidence stems from the phenotypic and genetic characterization of cdc42-6, a novel temperature-sensitive allele of CDC42. At the minimal restrictive temperature of 33°C, cdc42-6 cells were still able to polarize the actin cytoskeleton, but accumulated many vesicles within small-budded cells. The vesicles contained v-SNAREs and cargo characteristic of post-Golgi secretory vesicles, and were the same size as the post-Golgi secretory vesicles that accumulate in sec mutants known to block exocytosis. Furthermore, the vesicles accumulated very asymmetrically within the bud (and not the mother) portion of the cell, indicating that delivery of vesicles along polarized actin cables was not impaired. That the exocytosis defect was key to the inviability of the cdc42-6 cells at 33°C was shown by the finding that this lethality could be suppressed by overexpression of a number of components of the exocytic apparatus, including the plasma membrane t-SNAREs Sec9 and Sso2, the Sec9 binding protein Sro7, and the Rab GTPase Sec4. Furthermore, cdc42-6 displayed synthetic lethality with many other mutations involved in exocytosis. Together, these observations provide a compelling case for a role of Cdc42 in promoting exocytosis.
Intriguingly, we only observed significant vesicle accumulation in small-budded (and not large-budded) cdc42-6 cells, and in synchronized cell populations the secretory defect was most pronounced at the time of bud emergence and early bud growth. These observations suggest that the role of Cdc42 in exocytosis is only critical at the time of most highly polarized secretion. We speculate that when the polarized actin cables are delivering large numbers of secretory vesicles to a very small patch of the plasma membrane, cells may need to employ special mechanisms to maintain the ability of that small patch of membrane to continue docking and fusing the high throughput of vesicles. Cdc42 is ideally located to perform such a function, and this role explains why no secretory defect was noted for cdc42 mutants that depolarize the actin cytoskeleton, such as cdc42-1. Even if such mutants were defective in the exocytic function of Cdc42, in the absence of polarized actin no patch of the plasma membrane would have high-throughput vesicle fusion and the defect in this process would not be apparent. Only by isolating a mutant that retained the ability to polarize actin were we able to document the downstream role of Cdc42 in efficient vesicle docking and fusion.
Another surprising aspect of the cdc42-6 phenotype was its cargo-specific effect: whereas vesicles delivering Bgl2 to the surface accumulated intracellularly, no comparable accumulation was detected for invertase. This suggests that the Bgl2-containing vesicles manifest an increased sensitivity to the less active exocytic machinery of cdc42-6 mutants. This could result from a slightly lower amount of the v-SNAREs Snc1/2 or the rab GTPase Sec4 present on the Bgl2-containing vesicles compared with the invertase-containing vesicles. Such a difference would provide a simple explanation for the suppression of the secretory defect by elevated expression of Sec4 or SNAREs.
The cdc42-6 mutant showed an especially strong genetic interaction with rho3 mutants. As with Cdc42, Rho3 appears to play dual roles in actin organization and in exocytosis, and it was the identification of an allele, rho3-V51, with a specific defect in exocytosis that established these as independent functions. Increased dosage of RHO3 effectively suppressed cdc42-6, and likewise increased dosage of CDC42 suppressed rho3-V51. cdc42-6 was synthetically lethal with rho3-V51, and overexpression of the same panel of genes with exocytic functions was able to suppress both mutants. However, unlike cdc42-6, the rho3-V51 mutant displayed an equivalent level of vesicle accumulation throughout the cell cycle. This suggests that whereas both GTPases function during bud emergence, only Rho3 is required during later bud growth.
What is the mechanism by which Cdc42 and Rho3 regulate exocytosis? Previous work on Rho3 had shown that it interacts by two-hybrid analysis with the Exo70 component of the exocyst. More recently, studies have shown that the Sec3 component of the exocyst could interact by two-hybrid analysis with Rho1, Rho3, and Rho4. We have examined these exocyst components as possible effectors for Cdc42 and have found that recombinant forms of the relevant regions of Exo70 and Sec3 can indeed interact with Cdc42 in vitro, and that this interaction is GTP-dependent. However we have been unable, as of yet, to find Cdc42 in association with the exocyst by other methods such as native immunoprecipitation or coprecipitation following treatment with chemical cross-linkers. In addition, we observed no defect in exocyst localization in cdc42-6 mutant cells. It remains entirely possible that Cdc42, plays a role in exocyst localization, but it appears that such a role does not account for the defect in the cdc42-6 mutant, and the data suggest that Cdc42 is working as an allosteric regulator of some component of the vesicle docking and fusion machinery. Determining the precise identity of the effector(s) in this pathway will be of paramount importance in delineating the precise mechanism of this regulation. Conceivably, this could involve affecting the conformation of the exocyst complex, or activation of a protein kinase, or effects on SNAREs or SNARE-regulatory proteins.
To our knowledge, this report provides the first evidence linking Cdc42 directly to exocytosis independent of its role in actin polarization. However, previous studies in mammalian cells have suggested a role for Cdc42 in sorting and exit of cell surface proteins from the Golgi apparatus (Kroschewski et al., 1999; Musch et al., 2001), and a biochemical interaction has been documented between Cdc42 and the coatomer complex (Wu et al., 2000). It remains to be seen to what extent Cdc42 participates in exocytosis in mammalian cells. However, the high degree of conservation of the exocytic machinery in yeast and mammalian cells would suggest that such a role for Cdc42 might also be conserved.
Materials and methods
Yeast strains, reagents, and genetic techniques
Cells were grown in YPD media (1% Bacto yeast extract, 2% Bacto peptone, and 2% glucose), or minimal media (SD) with amino acids added as indicated. For all assays performed, 25°C was the permissive temperature, whereas 31 or 33°C was used as the restrictive temperature for cdc42-6. Control strains used 14 or 37°C as the restrictive temperature. Yeast transformations for suppression analysis were performed using the lithium acetate method as described previously (Ito et al., 1983). Standard methods of yeast genetics were used (Guthrie and Fink, 1991).
Sorbitol, sodium azide, sodium fluoride, N-ethylmaleimide, β-mercaptoethanol, O-dianasidine, glucose oxidase, α-factor, peroxidase, Triton X-100, and the protease inhibitor cocktail components p-aminoethylbenzenesulfonyl fluoride, leupeptin, antipain, aprotinin, and pepstatin were obtained from Sigma-Aldrich. Zymolyase (100T) was from Seikagaku Corporation. BSA and yeast nitrogen base were from USBiologicals. 37% formaldehyde, gluteraldehyde, and Spurr's resin were from Electron Microscopy Sciences. 125I-Protein A and 35S EasyTag Express Protein Labeling Mix was from NEN Life Science Products. Molecular mass standards and Tween 20 were from Bio-Rad Laboratories.
Isolation of novel temperature-sensitive alleles of CDC42 by random mutagenesis
cdc42-6 and other cdc42 alleles were generated by the same PCR mutagenesis and gap repair strategy described for isolation of pheromone-resistant cdc42-md alleles (Moskow et al., 2000), except that transformants were screened for temperature-sensitive growth rather than pheromone resistance. The alleles were then integrated at the genomic CDC42 locus by using homologous recombination to replace a cdc42::URA3 disruption with the mutant allele as described for effector loop cdc42 alleles (Gladfelter et al., 2001). Gene replacement was confirmed by PCR, and mutants were backcrossed to a wild-type strain to confirm that the cdc42 phenotypes (see text) segregated 2:2 in at least 10 tetrads.
The cdc42-6 allele was sequenced on both strands with two different flanking primers, and five mutations were identified that resulted in changes in the amino acid sequence. Mapping of the temperature-sensitive components of the cdc42-6 allele was done by preparing synthetic oligonucleotides to introduce three sets of mutations: (a) the cluster of three effector domain mutations (P29L, D31V, Y32H); (b) V9A mutation; and (c) K150E mutation. All three sets of individual mutations and all combinations were prepared by fusion PCR mutagenesis and transformed into DLY3067 (Mata, GAL1p-CDC42::LEU2, ura3-52; trp1-289; his2), where the chromosomal GAL1p-CDC42 is repressed so that the only source of expressed Cdc42 is from the introduced plasmid. The temperature-sensitivity of transformants was determined by replicating to YPD plates at 25, 33, and 37°C.
Invertase, Bgl2 secretion, and CPY pulse–chase assays
Invertase assays were performed as described in Adamo et al. (1999). For Bgl2 assays, wild-type, cdc42-6, and sec6-4 cells were grown overnight to mid-log phase at 25°C in YPD. Strains were shifted for 1 h to 33°C for cdc42-6 and wild-type and 37°C for sec6-4. NaN3 and NaF were then added to a final concentration of 20 mM. 25 ODs of cells were washed with a Tris/NaN3/NaF buffer and then spheroplasted and analyzed as described previously (Adamo et al., 1999). Quantitation was done on a STORM phosphoimager using ImageQuant software (Molecular Dynamics).
For Bgl2 experiments with α-factor arrest, cells were prepared as above except that mid-log cells were arrested in G1 phase by the addition of ∼5 μM α-factor for 2.5 h, until >80% of the cells were unbudded. The α-factor was then washed out and cells were released into fresh YPD for varying lengths of time (from 5 to 90 min), followed by a 30-min shift to 33°C. Cells were then analyzed as described above for Bgl2 secretion. When probing for CPY, cells were grown in minimal media supplemented with uracil. Cells were harvested and preshifted for 5 min to 33°C before the addition of 35S EasyTag Express Labeling Mix, labeled for 5 min, and then chased by adding 1/10 vol of a mix of 5% methionine/5% cysteine/1% BSA. At 5-, 10-, and 30-min intervals after the addition of the chase, aliquots were mixed with 10× stop buffer (200 mM NaN3, 200 mM NaF, 100 mM Tris, pH 8). The supernatant was removed for other analysis while proteins were TCA precipitated out of the cell pellet. The cell pellet was boiled in boiling buffer (10 mM Tris, pH7.5, 1 mM EDTA, 1% SDS) and diluted 20-fold with immunoprecipitation (IP) buffer. Insoluble material was removed and the samples were immunoprecipitated with CPY antibody overnight incubation on ice. 65 μl of a 1:1 protein A slurry was added and the beads were washed with IP buffer, then with IP buffer + 2 M urea, and finally with 1% β-mercaptoethanol. Samples were boiled in sample buffer and subjected to SDS-PAGE, stained, dried, and exposed to film.
Vesicle velocity gradients
Cells were grown overnight in YPD until mid-log phase and then shifted to the restrictive temperatures. sec6-4 was shifted to 37°C for 60 min while cdc42-6 and wild-type cells were shifted to 33°C for 60 min. 250 ODs of cells were treated with 20 mM NaN3/20 mM NaF, harvested, washed, and resuspended in a Tris/NaN3 buffer. Cells were then spheroplasted at 37°C for 30 min, gently harvested, and lysed in 1.8 ml lysis buffer (10 mM triethanolamine, 1 mM EDTA, 0.8 M sorbitol, 20 mM NaN3, and protease inhibitor cocktail). Lysates were spun at 10,000 g to remove mostly plasma membrane and Golgi fragments, and the supernatant was transferred to a new tube and spun for 1 h at 4°C at 100,000 g to pellet small membranes (such as vesicles), ribosomes, and large protein complexes. The pellet was resuspended in lysis buffer, microfuged for 10 min, and then 1/2 of the total was layered onto the sorbitol velocity gradients and analyzed as previously described.
Thin section electron microscopy
cdc42-6 and CDC42 cells were grown overnight in YPD media to mid-log phase. Half of the culture was shifted to the restrictive temperature, 33°C, for 1 h. Shifted and unshifted cells were processed for electron microscopy as previously described (Adamo et al., 1999).
α-Cdc42 antisera production and immunoblot analysis of cell lysates
A PCR fragment containing the COOH-terminal two thirds of the CDC42 coding sequence (residues 60–188) was subcloned into pGEX4Ti vector to make an in-frame fusion with GST. Recombinant protein was batch purified on glutathione-Sepharose (Amersham Pharmacia Biotech) and eluted by boiling in buffer containing 10 mM Tris, pH 7.5, 150 mM NaCl, 0.1% SDS, 0.5 mM DTT. Rabbit polyclonal antibodies were raised against this fusion protein by Cocalico, Inc. Immune sera was subjected to a three-step affinity purification protocol using a GST-Cdc42 (60–188) protein chemically crosslinked to glutathione-Sepharose beads as described previously (Lehman et al., 1999).
Whole cell glass bead lysates were prepared from yeast and boiled in 2× sample buffer. Samples were loaded onto a 12.5% polyacrylamide gel and subjected to SDS-PAGE analysis. Proteins were then transferred overnight to nitrocellulose and blots were probed with affinity-purified α-Cdc42 (1:150 dilution) or α-Sso2 (1:200 dilution). Protein A conjugated to 125I was used for autoradiography detection (New England Nuclear).
Indirect immunofluorescence and light microscopy
Cells were grown overnight to mid-log phase and then fixed immediately or shifted to the restrictive temperature for the indicated amount of time. Cells were processed as previously described for actin (Gladfelter et al., 2001) or Sec4 staining (Brennwald and Novick, 1993; Walch-Solimena et al., 1997), except that in the latter case washed cells were permeabilized for 5 min at room temperature (RT) in 0.5% SDS in a 0.1 M Hepes/1 M sorbitol buffer before the cells were affixed to the slides. Primary antibodies were Sec4 mAb (1:100 dilution), and affinity-purified polyclonals: Cdc42 (1:50 dilution), Myo2 (1:100 dilution), or Tpm1 (1:100 dilution), and incubations went for 1 h at RT. The secondary antibodies used were FITC-conjugated goat anti–mouse for detection of Sec4, FITC-conjugated goat anti–rabbit for Tpm1, and rhodamine-conjugated goat anti–rabbit for detection of Cdc42 and Myo2 (all from Jackson ImmunoResearch Laboratories), and were diluted 1:50 with PBT. Mounting media (90% glycerol with 4′,6-diamidino-2-phenylindol to visualize DNA and o-phenylenediamine to retard photobleaching) was added before a coverslip was sealed over the top. Epifluorescence microscopy was done using either a Zeiss Axioskop or a Nikon E600 microscope, equipped with a Princeton Instruments 512 × 512 back illuminated frame-transfer CCD camera and Metamorph (Universal Imaging Corp.) to capture images.
GFP tagging and localization
GFP experiments were performed using cells that carried a plasmid containing Cdc12, Exo70, Sec3, or Sec8 with a single GFP molecule attached in frame to the COOH-terminal end. Cells were grown in minimal media for overnight growth and shifted to YPD for at least 2 h before viewing or before being shifted to the restrictive temperature for 1 h. Cells were the fixed according to Finger et al. (1998), or were washed with 1 ml PBS and then resuspended in 2% glucose and viewed immediately after the shift. Unfixed cells were viewed for only a short period of time, as the microscope does not have a heated stage to maintain the temperature shift. Images were captured as described above.
Acknowledgments
We thank the members of the 1996 class of the Woods Hole Physiology course (Boston, MA), who isolated many cdc42 mutants, and the members of the 1997 class, who isolated many suppressors of those mutants. Tpm1 antibodies were a gift from Tony Bretscher (Cornell University, Ithaca, NY). We thank Dr. Guendalina Rossi for many helpful discussions and critical reading of the manuscript. The authors are especially grateful to Linda Burg-Freedman and Lee Cohen-Gould for assistance with electron microscopy, and to Veronica Lee for technical assistance.
This work was supported by grants from the Mathers Charitable Foundation, the Pew Scholars in Biomedical Sciences Program, and the National Institutes of Health (GM54712) to P.J. Brennwald, and by grants from the National Institutes of Health (GM53050 and GM62300) and American Cancer Society (RPG-98-046-CCG) to D.J. Lew.
D.J. Lew and P.J. Brennwald contributed equally to this work.
Footnotes
Abbreviations used in this paper: CPY, carboxypeptidase Y; IP, immunoprecipitation; RT, room temperature; SD, minimal medium.
References
- Adamo, J.E., G. Rossi, and P. Brennwald. 1999. The Rho GTPase Rho3 has a direct role in exocytosis that is distinct from its role in actin polarity. Mol. Biol. Cell. 10:4121–4133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams, A.E., D.I. Johnson, R.M. Longnecker, B.F. Sloat, and J.R. Pringle. 1990. CDC42 and CDC43, two additional genes involved in budding and the establishment of cell polarity in the yeast Saccharomyces cerevisiae. J. Cell Biol. 111:131–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennwald, P., and P. Novick. 1993. Interactions of three domains distinguishing the Ras-related GTP-binding proteins Ypt1 and Sec4. Nature. 362:560–563. [DOI] [PubMed] [Google Scholar]
- Brennwald, P., B. Kearns, K. Champion, S. Keranen, V. Bankaitis, and P. Novick. 1994. Sec9 is a SNAP-25-like component of a yeast SNARE complex that may be the effector of Sec4 function in exocytosis. Cell. 79:245–258. [DOI] [PubMed] [Google Scholar]
- Drubin, D.G., and W.J. Nelson. 1996. Origins of cell polarity. Cell. 84:335–344. [DOI] [PubMed] [Google Scholar]
- Feltham, J.L., V. Dotsch, S. Raza, D. Manor, R.A. Cerione, M.J. Sutcliffe, G. Wagner, and R.E. Oswald. 1997. Definition of the switch surface in the solution structure of Cdc42Hs. Biochemistry. 36:8755–8766. [DOI] [PubMed] [Google Scholar]
- Finger, F.P., and P. Novick. 1998. Spatial regulation of exocytosis: lessons from yeast. J. Cell Biol. 142:609–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finger, F.P., and P. Novick. 2000. Synthetic interactions of the post-Golgi sec mutations of Saccharomyces cerevisiae. Genetics. 156:943–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finger, F.P., T.E. Hughes, and P. Novick. 1998. Sec3p is a spatial landmark for polarized secretion in budding yeast. Cell. 92:559–571. [DOI] [PubMed] [Google Scholar]
- Gladfelter, A.S., J.J. Moskow, T.R. Zyla, and D.J. Lew. 2001. Isolation and characterization of effector-loop mutants of cdc42 in yeast. Mol. Biol. Cell. 12:1239–1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govindan, B., R. Bowser, and P. Novick. 1995. The role of Myo2, a yeast class V myosin, in vesicular transport. J. Cell Biol. 128:1055–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grindstaff, K.K., C. Yeaman, N. Anandasabapathy, S.C. Hsu, E. Rodriguez-Boulan, R.H. Scheller, and W.J. Nelson. 1998. Sec6/8 complex is recruited to cell-cell contacts and specifies transport vesicle delivery to the basal-lateral membrane in epithelial cells. Cell. 93:731–740. [DOI] [PubMed] [Google Scholar]
- Guo, W., A. Grant, and P. Novick. 1999. a. Exo84p is an exocyst protein essential for secretion. J. Biol. Chem. 274:23558–23564. [DOI] [PubMed] [Google Scholar]
- Guo, W., D. Roth, C. Walch-Solimena, and P. Novick. 1999. b. The exocyst is an effector for Sec4p, targeting secretory vesicles to sites of exocytosis. EMBO J. 18:1071–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, W., F. Tamanoi, and P. Novick. 2001. Spatial regulation of the exocyst complex by Rho1 GTPase. Nat. Cell Biol. 3:353–360. [DOI] [PubMed] [Google Scholar]
- Guthrie, C., and G. Fink. 1991. Guide to yeast genetics and molecular biology. In Methods in Enzymology. Academic Press, San Diego. [PubMed]
- Harsay, E., and A. Bretscher. 1995. Parallel secretory pathways to the cell surface in yeast. J. Cell Biol. 131:297–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazuka, C.D., D.L. Foletti, S.C. Hsu, Y. Kee, F.W. Hopf, and R.H. Scheller. 1999. The sec6/8 complex is located at neurite outgrowth and axonal synapse-assembly domains. J. Neurosci. 19:1324–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai, J., A. Toh-e, and Y. Matsui. 1996. Genetic analysis of the Saccharomyces cerevisiae RHO3 gene, encoding a rho-type small GTPase, provides evidence for a role in bud formation. Genetics. 142:359–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito, H., Y. Fukuda, K. Murata, and A. Kimura. 1983. Transformation of intact yeast cells treated with alkali cations. J. Bacteriol. 153:163–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson, D.I. 1999. Cdc42: An essential Rho-type GTPase controlling eukaryotic cell polarity. Microbiol. Mol. Biol. Rev. 63:54–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz, L., P.I. Hanson, J.E. Heuser, and P. Brennwald. 1998. Genetic and morphological analyses reveal a critical interaction between the C-termini of two SNARE proteins and a parallel four helical arrangement for the exocytic SNARE complex. EMBO J. 17:6200–6209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozminski, K.G., A.J. Chen, A.A. Rodal and D.G. Drubin. 2000. Functions and functional domains of the GTPase Cdc42p. Mol. Biol. Cell. 11:339–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroschewski, R., A. Hall, and I. Mellman. 1999. Cdc42 controls secretory and endocytic transport to the basolateral plasma membrane of MDCK cells. Nat. Cell Biol. 1:8–13. [DOI] [PubMed] [Google Scholar]
- Lehman, K., G. Rossi, J.E. Adamo, and P. Brennwald. 1999. Yeast homologues of tomosyn and lethal giant larvae function in exocytosis and are associated with the plasma membrane SNARE, Sec9. J. Cell Biol. 146:125–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, H., and A. Bretscher. 1989. Disruption of the single tropomyosin gene in yeast results in the disappearance of actin cables from the cytoskeleton. Cell. 57:233–242. [DOI] [PubMed] [Google Scholar]
- Matsui, Y., and A. Toh-e. 1992. Isolation and characterization of two novel ras superfamily genes in Saccharomyces cerevisiae. Gene. 114:43–49. [DOI] [PubMed] [Google Scholar]
- McNew, J.A., F. Parlati, R. Fukuda, R.J. Johnston, K. Paz, F. Paumet, T.H. Sollner and J.E. Rothman. 2000. Compartmental specificity of cellular membrane fusion encoded in SNARE proteins. Nature. 6801:153–159. [DOI] [PubMed] [Google Scholar]
- Misura, K.M., R.H. Scheller and W.I. Weis. 2000. Three-dimensional structure of the neuronal-Sec1-syntaxin 1a complex. Nature. 6776:355–362. [DOI] [PubMed] [Google Scholar]
- Mosch, H.U., T. Kohler, G.H. Braus. 2001. Different domains of the essential GTPase Cdc42p required for growth and development of Saccharomyces cerevisiae. Mol. Cell. Biol. 21:235–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moskow, J.J., A.S. Gladfelter, R.E. Lamson, P.M. Pryciak, and D.J. Lew. 2000. Role of Cdc42p in pheromone-stimulated signal transduction in Saccharomyces cerevisiae. Mol. Cell Biol. 20:7559–7571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mrsa, V., F. Klebl, and W. Tanner. 1993. Purification and characterization of the Saccharomyces cerevisiae BGL2 gene product, a cell wall endo-beta-1,3-glucanase. J. Bacteriol. 175:2102–2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musch, A., D. Cohen, G. Kreitzer, and E. Rodriguez-Boulan. 2001. cdc42 regulates the exit of apical and basolateral proteins from the trans-Golgi network. EMBO J. 20:2171–2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novick, P., and D. Botstein. 1985. Phenotypic analysis of temperature-sensitive yeast actin mutants. Cell. 40:405–416. [DOI] [PubMed] [Google Scholar]
- Pringle, J.R., E. Bi, H.A. Harkins, J.E. Zahner, C. De Virgilio, J. Chant, K. Corrado, and H. Fares. 1995. Establishment of cell polarity in yeast. Cold Spring Harb. Symp. Quant. Biol. 60:729–744. [DOI] [PubMed] [Google Scholar]
- Pruyne, D., and A. Bretscher. 2000. Polarization of cell growth in yeast. I. Establishment and maintenance of polarity states. J. Cell Sci. 113:365–375. [DOI] [PubMed] [Google Scholar]
- Pruyne, D., D.H. Schott, and A. Bretscher. 1998. Tropomyosin-containing actin cables direct the Myo2p-dependent polarized delivery of secretory vesicles in budding yeast. J. Cell Biol. 143:1931–1945. [DOI] [PubMed] [Google Scholar]
- Richman T.J., and D.I. Johnson. 2000. Saccharomyces cerevisiae Cdc42p GTPase is involved in preventing the recurrence of bud emergence during the cell cycle. Mol. Cell Biol. 20:8548–8559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richman, T.J., M.M. Sawyer, and D.I. Johnson. 1999. The Cdc42p GTPases is involved in a G2/M morphogenetic checkpoint regulating the apical-isotropic switch and nuclear division in yeast. J. Biol Chem. 274:16861–16870. [DOI] [PubMed] [Google Scholar]
- Robinson, N.G., L. Guo, J. Imai, E.A. Toh, Y. Matsui, and F. Tamanoi. 1999. Rho3 of Saccharomyces cerevisiae, which regulates the actin cytoskeleton and exocytosis, is a GTPase which interacts with Myo2 and Exo70. Mol. Cell Biol. 19:3580–3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothman, J.E., and G. Warren. 1994. Implications of the SNARE hypothesis for intracellular membrane topology and dynamics. Curr. Biol. 4:220–233. [DOI] [PubMed] [Google Scholar]
- Salminen, A., and P.J. Novick. 1987. A ras-like protein is required for a post-Golgi event in yeast secretion. Cell. 49:527–538. [DOI] [PubMed] [Google Scholar]
- Symons, M., and J. Settleman. 2000. Rho family GTPases: more than simple switches. Trends Cell Biol. 10:415–419. [DOI] [PubMed] [Google Scholar]
- TerBush, D.R., and P. Novick. 1995. Sec6, Sec8, and Sec15 are components of a multisubunit complex which localizes to small bud tips in Saccharomyces cerevisiae. J. Cell Biol. 130:299–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walch-Solimena, C., R.N. Collins, and P.J. Novick. 1997. Sec2p mediates nucleotide exchange on Sec4p and is involved in polarized delivery of post-Golgi vesicles. J. Cell Biol. 137:1495–1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, W.J., J.W. Erickson, R. Lin, and R.A. Cerione. 2000. The gamma-subunit of the coatomer complex binds Cdc42 to mediate transformation. Nature. 405:800–804. [DOI] [PubMed] [Google Scholar]
- Ziman, M., J.M. O'Brien, L.A. Ouellette, W.R. Church, and D.I. Johnson. 1991. Mutational analysis of CDC42Sc, a Saccharomyces cerevisiae gene that encodes a putative GTP-binding protein involved in the control of cell polarity. Mol. Cell Biol. 11:3537–3544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziman, M., D. Preuss, J. Mulholland, J.M. O'Brien, D. Botstein, and D.I. Johnson. 1993. Subcellular localization of Cdc42p, a Saccharomyces cerevisiae GTP-binding protein involved in the control of cell polarity. Mol. Biol. Cell. 4:1307–1316. [DOI] [PMC free article] [PubMed] [Google Scholar]