

Abstract
The stimulatory and inhibitory pathways initiated by engagement of stimulatory receptors such as the B cell receptor for antigen (BCR) and inhibitory receptors such as Fcγ receptors of the IIB1 type (FcγRIIB1) intersect in ways that are poorly understood at the molecular level. Because the tyrosine kinase Csk is a potential negative regulator of lymphocyte activation, we examined the effects of BCR and FcγRIIB1 engagement on the binding of Csk to phosphotyrosine-containing proteins. Stimulation of a B lymphoma cell line, A20, with intact anti-IgG antibody induced a direct, SH2-mediated association between Csk and a 62-kD phosphotyrosine-containing protein that was identified as RasGTPase-activating protein–associated p62 (GAP-A.p62). In contrast, stimulation of A20 cells with anti-IgG F(ab′)2 resulted in little increase in the association of Csk with GAP-A.p62. The effect of FcγRIIB1 engagement on this association was abolished by blockade of FcγRIIB1 with the monoclonal antibody 2.4G2. Furthermore, the increased association between Csk and GAP-A.p62 seen upon stimulation with intact anti-Ig was abrogated in the FcγRIIB1-deficient cell line IIA1.6 and recovered when FcγRIIB1 expression was restored by transfection. The differential effects of BCR and BCR-FcγRIIB1–mediated signaling on the phosphorylation of GAP-A.p62 and its association with Csk suggest that docking of Csk to GAP-A.p62 may function in the negative regulation of antigen receptor–mediated signals in B cells.
At the cellular level, the quality and magnitude of immune responses are determined by the summation of positive and negative signals. In B lymphocytes, for example, the relative levels of stimulatory and inhibitory signaling determine the amount and duration of antibody production and the type of antibody produced. Mediating such signals at the B cell surface are stimulatory receptors such as the BCR and inhibitory receptors such as Fcγ receptors of the IIB1 type (FcγRIIB1). Although aspects of proximal signaling through the BCR and FcγRIIB1 have been defined, the intersection of stimulatory and inhibitory pathways initiated by engagement of these receptors is poorly understood at the biochemical level.
The BCR consists of membrane-bound immunoglobulin (sIg) and associated Ig-α and Ig-β heterodimers. Engagement of the B cell receptor for antigen (BCR)1 initiates a cascade of tyrosine phosphorylation, resulting in the activation of Ras- and phospholipase-dependent signal transduction pathways and culminating in proliferative and differentiative responses (reviewed in reference 1). BCR stimulation induces immediate activation of the Src-related tyrosine kinases Lyn, Fyn, and Blk, followed within minutes by activation of the non–Src-related tyrosine kinases Btk and Syk (2). Genetic evidence clearly implicates Syk (3, 4) and Btk (5, 6) as essential for normal B cell proliferation in response to BCR engagement; no single Src-related kinase, however, is essential for BCR signaling in the mouse (7–9). In contrast, ablation of Lyn in the chicken B cell line DT40, which expresses a restricted repertoire of Src-related kinases, abrogates signaling through the BCR (10), supporting the view that Src-related kinases play essential, if redundant, roles in B cell activation.
Although the mechanisms that underlie negative regulation of BCR signaling are incompletely understood, several consequences of BCR stimulation, including phosphoinositide hydrolysis, increased free intracellular calcium, cellular proliferation, and immunoglobulin secretion, can be inhibited by engagement of FcγRIIB1 (11, 12). As a consequence of this inhibitory effect, intact anti-Ig antibodies, which engage both the BCR and FcγRIIB1, are relatively ineffective polyclonal B cell activators in comparison to F(ab′)2 fragments, which cannot engage FcγRIIB1 (13, 14). A conserved 13–amino acid motif in the cytoplasmic portion of FcγRIIB1 (the immunoreceptor tyrosine-based inhibitory motif) is required for inhibition (15). The immunoreceptor tyrosine-based inhibitory motif of FcγRIIB1 represents a docking site for the SH2 domains of at least two proteins, the tyrosine phosphatase SHP1 (16) and the inositide phosphatase SHIP (17), either of which may mediate inhibitory effects.
The COOH-terminal Src kinase (Csk) represents another potential negative regulator of lymphocyte activation. Csk specifically phosphorylates a conserved tyrosine residue near the COOH terminus of Src and related kinases, thereby inhibiting catalytic activity (18). Correspondingly, cells derived from Csk-deficient mouse embryos, which die at mid-gestation, exhibit increased activity of Src and related kinases (19, 20). In response to extracellular signals, the activity of Csk is known to be modulated by changes in its intracellular localization (21). In T lymphocytes, overexpression of Csk suppresses phosphorylation of tyrosine kinase substrates and lymphokine secretion in response to antigenic stimulation, possibly by inhibition of the Src-related kinases Lck and Fyn, which have been implicated as mediators of signaling through the T cell receptor (22). Deletion of the SH2 or SH3 domains of Csk abrogates the inhibition of antigen receptor signaling in T cells (23); this result is consistent with the ability of similar mutations to suppress Csk-induced inactivation of Src in fibroblastoid cells (24). The effect of an SH2-inactivating mutation could be corrected by targeting Csk to the plasma membrane, suggesting that with respect to inhibition of antigen receptor signaling, the SH2 domain of Csk provides an essential localization function (21, 23). One specific means of localization was suggested by the recent observation (25) that in cells expressing activated forms of Src, the SH2 domain of Csk interacts with the Ras GTPase-activating protein (GAP)-associated p62 protein (GAP-A.p62); this complex is found in subcellular membrane or cytoskeletal fractions. Thus, GAP.A-p62 may represent a docking protein for Csk, RasGAP, and possibly other ligands.
Csk, therefore, may be situated at a point of intersection between positive and negative signaling in lymphocytes. Because FcγRIIB1 is known to inhibit B cell stimulation through the BCR, we wished to determine whether engagement of FcγRIIB1 leads to events that alter Csk's association with other proteins; such changes could modify stimulatory signaling through the BCR. To test this hypothesis, we examined the effects of BCR and FcγRIIB1 engagement on the binding of Csk to phosphotyrosine-containing proteins in a B lymphoma cell line, A20. Stimulation of A20 cells with intact anti-IgG antibody induced a direct, SH2-mediated association between Csk and a 62-kD phosphotyrosine-containing protein which was identified as GAP-A.p62. In contrast, stimulation of A20 cells with anti-IgG F(ab′)2 resulted in less of an increase in association between Csk and GAP-A.p62. Treatment of A20 cells with the monoclonal antibody 2.4G2, which blocks FcγRIIB, abolished the ability of intact anti-IgG antibodies to induce association of Csk with GAP-A.p62. Furthermore, the differential effect of intact anti-IgG and F(ab′)2 on association of Csk with GAP-A.p62 was abolished in the FcγRIIB-deficient, A20-derived mutant cell line, IIA1.6, and was restored in IIA1.6 cells transfected with wild-type FcγRIIB1. Taken together, these observations indicate that BCR- and FcγRIIB1-mediated signaling have differential effects on the phosphorylation of GAP-A.p62 and its association with Csk. These findings suggest that Csk functions in the negative regulation of antigen receptor-mediated signals in B cells.
Materials and Methods
Cell Lines.
A20 cells were obtained from the American Type Culture Collection (Rockville, MD). Cells were cultured in RPMI 1640 media (GIBCO BRL, Gaithersburg, MD) supplemented with 5% fetal bovine serum and 50 μmol 2-mercaptoethanol. IIA1.6 cells were a gift from Charles Janeway (Yale University, New Haven, CT) and John Cambier (National Jewish Center, Denver, CO). They were cultured in IMDM supplemented with 5% fetal bovine serum. IIA1.6 cells transfected with FcγIIB1 (CT314; a gift from J. Cambier) were grown in media supplemented with 0.5 mg/ml G418. All cells were grown in 5% CO2 at 37°C.
Antibodies.
The phosphotyrosine-specific mAb PY20, an anti-Csk mAb, and antibodies against p125Fak were purchased from Transduction Laboratories (Lexington, KY). The antiphosphotyrosine mAb 4G10 was purchased from Upstate Biotechnology Inc. (Lake Placid, NY). Affinity-purified, polyclonal, agarose-conjugated and unconjugated rabbit antibodies against Csk and monoclonal, agarose-conjugated antibodies against RasGAP and Csk were purchased from Santa Cruz (Santa Cruz, CA). Polyclonal rabbit antibodies against GAP were a gift from Dr. Tony Pawson (Mount Sinai Hospital, Toronto, Canada); those against Sam68 were provided by Andrey Shaw (University of Washington, St. Louis, MO). mAb 2C4 against GAP-A.p62 was provided by Richard Roth (Stanford University, Stanford, CA). Antibodies against paxillin were purchased from Zymed Labs., Inc. (S. San Francisco, CA). mAb 2.4G2 was purchased from PharMingen (San Diego, CA).
Construction and Isolation of Glutathione S Transferase Fusion Proteins.
A cDNA clone encoding Csk was provided by Akira Imamoto (Fred Hutchinson Cancer Research Center, Seattle, WA). DNA fragments encoding SH2 (amino acids 77–180), SH3 (amino acids 14–68), and SH3/2 (amino acids 14–180) of Csk were amplified by the PCR. These fragments were cloned between the BamHI and EcoRI restriction sites of the Escherichia coli expression vector pGEX-2T (Pharmacia, Piscataway, NJ) to produce the plasmids pGEX-2T-CskSH2, pGEX-2T-CskSH3, and pGEX-2T-CskSH3/2. Cassettes for expression of Csk mutants R107K and S109C were generated by PCR using 3′ Csk antisense primers specifying the mutations of interest (R107K: 5′-GTT CCT CGT GAA GGA AAG CAC C-3′; S109C: 5′-CGT GCG GGA ATG CAC CAA CTA CCC-3′). The resulting fragments were cloned into pGEX-2T as above. All constructions were verified by nucleotide sequencing.
Plasmids encoding glutathione S transferase (GST) fusion proteins were introduced into E. coli strain DH5α. Transfectants were cultured overnight at 37°C in Luria broth supplemented with ampicillin at 50 μg/ml. Overnight cultures were diluted 1:10 into fresh medium supplemented with ampicillin and grown at 37°C to an OD595 of 0.6–1.0; expression of fusion proteins was then induced by addition of isopropyl-1-thio-β-d-galactopyranoside to 0.1 mM. After 4 h, bacteria were pelleted and resuspended in ice-cold PBS supplemented with protease inhibitors and Triton X-100 to a final concentration of 1%. The mixtures were incubated for 20–30 min on ice and lysis was completed by sonication (4 cycles of 10 pulses each with a Branson [Danbury, CT] microtip sonifier at output 2-3, duty cycle 50%). Lysates were clarified by centrifugation at 10,000 g for 15 min at 4°C. Supernatants were incubated with 500 μl of a 50% slurry of glutathione–agarose beads and incubated for 15 min at room temperature. Beads were washed five times with PBS. The amount of bound protein was estimated by SDS-PAGE and Commassie staining. To prepare affinity matrices for use in binding assays, the amount of bacterial lysate used in each adsorption was adjusted to produce beads of similar substituent density. Beads were stored on ice as a 50% slurry in PBS supplemented with 1 mM PMSF, 10% glycerol, and were used within one week of preparation.
Cell Stimulation.
Before stimulation, cells were washed in PBS, resuspended in serum-free RPMI 1640 medium, and incubated at 37°C for 20 min. Cells (108/ml) were stimulated with F(ab′)2 (30 μg/ml) or intact (50 μg/ml) anti-IgG antibodies (Jackson ImmunoResearch Labs., Inc., West Grove, PA) in serum-free RPMI for 1 min at 37°C unless otherwise indicated. In 2.4G2 blockade experiments, cells were incubated with medium or 50 μg/ml 2.4G2 antibodies for 15 min at room temperature before stimulation with anti-IgG antibodies.
Immunoprecipitation.
Pellets of unstimulated or stimulated cells were lysed in 1% Triton lysis buffer (150 mM NaCl, 20 mM Tris-HCl [pH 7.6], 10 μg/ml of aprotinin, 1 mM EDTA, 1 mM sodium orthovanadate, and 1 mM phenylmethylsulfonyl fluoride, 10% glycerol, and 1% Triton X-100). Lysates were cleared by centrifugation at 13,000 g for 15 min. Cell lysates were precleared with normal rabbit Ig or normal mouse Ig and Sepharose A (Repligen Corp., Cambridge, MA), or normal rabbit serum covalently coupled to Sepharose (Sigma Chemical Co., St. Louis, MO). Proteins of interest were precipitated at 4°C using affinity purified, agarose-conjugated antibodies (10 μg) for 1 h. Immunocomplexes were washed five times in lysis buffer.
In Vitro Binding Assays.
Cell lysates were incubated with 3–10 μg of GST fusion proteins at 4°C for 2–4 h and washed four times with lysis buffer. Some samples were boiled in lysis buffer supplemented with 1% SDS for 5 min, diluted 10-fold in 1% Triton lysis buffer, and incubated with GST fusion proteins as indicated above. For phospho–amino acid competition, twofold serial dilutions of cell lysates were incubated with GST-CskSH2 beads in the presence of 50 mM l-phosphotyrosine or 25 mM each of l-phosphoserine and l-phosphothreonine.
Immunoblot Assays.
Proteins were eluted from beads by boiling in SDS sample buffer, separated by SDS–polyacrylamide gel electrophoresis, and transferred to nitrocellulose. Filters were blocked with 5% nonfat dry milk in PBS, 0.1% Tween 20 for 1 h, and incubated with primary antibodies for 1–5 h at room temperature. Antibodies were used at dilutions recommended by suppliers. Horseradish peroxidase–conjugated goat anti–mouse or anti–rabbit antibodies were used to detect primary antibodies and visualized by an enhanced chemiluminiscence assay (ECL; Amersham Corp., Arlington Heights, IL).
In Vitro Kinase Assay and Phosphopeptide Mapping.
After precipitation as described above, complexes were washed three times in lysis buffer, two times in kinase buffer (20 mM Tris-HCl [pH 7.6], 10 mM MgCl2, 5 mM MnCl2, 1 mM 2-mercaptoethanol), and incubated in 100 μl of kinase buffer supplemented with 10 μCi of [γ-32P]ATP for 30 min at room temperature. After washing, labeled proteins were separated by SDS–polyacrylamide gel electrophoresis. Phosphopeptide mapping was performed as described previously (26). In brief, 32P-labeled 62-kD bands from Csk and RasGAP immunoprecipitates were digested without previous dilution by placing gel slices containing these bands in the sample wells of a second, 15% SDS polyacrylamide gel containing a 5.5-cm stacking layer and then overlaying each slice with 10 μl V-8 protease solution (20 μg/ml). Digestion was carried out in the stacking gel during subsequent electrophoresis at 20 mA. After the bromophenol blue dye reached the bottom of the stacking gel, current was turned off for 30 min, after which time electrophoresis was resumed.
Results
Association of Csk with Phosphotyrosine-containing Proteins from A20 B Cells Stimulated with Intact Anti-IgG or F(ab′)2.
As a first step toward identifying proteins associated with Csk under conditions of BCR and FcγRIIB stimulation, A20 cells were incubated with intact anti-IgG antibody or with F(ab′)2 fragments of similar specificity, and phosphotyrosine-containing proteins were detected in anti-Csk immunoprecipitates by immunoblotting. Several phosphotyrosine-containing species, including proteins of 62, 97, and 125 kD (p62, p97, and p125, respectively), were observed to immunoprecipitate with Csk (Fig. 1). The amount of p62 detectable in Csk immunoprecipitates by antiphosphotyrosine immunoblotting increased significantly after stimulation, indicating an elevation in phosphotyrosine content, association with Csk, or both. However, the increase in antiphosphotyrosine reactivity of p62 was substantially greater after stimulation with intact antibodies than with F(ab′)2. The antiphosphotyrosine signals associated with p97 and p125, in contrast, showed relatively little change after stimulation with anti-IgG antibodies.
Figure 1.

Stimulation-dependent association of p62 and Csk in vivo. A20 cells were unstimulated (lanes 1–3), stimulated with intact anti–mouse IgG (lanes 4–6), or stimulated with F(ab′)2 (lanes 7–9). Precleared cell lysates were incubated with normal rabbit immunoglobulin (lanes 2, 5, and 8) or affinity-purified rabbit antibodies against Csk (lanes 3, 6, and 9). One-tenth of the total amount of lysate used in each experiment was fractionated in parallel (lanes 1, 4, and 7). Immunoprecipitated proteins were fractionated on a 10% SDS– polyacrylamide gel, were transferred to nitrocellulose, and immunoblotted with an antiphosphotyrosine antibody (PY20). The membrane was stripped and Csk was detected by immunoblotting.
The SH2 Domain of Csk Binds p62 Directly through a Specific, Phosphotyrosine-dependent Interaction.
To identify the region(s) of Csk that bind p62, the SH2 and SH3 domains of Csk were fused individually or in combination to GST. The resulting reagents (GST-CskSH2, GST-CskSH3, and GST-SH3/2) were assayed for binding to phosphotyrosine-containing proteins from lysates of unstimulated A20 cells or A20 cells that had been stimulated with intact anti-IgG. After stimulation, p62 was the most prominent of the phosphotyrosine-containing proteins retained by GST-CskSH2 (Fig. 2 A). Detection of p62 reached a maximum within 2 min of stimulation, but remained detectable at 60 min after stimulation (data not shown). The p62 antiphosphotyrosine signal associated with GST-CskSH3/2 was greater than that associated with GST-CskSH2 alone, consistent with previous observations indicating that flanking SH3 domains influence the affinity of SH2 domains for phosphopeptides. Neither GST-CskSH3 nor GST alone exhibited binding to phosphotyrosine-containing proteins from A20 cells. In separate experiments in which cells were stimulated with an anti-IgG F(ab′)2 fragment, p62 was also present, but the antiphosphotyrosine signal associated with p62 was weak in comparison to that observed after stimulation with intact antibodies. As was observed after stimulation with intact anti-IgG, GST-CskSH3 and GST alone failed to retain tyrosine phosphorylated proteins from A20 cells stimulated with F(ab′)2 (data not shown).
Figure 2.


Association of Csk with p62 is mediated by a direct, phosphotyrosine-dependent interaction with the Csk SH2 domain. (A) A20 cells were unstimulated or stimulated for 1, 2, or 10 min with intact anti– mouse IgG. Cells were lysed, split into four samples, and soluble proteins were assayed for binding to GST-CskSH2 (lanes 1–4), GST-CskSH3 (lanes 5–8), GST-CskSH3/SH2 (lanes 9–12), or GST (lanes 13–16). (B) Cells were unstimulated or stimulated for 1 min with intact anti–mouse IgG. SDS was added to a final concentration of 1%. Samples were either boiled or left on ice, and then diluted 10-fold in lysis buffer. Lysates were incubated with GST-CskSH3/2 (lanes 3–6) or GST (lanes 1 and 2). (C) Twofold serially diluted lysates of cells stimulated with intact anti–mouse IgG were incubated with GST-CskSH2 in the absence (lanes 1–5) or presence of 25 mM each phosphoserine (pSer) and phosphothreonine (pThr) (lanes 6–10), or in the presence of 50 mM phosphotyrosine (pTyr) (lanes 11–15). (D) Cells were stimulated for 1 min with intact anti–mouse IgG. Lysates were diluted serially twofold and incubated with equal amounts of GST-CskSH2 (lanes 1–4), GST-CskSH2 R107K (lanes 5–8), or GST-CskSH2 S109C (lanes 9–12). Proteins bound to GST fusions were precipitated as described in Materials and Methods, fractionated by SDS–polyacrylamide gel electrophoresis, and detected by immunoblotting with an antiphosphotyrosine antibody (4.G10).
SH2 domains are able to recognize extended phosphopeptide sequences that persist after disruption of protein ternary structure by denaturation. This property allowed us to determine whether the interaction between the Csk SH2 domain and p62 was direct. Lysates of unstimulated A20 cells or A20 cells stimulated with intact anti-IgG were denatured by boiling in 1% SDS, diluted 10-fold in lysis buffer, and assayed for binding to GST-CskSH3/SH2 (Fig. 2 B) or GST-CskSH2 (data not shown). Both fusion proteins retained p62 even after denaturation of A20 proteins in SDS, suggesting that their interaction with p62 is direct. Notably, p62 was the only protein from stimulated cells observed to bind to the Csk SH2 domain after SDS denaturation. Similar results were obtained after stimulation of A20 cells with an anti-IgG F(ab′)2 fragment (data not shown).
Two approaches were taken to examine the phosphotyrosine dependence of the interaction between p62 and the Csk SH2 domain. When a stimulated A20 cell lysate was serially diluted and incubated with GST-CskSH2 in the presence of free phosphotyrosine, retention of p62 was reduced; this reduction was specific, as an equimolar mixture of phosphoserine and phosphothreonine had no effect (Fig. 2 C). The specificity of competition by free phosphotyrosine suggests that binding of p62 to the Csk SH2 domain is phosphotyrosine dependent. The conserved FLVRES motif (amino acid residues 104–109 of Csk) comprises part of the phosphotyrosine binding site of SH2 domains, and mutations within this motif abolish or greatly reduce binding to phosphotyrosine-containing proteins (27–29). To test the specificity of p62 binding to the Csk SH2 domain, R107K and S109C mutations were introduced. A lysate of A20 cells, stimulated with intact anti-IgG, was serially diluted and assayed for binding to wild-type and mutant GST-CskSH2 domains. Although the S109C mutation abolished binding of the Csk SH2 domain to p62, the R107K mutation had little effect (Fig. 2 D).
Csk-associated p62 also Associates with RasGAP.
The ability of the R107K mutant to bind p62 was unexpected and reminiscent of the binding of GAP-A.p62 to the Csk SH2 domain, which is known to persist after mutation of R107 (25). To test whether the 62-kD species might represent GAP-A.p62, A20 cells were stimulated with intact anti-IgG; tyrosine phosphorylated co-migrating 62-kD proteins were detected in RasGAP, Csk, and Csk SH2 precipitates (Fig. 3 A). Next, A20 cells were stimulated with intact anti-IgG and lysates were immunodepleted using antibodies specific for Csk, RasGAP, the 62-kD phosphoprotein recognized by mAb 2C4 (30), or Sam68, a protein initially identified as related to RasGAP-associated p62 (31). Immunodepleted or undepleted lysates were then assayed for protein binding to GST (data not shown) or GST-CskSH2. The amount of tyrosine phosphorylated p62 recovered from GST-CskSH2 after immunodepletion of RasGAP was reduced significantly in comparison to a control immunodepletion with nonimmune antibody (Fig. 3 B). The recovery of p62 was also reduced after immunodepletion of Csk, as expected. In contrast, immunodepletion of Sam68 had no effect on the recovery of p62 from the GST-CskSH2 eluate. Because the 2C4 antibody does not detect its ligand on immunoblot, we were not able to estimate how much of the 2C4-binding p62 species was depleted in this experiment. These results indicate that p62 interacts with RasGAP in A20 cell lysates, and are consistent with the interpretation that the Csk-associated p62 is related to or identical to GAP-A.p62.
Figure 3.
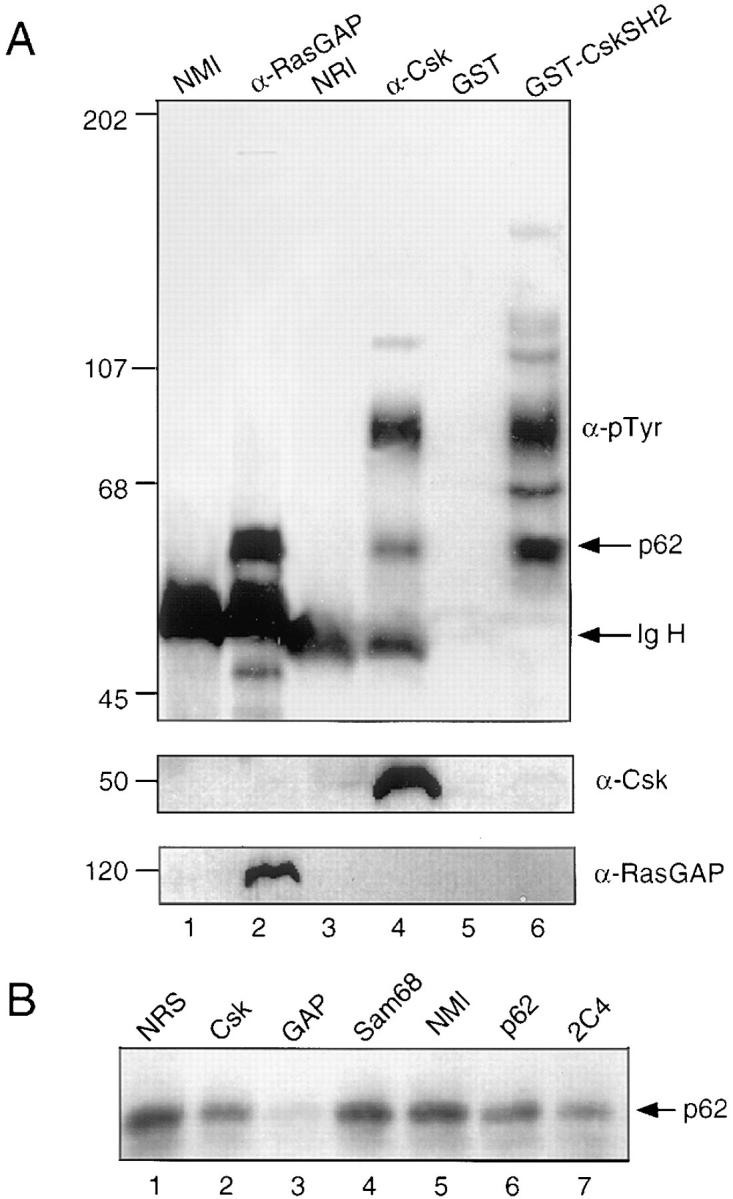

The 62-kD protein bound to Csk is associated with RasGAP. (A) Lysates of A20 cells stimulated with intact anti-IgG were incubated with agarose-conjugated normal mouse antibodies (lane 1), mAbs against RasGAP (lane 2), normal rabbit antibodies (lane 3), or affinity-purified antibodies against Csk (lane 4). Alternatively, lysates were incubated with GST (lane 5) or GST-CskSH2 (lane 6). (B) Lysates were immunodepleted with normal rabbit serum (lane 1), antibodies against Csk (lane 2), RasGAP (lane 3), Sam68 (lane 4), normal mouse immunoglobulin (lane 5) or p62 (Transduction Laboratories; lane 6, 2C4; lane 7). Depleted lysates were then precipitated with GST-CskSH2 (lanes 1–6). (C) RasGAP was precipitated as described above (lanes 2–4). The sample was boiled in lysis buffer containing 1% SDS. One-third was reserved for analysis of the RasGAP immunoprecipitate. The remaining two-thirds of the sample was diluted 10-fold in lysis buffer, split into two aliquots, and incubated either with GST (lane 3) or GST-CskSH2 (lane 4). A control immunoprecipitation was performed with normal mouse immunoglobulin (lane 1). All precipitates (A–C) were separated by 9% SDS-PAGE and analyzed by immunoblotting with antiphosphotyrosine antibodies. Membranes were stripped and reprobed either with mAbs against Csk or rabbit serum against RasGAP. (D) RasGAP and Csk immunoprecipitates were subjected to an in vitro kinase assay; 32P-labeled p62 was isolated from each precipitate by gel electrophoresis, excised, and analyzed by partial V-8 proteolysis as described in Materials and Methods. Phosphopeptides were visualized by autoradiography.
We used the ability of SH2 domains to recognize extended phosphopeptide sequences to determine whether the Csk SH2 domain could bind directly to GAP-A.p62. We were able to show that the Csk SH2 domain can recognize p62 from denatured RasGAP immunocomplexes, further supporting the interpretation that GAP-A.p62 represents the same species as Csk-associated p62 (Fig. 3 C). To demonstrate this more directly, we generated 32P-labeled RasGAP- and Csk-associated p62 in immunoprecipitates by an in vitro kinase assay, and compared partial V-8 protease digestion products by SDS–gel electrophoresis (Fig. 3 D). The proteins yielded similar distributions of phosphopeptides, indicating that these two species are related proteins.
Engagement of FcγRIIB1 Induces an Increase in the Association of Tyrosine Phosphorylated p62 with Csk.
Intact anti-IgG induced a greater increase in the antiphosphotyrosine reactivity of Csk-associated p62 than stimulation with F(ab′)2. We considered whether engagement of FcγRIIB1 engagement, as would occur upon stimulation with intact antibody, could account for this difference. To test this, we stimulated A20 cells in the presence or absence of the 2.4G2 mAb, which blocks signaling through the FcγRIIB receptor (32). Treatment of A20 cells with 2.4G2 alone induced an increase in SH2-associated, tyrosine-phosphorylated p62 similar to that observed upon stimulation by F(ab′)2. Strikingly, in the presence of 2.4G2, the amount of tyrosine phosphorylated p62 that bound GST-CskSH2 in response to stimulation with intact anti-IgG was reduced to the level observed after stimulation with F(ab′)2 (Fig. 4 A).
Figure 4.


Blockade or absence of FcγRIIB1 abolishes the increase in Csk-associated p62 after treatment with intact antibodies. (A) A20 cells were unstimulated (lanes 1, 4, 7, and 10), stimulated by F(ab′)2 (lanes 2, 5, 9, and 11), or stimulated with intact anti– mouse IgG (lanes 3, 6, 10, and 12) in the absence (lanes 1–3 and 7–9) or presence (lanes 4–6 and 10–12) of an FcγRIIB blocking reagent (mAb 2.4G2). Cells were lysed and soluble proteins were incubated with GST (lanes 1–6) or GST-CskSH2 (lanes 7–12). (B) Cells were unstimulated, stimulated with F(ab′)2, or stimulated with intact anti–mouse IgG. Lysates were incubated with GST (lanes 1–6) or GST-CskSH2 (lanes 7–12). GST and GST-CskSH2–bound proteins were separated on a 10% polyacrylamide gel, transferred to nitrocellulose, and detected by immunoblotting with an antiphosphotyrosine antibody (PY20). (B, top) A20 cells; (middle) the FcγRIIB1-deficient cell line IIA1.6; (bottom) IIA1.6 cells in which expression of FcγRIIB1 had been restored by transfection.
To confirm the involvement of FcγRIIB1 in the response of p62 to stimulation with intact antibody we used two cell lines derived from A20: IIA1.6, which lacks FcγRIIB, and CT314, a IIA1.6 derivative in which expression of wild-type FcγRIIB1 has been restored by transfection (33). In agreement with results of the FcγRIIB1 blockade experiment, IIA1.6 cells failed to exhibit a difference in the recovery of tyrosine phosphorylated p62 from GST-CskSH2 beads after stimulation with F(ab′)2 relative to stimulation with intact anti-IgG (Fig. 4 B, middle). This difference was reestablished in IIA1.6 cells transfected with FcγRIIB1 (Fig. 4 B, bottom). Taken together, these results strongly suggest that the observed difference in the association of tyrosine-phosphorylated p62 with the Csk SH2 domain after stimulation of A20 cells with intact anti-IgG or F(ab′)2 is the result of differential phosphorylation of p62 after BCR or FcγRIIB1 engagement.
Discussion
Although the ability of FcγRIIB1 to oppose stimulation of B cell responses by engagement of the BCR is well documented, the mechanism by which FcγRIIB1 antagonizes the BCR is incompletely understood. The participation of Src-related kinases in BCR-mediated signaling and the ability of Csk to suppress Src-related kinase activity in resting B cells suggested the possible participation of Csk in antagonism of BCR signaling by FcγRIIB1. In response to extracellular signals, the activity of Csk is known to be modulated by changes in its intracellular localization (21, 23). Recruitment of Csk into a signaling pathway emanating from FcγRIIB1 would therefore be expected to involve FcγRIIB1-induced changes in the association of Csk with other proteins or in the phosphorylation state of proteins known to interact with Csk. In this communication we have demonstrated that signaling through FcγRIIB1 induces tyrosine phosphorylation of a 62-kD protein (p62) that associates with the SH2 domain of Csk. In comparison, engagement of the BCR has relatively little effect on phosphorylation of p62 or on the ability of p62 to bind Csk. Thus, the effect of FcγRIIB1 engagement on p62 is relatively specific.
By its association with RasGAP, the 62-kD, Csk-associated protein phosphorylated in response to FcγRIIB1 engagement is likely to represent GAP-A.p62. This interpretation is in agreement with the recent observation that Csk is associated with GAP-A.p62 in murine fibroblasts stably expressing v-Src or an activated c-Src mutant (24). A cDNA previously reported to encode GAP-A.p62 has been shown since to encode a distinct protein, Sam68, a major Src substrate in mitotic cells (34, 35). Sam68 does not appear to represent the major phosphotyrosine-containing protein associated with GAP (31). Although an antibody against Sam68 detected small amounts of as yet unidentified phosphotyrosine-containing proteins in CskSH2 precipitates, antibodies against Sam68 were unable to precipitate Csk from lysates of stimulated A20 B cells (data not shown).
In addition to GAP-A.p62, we were able to detect two other phosphotyrosine-containing proteins, p125 and p97, that associated with the Csk SH2 domain in vivo. Unlike p62, the association of p125 and p97 with Csk SH2 did not increase after stimulation of A20 cells through FcγRIIB1 or the BCR. The 125-kD protein was not reactive with antibodies against p125Fak, nor was it immunoprecipitated by antibodies against RasGAP. Although, under the conditions described above, we observed little or no binding of p125 to CskSH2 in vitro, some binding of p125 could be observed when large amounts of A20 cell lysates were used (data not shown). This binding was apparently indirect because association of p125, unlike that of p62, was not reestablished after dissociation and reprecipitation (data not shown). Association of p125 with the Csk SH2 domain is therefore likely to be mediated by one or more additional proteins, perhaps including GAP-A.p62. Binding of p97 to CskSH2 was observed inconsistently. The identities of p125 and p97 are unknown, and the significance of their binding to Csk SH2 is unclear. Interestingly, in contrast to previous reports (25, 36), we were not able to detect association of RasGAP and Csk in these experiments.
Although complexes containing Csk and GAP-A.p62 may function in the regulation of Src-related kinase activity, it has been difficult to establish such a relationship. In addition to the changes documented here in response to FcγRIIB1 stimulation of B cells, association of GAP-A.p62 with Csk has been documented in v-Src–transformed cells, in resting T cells, and in T cells stimulated with anti-CD3 (25, 36). Such an association is absent in unstimulated B cells in which Lyn activity is apparently suppressed by Csk (37). Thus, association of p62 with Csk is not correlated with general suppression of Src-related kinase activity. This is underscored by the ability of intact anti-Ig antibody or F(ab′)2 fragments to induce similar levels of Src-related kinase activity in B cells (38). It has been proposed that the association of GAP-A.p62 and Csk functions to redistribute Csk to sites of c-Src activation (25). If so, it is possible that at any one time, only a fraction of total cellular Src-related kinase activity is present in such complexes and negatively regulated by Csk. The regulation of a portion of cellular Src-related kinase activity by recruitment into a complex containing GAP-A.p62 and Csk might not, under this scenario, be detectable by examination of bulk activity; this may explain the failure of Csk overexpression to reduce general Src activity in fibroblastoid cells (39). Csk could possibly be involved in suppression of Ras activity after engagement of FcγRIIB1 (40), since it has been shown that overexpression of Csk modulates Ras activity indirectly by suppression of Src activity (41). Alternatively, Csk may act on substrates other than Src (42).
One common feature of proteins that associate with p62, directly or indirectly, is their involvement in cytoskeletal rearrangements. In addition to RasGAP (43), Csk, and Src (44), these proteins include the γ isoform of phospholipase C (45). RasGAP can associate with p190, a molecule with GAP activity toward the small GTP-binding proteins Rho and Rac (46). Rho and Rac, in turn, participate in the assembly of cytoskeletal structures and membrane ruffling in response to growth factors (47, 48). RasGAP has also been shown to regulate activity and membrane association of the γ isoform of phospholipase C (49); one of its products, diacylglycerol, stimulates formation of actin nucleation sites at plasma membranes (50) and binds the actin-severing proteins gelsolin and villin (51). Tyrosine-phosphorylated GAP-A.p62 is inferred to participate in the stabilization of RasGAP interactions with the plasma membrane (52). Our observation that association of p62 and RasGAP with Csk SH2 is inefficiently induced by BCR engagement is consistent with the observation that RasGAP does not distribute to the membrane after BCR stimulation (53). Csk associates with p125Fak and contributes to its dephosphorylation (54). Moreover, overexpression of Csk is associated with reorganization of αvβ5 integrin and interference with cell spreading (39). Src itself has been shown to regulate epidermal growth factor–dependent cytoskeletal reorganization through phosphorylation of the p190 Rho/Rac GAP in murine fibroblasts (55). Src-related kinases also function in the organization of focal adhesion plaques. In cells from Csk-deficient mice, several adhesion proteins were found to be hyperphosphorylated through the action of Src and Fyn (56). Although relatively little is known about cytoskeletal rearrangements in B cells after activation, our evidence suggests that differences in the amount of p62 bound to Csk or RasGAP could effect differential regulation of cytoskeletal assembly after BCR or BCR-FcγRIIB1 stimulation.
Acknowledgments
We would like to thank Drs. John Cambier, Charles Janeway, Jr., Tony Pawson, Jonathan Cooper, Richard Roth, and Andrey Shaw for providing reagents and Drs. Thorunn Rafnar and Weiyi Yang for advice.
This work was supported by grants RO1 AI29575 and ACS IM736 (to J.P. Schneck) and by the Howard Hughes Medical Institute (to S. Desiderio).
Footnotes
1 Abbreviations used in this paper: BCR, B cell receptor for antigen; Csk, COOH-terminal Src kinase; GAP, GTPase-activating protein; GAP-A.p62, RasGAP-associated p62 protein; GST, glutathione S transferase.
Drs. Desiderio and Schneck are co-senior authors.
References
- 1.Pleiman CM, D'Ambrosio D, Cambier JC. The B-cell antigen receptor complex: structure and signal transduction. Immunol Today. 1994;15:393–409. doi: 10.1016/0167-5699(94)90267-4. [DOI] [PubMed] [Google Scholar]
- 2.Saouaf SJ, Mahajan S, Rowley RB, Kut SA, Fargnoli J, Burkhardt AL, Tsukada S, Witte ON, Bolen JB. Temporal differences in the activation of three classes of non-transmembrane protein tyrosine kinases following B-cell antigen receptor surface engagement. Proc Natl Acad Sci USA. 1994;91:9524–9528. doi: 10.1073/pnas.91.20.9524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Turner M, Mee PJ, Costello PS, Williams O, Price AA, Duddy LP, Furlong MT, Geahlen RL, Tybulewicz VL. Perinatal lethality and blocked B-cell development in mice lacking the tyrosine kinase Syk. Nature (Lond) 1995;378:298–302. doi: 10.1038/378298a0. [DOI] [PubMed] [Google Scholar]
- 4.Cheng AM, Rowley B, Pao W, Hayday A, Bolen JB, Pawson T. Syk tyrosine kinase required for mouse viability and B-cell development. Nature (Lond) 1995;378:298–302. doi: 10.1038/378303a0. [DOI] [PubMed] [Google Scholar]
- 5.Khan WN, Alt FW, Gerstein RM, Malynn BA, Larsson I, Rathbun G, Davidson L, Muller S, Kantor AB, Herzenberg LA, et al. Defective B cell development and function in Btk-deficient mice. Immunity. 1995;3:283–299. doi: 10.1016/1074-7613(95)90114-0. [DOI] [PubMed] [Google Scholar]
- 6.Kerner JD, Appleby MW, Mohr RN, Chien S, Rawlings DJ, Maliszewski CR, Witte ON, Perlmutter RM. Impaired expansion of mouse B cell progenitors lacking Btk. Immunity. 1995;3:301–312. doi: 10.1016/1074-7613(95)90115-9. [DOI] [PubMed] [Google Scholar]
- 7.Hibbs ML, Tarlinton DM, Armes J, Grail D, Hodgson G, Maglitto R, Stacker SA, Dunn AR. Defects in the immune system of Lyn-deficient mice, culminating in autoimmune disease. Cell. 1995;83:301–311. doi: 10.1016/0092-8674(95)90171-x. [DOI] [PubMed] [Google Scholar]
- 8.Nishizumi H, Taniuchi I, Yamanashi Y, Kitamura D, Ilic D, Mori S, Watanabe T, Yamamoto T. Impaired proliferation of peripheral B cells and indication of autoimmune disease in lyn-deficient mice. Immunity. 1995;3:549–560. doi: 10.1016/1074-7613(95)90126-4. [DOI] [PubMed] [Google Scholar]
- 9.Molina TJ, Kishihara K, Siderovski DP, van Ewijk W, Narendran A, Timms E, Wakeham A, Paige CJ, Hartmann KU, Veillette A. Profound block in thymocyte development in mice lacking p56lck . Nature (Lond) 1992;357:161–164. doi: 10.1038/357161a0. [DOI] [PubMed] [Google Scholar]
- 10.Takata M, Sabe H, Hata A, Inazu T, Homma Y, Nukada T, Yamamura H, Kurosaki T. Tyrosine kinases Lyn and Syk regulate B cell receptor–coupled Ca2+mobilization through distinct pathways. EMBO (Eur Mol Biol Org) J. 1994;13:1341–1349. doi: 10.1002/j.1460-2075.1994.tb06387.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bijsterbosch MK, Klaus GG. Crosslinking of surface immunoglobulin and Fc receptors on B lymphocytes inhibits stimulation of inositol phospholipid breakdown via the antigen receptors. J Exp Med. 1985;162:1825–1836. doi: 10.1084/jem.162.6.1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wilson HA, Greenblatt D, Taylor CW, Putney JW, Tsien RY, Finkelman FD, Chused TM. The B lymphocyte calcium response to anti-Ig is diminished by membrane immunoglobulin cross-linkage to the Fcγ receptor. J Immunol. 1987;138:1712–1718. [PubMed] [Google Scholar]
- 13.Phillips NE, Parker DC. Cross-linking of B lymphocyte Fc gamma receptors and membrane immunoglobulin inhibits anti-immunoglobulin–induced blastogenesis. J Immunol. 1984;132:627–632. [PubMed] [Google Scholar]
- 14.Phillips NE, Parker DC. Subclass specificity of Fcγ receptor–mediated inhibition of mouse B cell activation. J Immunol. 1985;134:2835–2838. [PubMed] [Google Scholar]
- 15.Muta T, Kurosaki T, Misulovin Z, Sanchez M, Nussenzweig MC, Ravetch JV. A 13–amino-acid motif in the cytoplasmic domain of Fc gamma RIIB modulates B-cell receptor signalling. Nature (Lond) 1994;369:340–343. doi: 10.1038/369340a0. [DOI] [PubMed] [Google Scholar]
- 16.D'Ambrosio D, Hippen KL, Minskoff SA, Mellman I, Pani G, Siminovitch KA, Cambier JC. Recruitment and activation of PTP1C in negative regulation of antigen receptor signaling by Fc gamma RIIB1. Science (Wash DC) 1995;268:263–264. doi: 10.1126/science.7716523. [DOI] [PubMed] [Google Scholar]
- 17.Ono M, Bolland S, Tempst P, Ravetch JV. Role of the inositol phosphatase SHIP in negative regulation of the immune system by the receptor FcγRIIB. Nature (Lond) 1996;383:263–266. doi: 10.1038/383263a0. [DOI] [PubMed] [Google Scholar]
- 18.Nada S, Okada M, MacAuley A, Cooper J, Nakagawa H. Cloning of a complementary DNA for a protein-tyrosine kinase that specifically phosphorylates a negative regulatory site of p60c-src. Nature (Lond) 1991;351:69–72. doi: 10.1038/351069a0. [DOI] [PubMed] [Google Scholar]
- 19.Imamoto A, Soriano P. Disruption of the Csk gene, encoding a negative regulator of Src family tyrosine kinases, leads to neural tube defects and embryonic lethality in mice. Cell. 1993;73:1117–1124. doi: 10.1016/0092-8674(93)90641-3. [DOI] [PubMed] [Google Scholar]
- 20.Nada S, Yagi T, Takeda H, Tokunaga T, Nakagawa H, Ikawa Y, Okada M, Aizawa S. Constitutive activation of Src family kinases in mouse embryos that lack Csk. Cell. 1993;73:1125–1135. doi: 10.1016/0092-8674(93)90642-4. [DOI] [PubMed] [Google Scholar]
- 21.Howell BW, Cooper JA. Csk suppression of Src involves movement of Csk to sites of Src activity. Mol Cell Biol. 1994;14:5402–5411. doi: 10.1128/mcb.14.8.5402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chow LM, Fournel M, Davidson D, Veillette A. Negative regulation of T-cell receptor signalling by tyrosine protein kinase p50csk . Nature (Lond) 1993;365:156–160. doi: 10.1038/365156a0. [DOI] [PubMed] [Google Scholar]
- 23.Cloutier JF, Chow LM, Veillette A. Requirement of the SH3 and SH2 domains for the inhibitory function of tyrosine protein kinase p50cskin T lymphocytes. Mol Cell Biol. 1995;15:5937–5944. doi: 10.1128/mcb.15.11.5937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sabe H, Hata A, Okada M, Nakagawa H, Hanafusa H. Analysis of the binding of the Src homology 2 domain of Csk tyrosine-phosphorylated proteins in the suppression and mitotic activation of c-src. Proc Natl Acad Sci USA. 1994;91:3984–3988. doi: 10.1073/pnas.91.9.3984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Neet K, Hunter T. The nonreceptor protein-tyrosine kinase CSK complexes directly with the GTPase-activating protein-associated p62 protein in cells expressing v-Src or activated c-Src. Mol Cell Biol. 1995;15:4908–4920. doi: 10.1128/mcb.15.9.4908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cleveland DW, Fischer SG, Kirschner MW, Laemmli UK. Peptide mapping by limited proteolysis in sodium dodecyl sulfate and analysis by gel electrophoresis. J Biol Chem. 1977;252:1102–1106. [PubMed] [Google Scholar]
- 27.Bibbins KB, Boeuf H, Varmus H. Binding of the Src SH2 domain to phosphopeptides is determined by residues in both the SH2 domain and the phosphopeptides. Mol Cell Biol. 1993;13:7278–7287. doi: 10.1128/mcb.13.12.7278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Panchamoorthy G, Fukazawa T, Stolz L, Payne G, Reedquist K, Shoelson S, Zhou S, Cantley L, Walsh C, Band H. Physical and functional interactions between SH2 and SH3 domains of the Src family protein tyrosine kinase p59fyn . Mol Cell Biol. 1994;14:6372–6385. doi: 10.1128/mcb.14.9.6372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mayer BJ, Jackson PK, Van Etten RA, Baltimore D. Point mutations in the abl SH2 domain coordinately impair phosphotyrosine binding in vitro and transforming activity in vivo. Mol Cell Biol. 1992;12:609–618. doi: 10.1128/mcb.12.2.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ogawa W, Hosomi Y, Shii K, Roth R. Evidence for two distinct 60-kilodalton substrates of the SRC tyrosine kinase. J Biol Chem. 1994;269:29602–29608. [PubMed] [Google Scholar]
- 31.Lock P, Fumagalli S, Polakis P, McCormick F, Courtneidge SA. The human p62 cDNA encodes Sam68 and not the RasGAP-associated p62 protein. Cell. 1996;84:23–24. doi: 10.1016/s0092-8674(00)80989-7. [DOI] [PubMed] [Google Scholar]
- 32.Unkeless JC. Characterization of a monoclonal antibody directed against mouse macrophage and lymphocyte Fc receptors. J Exp Med. 1979;150:580–596. doi: 10.1084/jem.150.3.580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jones B, Tite JP, Janeway CA., Jr Different phenotypic variants of the mouse B cell tumor A20/2J are selected by antigen- and mitogen-triggered cytotoxicity of L3T4-positive, I-A–restricted T cell clones. J Immunol. 1986;136:348–356. [PubMed] [Google Scholar]
- 34.Fumagalli S, Totty NF, Hsuan JJ, Courtneidge SA. A target for Src in mitosis. Nature (Lond) 1994;368:871–874. doi: 10.1038/368871a0. [DOI] [PubMed] [Google Scholar]
- 35.Taylor SJ, Shalloway D. An RNA-binding protein associated with Src through its SH2 and SH3 domains in mitosis. Nature (Lond) 1994;368:867–871. doi: 10.1038/368867a0. [DOI] [PubMed] [Google Scholar]
- 36.Catipovic B, Schneck JP, Brummet ME, Marsh DG, Rafnar T. Csk is constitutively associated with a 60-kDa tyrosine-phosphorylated protein in human T cells. J Biol Chem. 1996;271:9698–9703. doi: 10.1074/jbc.271.16.9698. [DOI] [PubMed] [Google Scholar]
- 37.Hata A, Sabe H, Kurosaki T, Takata M, Hanafusa H. Functional analysis of Csk in signal transduction through the B-cell antigen receptor. Mol Cell Biol. 1994;14:7306–7313. doi: 10.1128/mcb.14.11.7306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sarkar S, Schlottmann K, Cooney D, Coggeshall K. Negative signaling via FcγRIIB1 in B cells blocks phospholipase Cγ2 tyrosine phosphorylation but not Syk or Lyn activation. J Biol Chem. 1996;271:20182–20186. doi: 10.1074/jbc.271.33.20182. [DOI] [PubMed] [Google Scholar]
- 39.Bergman M, Joukov V, Virtanen I, Alitalo K. Overexpressed Csk tyrosine kinase is localized in focal adhesions, causes reorganization of αvβ5 integrin, and interferes with HeLa cell spreading. Mol Cell Biol. 1995;15:711–722. doi: 10.1128/mcb.15.2.711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sármay G, Koncz G, Gergely J. Human type II Fc receptors inhibit B cell activation by interacting with the p21ras-dependent pathway. J Biol Chem. 1996;271:30499–30504. doi: 10.1074/jbc.271.48.30499. [DOI] [PubMed] [Google Scholar]
- 41.Luttrell LM, Hawes BE, van Biesen T, Luttrell DK, Lansing TJ, Lefkowitz RJ. Role of c-Src tyrosine kinase in G protein–coupled receptor and Gβγ subunit–mediated activation of mitogen-activated protein. J Biol Chem. 1996;271:19443–19450. doi: 10.1074/jbc.271.32.19443. [DOI] [PubMed] [Google Scholar]
- 42.Autero M, Saharinen J, Pessa-Morikawa T, Soula-Rothhut M, Oetken C, Gassmann M, Bergman M, Alitalo K, Burn P, Gahmberg CG, et al. Tyrosine phosphorylation of CD45 phosphotyrosine phosphatase by p50csk kinase creates a binding site for p56lcktyrosine kinase and activates the phosphatase. Mol Cell Biol. 1994;14:1308–1321. doi: 10.1128/mcb.14.2.1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ellis C, Moran M, McCormick F, Pawson T. Phosphorylation of GAP and GAP-associated proteins by transforming and mitogenic tyrosine kinases. Nature (Lond) 1990;343:377–381. doi: 10.1038/343377a0. [DOI] [PubMed] [Google Scholar]
- 44.Cicchetti P, Mayer BJ, Thiel G, Baltimore D. Identification of a protein that binds to the SH3 region of Abl and is similar to Bcr and GAP-rho. Science (Wash DC) 1992;257:803–806. doi: 10.1126/science.1379745. [DOI] [PubMed] [Google Scholar]
- 45.Maa M, Leu T, Trandel BJ, Chang J, Parsons SJ. A protein that is highly related to GTPase-activating protein-associated p62 complexes with phospholipase Cγ. Mol Cell Biol. 1994;14:5466–5473. doi: 10.1128/mcb.14.8.5466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Settleman J, Albright CF, Foster LC, Weinberg RA. Association between GTPase activators for Rho and Ras families. Nature (Lond) 1992;359:153–154. doi: 10.1038/359153a0. [DOI] [PubMed] [Google Scholar]
- 47.Ridley AJ, Paterson HF, Johnston CL, Diekmann D, Hall A. The small GTP-binding protein rac regulates growth factor–induced membrane ruffling. Cell. 1992;70:401–410. doi: 10.1016/0092-8674(92)90164-8. [DOI] [PubMed] [Google Scholar]
- 48.Ridley AJ, Hall A. The small GTP-binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell. 1992;70:389–399. doi: 10.1016/0092-8674(92)90163-7. [DOI] [PubMed] [Google Scholar]
- 49.Valius M, Secrist JP, Kazlauskas A. The GTPase-activating protein of Ras suppresses platelet-derived growth factor β receptor signaling by silencing phospholipase C-γ1. Mol Cell Biol. 1995;15:3058–307. doi: 10.1128/mcb.15.6.3058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shariff A, Luna EJ. Diacylglycerol-stimulated formation of actin nucleation sites at plasma membranes. Science (Wash DC) 1992;256:177–178. doi: 10.1126/science.1373523. [DOI] [PubMed] [Google Scholar]
- 51.Janmey PA, Lamb J, Allen PG, Matsudaira PT. Phosphoinositide-binding peptides derived from the sequences of gelsolin and villin. J Biol Chem. 1992;267:11818–11823. [PubMed] [Google Scholar]
- 52.Moran MF, Polakis P, McCormick F, Pawson T, Elli SC. Protein-tyrosine kinases regulate the phosphorylation, protein interactions, subcellular distribution, and activity of p21ras GTPase-activating protein. Mol Cell Biol. 1991;11:1804–1812. doi: 10.1128/mcb.11.4.1804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Boyer MJ, Gutmann DH, Collins FS, Bar-Sagi D. Crosslinking of the surface immunoglobulin receptor in B lymphocytes induces a redistribution of neurofibromin but not p120-GAP. Oncogene. 1994;9:349–357. [PubMed] [Google Scholar]
- 54.Tobe K, Sabe H, Yamamoto T, Yamauchi T, Asai S, Kaburagi Y, Tamemoto H, Ueki K, Kimura H, Akanuma Y, et al. Csk enhances insulin-stimulated dephosphorylation of focal adhesion proteins. Mol Cell Biol. 1996;16:4765–4772. doi: 10.1128/mcb.16.9.4765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chang JH, Gill S, Settleman J, Parsons SJ. c-Src regulates the simultaneous rearrangement of actin cytoskeleton, p190RhoGAP, and p120RasGAP following epidermal growth factor stimulation. J Cell Biol. 1995;130:355–368. doi: 10.1083/jcb.130.2.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Thomas SM, Soriano P, Imamoto A. Specific and redundant roles of Src and Fyn in organizing cytoskeleton. Nature (Lond) 1995;376:267–271. doi: 10.1038/376267a0. [DOI] [PubMed] [Google Scholar]