

Abstract
The tumor suppressor protein p53 is overexpressed in close to 50% of all human malignancies. The p53 protein is therefore an attractive target for immunotherapy. Cytotoxic T lymphocytes (CTLs) recognizing a murine wild-type p53 peptide, presented by the major histocompatibility complex class I molecule H-2Kb, were generated by immunizing p53 gene deficient (p53 −/−) C57BL/6 mice with syngeneic p53-overexpressing tumor cells. Adoptive transfer of these CTLs into tumor-bearing p53 +/+ nude mice caused complete and permanent tumor eradication. Importantly, this occurred in the absence of any demonstrable damage to normal tissue. When transferred into p53 +/+ immunocompetent C57BL/6 mice, the CTLs persisted for weeks in the absence of immunopathology and were capable of preventing tumor outgrowth. Wild-type p53-specific CTLs can apparently discriminate between p53-overexpressing tumor cells and normal tissue, indicating that widely expressed autologous molecules such as p53 can serve as a target for CTL-mediated immunotherapy of tumors.
The efficacy of virus-specific CTLs to eradicate virus-induced tumors has been well documented (1–5). However, since the majority of tumors are not virus- induced, characterization of tumor-associated antigens encoded by cellular genes is important for the development of new immunotherapeutic strategies. Target antigens on nonvirally-induced tumors recognized by CTLs were recently identified, notably in patients with melanoma (6–18). The fact that these antigens are lineage- or tumor-specific, limits the use of these targets in immunotherapy to a small group of cancers. On the other hand, the expression of some of these antigens on normal melanocytes demonstrates that self antigens can serve as targets for CTL-mediated destruction of tumors.
Mutations in the gene encoding the tumor suppressor protein p53 are found in ∼50% of all human malignancies (19). Recently, a direct link has been established between mutational hot spots in the p53 gene leading to its overexpression, and carcinogenic metabolites derived from agents in cigarette smoke (20). In normal cells, p53 induces a cell cycle arrest, allowing DNA to be checked for irregularities, thereby guarding the integrity of the genome (21). Mutation of p53 abolishes its function as a suppressor of the cell cycle, promoting the escape of transformed cells from the normal restriction of controlled growth. Since these mutations, causing overexpression of p53, are present in a wide variety of cancers (22–25), a large group of patients would benefit from p53 directed immunotherapy. One could consider mutant p53 sequences as target antigens for tumor-specific CTLs. However, p53 mutations occur at many different sites in the p53 molecule, necessitating identification of the site of mutation in each patient before therapy. Furthermore, not all mutations are contained in MHC-binding CTL epitopes. If, in contrast, wild-type (wt)1 p53 sequences are used, the entire sequence of the p53 protein is available for properly processed immunogenic T cell epitopes. We hypothesized that the altered expression of p53, seen in many cancers, leads to modified processing and presentation of wt p53-derived peptides by MHC class I molecules. Recently, wt p53 peptide-specific CTL were generated from human and murine responding lymphocytes, some of which recognized p53-overexpressing tumors in vitro (26–34).
Here, we report the in vivo eradication of established p53-overexpressing tumors in C57BL/6 p53 (+/+) nude mice by a well-defined wt p53-specific CTL clone in the absence of any demonstrable immunopathology. These CTLs, generated in p53-deficient mice and recognizing the murine wt p53-derived epitope AIYKKSQHM (amino acids [aa] 158–166) presented by the MHC class I molecule H-2Kb, were also capable of preventing the outgrowth of a more aggressive p53-overexpressing tumor in immunocompetent p53 (+/+) C57BL/6 mice.
Materials and Methods
Mice.
C57BL/6 (B6, H-2b) mice were obtained either from the Netherlands Cancer Institute (Amsterdam, The Netherlands) or from IFFA Credo (Larbresle, France), C57BL/6 nu/nu (B6 nude, H-2b) mice were obtained from Bomholtgard (Ry, Denmark), and the p53 knockout (p53 −/−; H-2b) mice were obtained from GenPharm (Mountain View, CA; 35) and held under specific pathogen-free conditions. Since offspring could only be obtained by crossing a p53 heterozygous female (p53 +/−) with a p53 male (p53 −/−), the mice had to be analyzed for their p53 status by PCR analysis. For the PCR analysis two primer sets were used. Primer set A consists of a forward primer binding to the neomycin resistance gene (5′ GCA TCG CCT TCT ATC GCC TTC TTG AC 3′, neo fwd), with which the wt p53 gene was destroyed, and a reverse primer that binds to a sequence in exon 5 (5′ ATC ACC ATC GGA GCA GCG CTC ATG 3′, p53 exon 5 rev). Primer set A gives a band of 120 bp only when the neomycin gene is present. Primer set B consists of a forward primer binding to a sequence in intron 4 of the wt p53 gene (5′ CAG TCC TCT CTT TGC TGG CTC GCT CT 3′, p53 intron 4 fwd), which is deleted by the insertion of the neomycin resistance gene in the wt p53 gene. This sequence is only present in an intact wt p53 gene. The same reverse primer used in primer set A was used for primer set B. Primer set B gives a band of 180 bp only when the wt p53 gene is present.
Cell Lines.
Mouse embryo cells (MECs) of C57BL/6 origin (B6MECs) were transformed by transfection with the following oncogenes: Xhoc3, Adenovirus type 5 E1 (2); C3, HPV16 E + L and EJras (36); 4J, mut.p53 + H-ras; 5A and 5D, mut.p53 + N-ras; and 6J3, mut.p53 + fos (van Hall, T., M.P.M. Vierboom, C.J.M. Melief, and R. Offringa, manuscript in preparation). The tumor cell lines 4J, 5A, 5D, and 6J3 express high levels of a murine p53 containing a mutation at position 135 (37). Cell line EL-4 originates from an H-2b thymoma (38). The cell line “Koko” was generated from a solid tumor that arose spontaneously in a p53 knockout mouse. The following CTL clones, all of C57BL/6 origin, were used: anti-MCF1233 CTL clone BTM (provided by F. Ossendorp, University Hospital Leiden, Leiden, The Netherlands; reference 39), anti-Ad5E1A CTL clone 5 (2), and anti-Ad5 E1B CTL clone 100B6 (5). Mouse cell lines were cultured in Iscove's modified Dulbecco medium (Gibco Biocult, Glasgow, UK) supplemented with 8% FCS, penicillin (100 IU/ml), and β-mercaptoethanol (2 × 10−5 M) at 37°C in humidified air containing 5% CO2.
Generation of Tumor Cell Line–specific CTL Cultures.
Tumor cell line–specific CTL cultures were generated as described previously (2, 39). In brief, spleen cells from B6 and p53 −/− mice were taken 3 wk after the second immunization with 107 irradiated 4J cells treated with IFN-γ (20 U/ml for 48 h) and brought into culture after enriching for T cells via passage over a nylon wool column. 5 × 104 responder cells were cocultured with 5 × 103 irradiated and IFN-γ–treated 4J cells in a total volume of 100 μl/ well in 96-well U-bottomed plates. Bulk cultures were restimulated in vitro with irradiated and IFN-γ–treated 4J cells once a week, for 6 wk, which resulted in the CTL line 8. CTL clones, of which one was clone 1H11, were obtained by limiting dilution of the 4J-specific bulk culture at wk 3 as described (2). Long-term cultures were grown in medium with 10% FCS and 1.5% culture supernatant from PMA/Con A–stimulated rat spleen cells (40) and 25 CU (150 IU) rIL-2/ml (Eurocetus, Amsterdam, The Netherlands). All CTL clones and lines obtained had the marker profile Thy-1+, CD4−, and CD8+. All of the in vitro and in vivo experiments were done with CTL clone 1H11 unless indicated otherwise.
Peptides.
Peptides were generated by solid phase strategies on an Abimed AMS 422 multiple peptide synthesizer (Abimed, Langenfeld, Germany) by repeated cycles in which addition of Fmoc protected aa to a resin of polystyrene was alternated with an Fmoc-deprotection procedure using Fmoc chemistry. The purity of the peptides was determined by reverse phase HPLC and was found to be routinely >90% pure. Peptides were dissolved in DMSO (final DMSO concentration 0.25%) and diluted in 0.9% NaCl to a peptide concentration of 2 mg/ml and stored at −80°C.
MHC Class I Peptide Binding Assay.
The MHC class I peptide binding assay using the processing defective cell line RMA-S and the peptide competition assay were performed as described (41). In brief, RMA-S cells were cultured for 36 h at 26°C in culture medium in the presence of 2% human pool serum to increase the cell surface expression of “empty” class I molecules (42). In 96-well plates, RMA-S cells (2.5 × 105/well) were cultured in serum-free medium (40 μl/well) and peptide (10 μl/well) for 4 h at 37°C. Subsequently, the cells were washed and stained for analysis on a FACscan® flow cytometer (Becton Dickinson, Mountain View, CA) using, in the first step, a monoclonal antibody against H-2Kb (B8.24.3; reference 43) and, in the second step, FITC-labeled goat anti–mouse F(ab′)2 fragments.
Peptide Competition Cytotoxicity Assay.
For the competition assay, competitor peptides were aliquoted in triplicate in 96-well U-bottom plates at 10 times the final concentration, 10 μl/well. To each well, 10 μl of 50 pM of the reference peptide MCF1233 574–581 (KSPWFTTL; reference 39) was added. Triplicate wells were included that contained either no peptide or reference peptide only. EL-4 target cells (103 cells/well) were labeled with Europium (Eu3+) and added to the peptides (40 μl/well). After 60 min of incubation, 5 × 103 CTL clone BTM cells (39) were added to the wells for another 4 h of incubation before supernatants were collected. By interpolation of the data obtained with each competitor peptide at different concentrations, we calculated the peptide concentration that inhibits 50% of the maximal CTL lysis in the presence of the reference peptide alone (IC50).
Transporter Associated with Processing Translocation Assay.
This assay was performed as described (44, 45). In brief, RMA cells were washed in incubation buffer and permeabilized during 10 min at 37°C with 2.5 IU/ml streptolysin O. Subsequently, radioiodinated reporter peptide (TVNKTERAY) was added to the permeabilized cells together with titrated amounts of competitor peptide and ATP during 5 min at 37°C. Glycosylated reporter peptide was recovered with Con A Sepharose and quantified by γ counting. The IC50 was calculated by determining the concentration of competitor peptide that decreased the maximal amount of recovered glycosylated reporter peptide by half.
Eu3+ Release Cytotoxicity Assay.
Experimental procedures to measure cell-mediated cytotoxicity were performed in an Eu3+ release assay as described elsewhere (46). In brief, varying numbers of effector cells (harvested on Ficoll if necessary) were added to 103 Eu3+ labeled target cells in 100 μl of culture medium in 96-well U-bottomed plates. After 4 h incubation time, 20 μl culture supernatant was collected and mixed with 200 μl Enhancer Solution (Wallac, Turku, Finland). Measurement of the samples took place in a fluorometer (1234 Delfia; Wallac). The mean percentage specific lysis of triplicate wells was calculated as follows: percent specific lysis = ([cpm experimental release − cpm spontaneous release]/[cpm maximum release − cpm spontaneous release]) × 100. The spontaneous release of the Eu3+-labeled target cells was <30% in all experiments. The figures shown are representative for experiments done in duplicate.
Transfection of COS-7 Cells.
Transient transfection of COS-7 cells was performed as described elsewhere (5, 12). In short, 100 ng of plasmid pcDNA/Amp-p53 containing mut.p53 (37) together with 100 ng of plasmid pcDNAI/Amp-Db or pcDNAI/ Amp-Kb were transfected by the DEAE-dextran-chloroquine method into 104 COS-7 cells (47). Plasmid pcDNA/Amp-p53 harbors the p53 and plasmids pcDNAI/Amp-Db and pcDNAI/ Amp-Kb harbor the H-2Db and H-2Kb genes, respectively. The COS-7 cells were incubated in 100 μl IMDM containing 10% FCS for 48–72 h at 37°C, after which 1,500 CTLs in 25 μl IMDM containing 50 CU rIL-2 were added. Upon specific stimulation of the CTL, TNF-α is released in the supernatant, as measured in a bioassay with the TNF-α–sensitive cells WEHI-164 clone 13 (5). Percent WEHI cell death was calculated by the following formula: percent specific lysis = (1 − [OD 550–650 in sample wells/OD 550–650 in wells containing untransfected COS-7 cells and CTLs]) × 100.
Cold Target Inhibition Cytotoxicity Assay.
Koko cells were loaded with peptide at a concentration of 50 μg/ml for 2 h. Cells were washed five times to remove unbound peptide. Effector cells were preincubated with 5 × 104 unlabeled “blocking” cells (50 times excess) for 60 min at 37°C, before addition of labeled cells. After a subsequent 4-h incubation period with labeled cells, the supernatant was collected.
Adoptive Transfer of Anti-p53–specific CTLs.
wt p53–specific CTL clone 1H11 (2.0 × 107) was intravenously injected in either tumor-bearing or nonchallenged p53-competent C57Bl/6 nude mice and C57Bl/6 immunocompetent mice in combination with 105 CU rIL-2 administered subcutaneously in IFA (on the day of adoptive transfer and one week later). Control mice received nothing or an irrelevant clone together with rIL-2. The recovery of the intravenously injected anti-p53 CTL clone 1H11 from the spleen was tested at the indicated time point in an Eu3+ release assay.
Winn Type Assay.
The tumor cell 5D was injected in the peritoneum at indicated doses. On the same day 2.0 × 107 wt p53-specific CTL clone 1H11 or a control clone 9.5, recognizing the HPV16 E7–derived epitope RAHYNIVTF (36), was injected intraperitoneally together with 105 CU rIL-2 administered subcutaneously in IFA (on the day of adoptive transfer and one week later).
Results
Generation of wt p53-specific CTL.
wt p53-specific CTLs were generated by immunizing C57BL/6 p53-deficient (p53 −/−) mice with p53-overexpressing 4J tumor cells. The tumor cell 4J expresses high levels of mutant p53 at the RNA level as tested on a Northern blot, and at the protein level p53 as shown by cytospin staining (data not shown). Spleen cells of mice immunized and boosted with irradiated IFN-γ–treated 4J cells were restimulated in vitro with 4J, and the antigen specificity of the resulting bulk CTL cultures was analyzed. The CTL specifically lysed 4J, but not the tumor cell line Koko derived from a p53 −/− mouse (Fig. 1 A). Specific recognition of p53 was tested by incubating these CTLs with COS-7 cells transiently transfected with cDNAs encoding one of the two MHC class I molecules, H-2Kb or H-2Db with or without mutant p53. Specific recognition was assayed by TNF-α production. Fig. 1 B shows that the CTL bulk cultures recognize COS-7 cells transfected with mutant p53 in an MHC class I H-2Kb–restricted manner. Several CTL clones, displaying the same specificity, were isolated from this bulk culture by limiting dilution. Subsequent experiments were performed with CTL clone 1H11, which is a representative of the clones recognizing p53.
Figure 1.

p53 specificity and Kb restriction of bulk culture of tumor-specific CTLs. (A) Recognition of 4J cells (filled squares) and not of Koko cells (open circles) by a 2-wk-old CTL bulk culture, named line 8, in a Eu3+ release cytotoxicity assay (46; Genzyme, Cambridge, MA). (B) CTL bulk culture line 8, specifically recognizing 4J, recognizes COS-7 cells transfected with plasmids containing the genes of H-2Kb and mutant p53 (37).
Identification of a Murine wt p53 CTL Epitope.
To identify the p53 epitope recognized by these CTLs, the murine wt p53 aa sequence (48) was screened for the presence of peptides matching the H-2Kb peptide binding motif (49). A total of 17 peptides were selected and tested for their capacity to bind and stabilize H-2Kb using the RMA-S cell binding assay (50). Seven peptides were identified that bound to H-2Kb (Table 1, UC50 column). The ability of these peptides to compete with a known H-2Kb–binding epitope MCF1233 574–581, thereby inhibiting the recognition of this peptide by CTL clone BTM, demonstrates direct binding to the murine MHC class I molecule H-2Kb (41). Table 1, CC5 0 column). Translocation by transporters associated with processing, as shown by their capacity to compete for transport of a reference peptide in a transporter associated with processing translocation assay (44), indicates that these peptides, when properly processed in the cytoplasm, can enter the endoplasmic reticulum lumen where they bind to the MHC class I molecules (Table 1, IC50 column). These peptides were subsequently tested for their ability to sensitize Eu3+-labeled Koko cells for lysis by the p53-specific CTL clone 1H11. Only the peptide AIYKKSQHM, derived from the wt sequence of p53 (aa 158–166), was recognized by the CTLs (Fig. 2 A). Titrated amounts of length variants of this peptide were tested to establish the optimal length. The peptide AIYKKSQHM (aa 158–166), recognized at a concentration range of 0.1–1 pM (Fig. 2 B), was the optimal length peptide and one of the best binders (Table 1). Koko cells loaded with the peptide AIYKKSQHM were able to specifically block the recognition of the p53-overexpressing tumor cell lines 4J (Fig. 2 C) and 5D (data not shown) in a cold target inhibition cytotoxicity assay, demonstrating that this peptide is the naturally processed epitope presented by 4J and 5D.
Table 1.
MHC Class I Kb-binding, Peptide Competition, and TAP-dependent Transport of Seven wt p53 Peptides
| Kb binding peptides | UC50 | CC50 | IC50 | |||
|---|---|---|---|---|---|---|
| μg/ml | μg/ml | μg/ml | ||||
| aa 119–127: VMCYSPPL | 18 | 12 | 2.5 | |||
| aa 122–130: TYSPLNKL | 20 | 14 | 6 | |||
| aa 123–131: YSPPLNKLF | 4 | 0.5 | >100 | |||
| aa 127–134: LNKLFCQL | 4 | 2 | 7 | |||
| aa 158–166: AIYKKSQHM | 2.7 | 0.3 | 4.5 | |||
| aa 222–229: AGSEYTTI | 100 | 18 | 70 | |||
| aa 227–234: TTIHYKYM | 7 | 1.1 | 6 | |||
| SV9: FAPGNYPAL | 0.1 | 0.2 | ND |
Seven wt p53-derived were characterized in an MHC class I binding assay (column 1; reference 42), peptide competition cytotoxicity assay (column 2; reference 41), and TAP-dependent translocation assay (column 3; reference 44). (UC50) Peptide concentration resulting in 50% of the maximal upregulation of H-2Kb in the presence of the known H-2Kb-binding peptide Sendai virus NP 324–332 (61). (CC50) Peptide concentration that inhibits 50% of the maximal lysis by CTL clone BTM recognizing the reference peptide KSPWFTTL derived from the MCF1233 virus (39). (IC50) Concentration of competitor peptide that decreased the maximal amount of recovered glycosylated reporter peptide by 50% (44, 45). Indicated in bold are so-called anchor residues in the H-2Kb–binding motif described by Falk et al. (49).
Figure 2.

Peptide specificity (A–C) and sensitivity (B) of p53-specific CTL clone 1H11 on peptide-pulsed target cells (A and B) and tumor cells (C). (A) A representative clone, 1H11, derived from line 8 by limiting dilution, was tested for lytic activity of Eu3+-labeled p53 −/− Koko cells pulsed with one of seven H-2Kb binding wt p53 peptides (Table 1). Targets were unloaded Koko cells (open circles) or Koko cells loaded with peptides (given in aa sequence): VMCTYSPPL (open triangles), TYSPPLNKL (filled triangles), YSPPLNKLF (open squares), LNKLFCQL (filled squares), AIYKKSQHM (filled circles), AGSEYTTI (open diamonds), TTIHYKYM (filled diamonds). As a control, Koko cells pulsed with Kb-binding MCF1233-derived peptide KSPWFTTL (39) was taken along (asterisks). (B) Koko cells were incubated with titrated amounts of length variants of the wt mouse p53 epitope AIYKKSQHM (aa 158–166). CTL clone 1H11 was added after a preincubation of 30 min at an E/T ratio of 10:1. The following length variants were tested: AIYKKSQHM (closed circles; aa 158–166), AIYKKSQHMT (open triangles; aa 158–167), AIYKKSQH (open squares; aa 158–165); IYKKSQHM (open circles; aa 159–166); AMAIYKKS (closed triangles; aa 156–163). (C) Cold target blocking of the p53-overexpressing tumor cell 4J was tested by preincubation of the CTL clone 1H11 with the following unlabeled cells: no cells (open squares), koko cells (open circles), Koko cells pulsed with the wt p53 epitope AIYKKSQHM (closed circles; aa 158–166) and 4J cells (closed triangles). After 60 min, labeled 4J cells were added.
Mutation-induced Overexpression of p53 Is Not Required for Recognition of Tumor Cells in Vitro.
To establish whether this epitope is commonly expressed, mutant p53-transformed cells and other tumor cells, with no known p53 overexpression, were tested for recognition. The lysis of HPV16 transformed tumor cell line C3 (36; Fig. 3 A) and the thymoma EL-4 (data not shown) by CTL clone 1H11 was comparable to the lysis of other p53-overexpressing lines 4J, 5A, 5D, and 6J3 (Fig. 3 A). Strikingly, nontransformed B6MEC and Con A–stimulated T cell blasts were efficiently recognized by CTL clone 1H11 (Fig. 3 A), demonstrating the potential cross-reacting ability of these p53-specific CTLs to nonmalignant cells. On the other hand, freshly isolated thymocytes (Fig. 3 B) and freshly isolated spleen cells (data not shown) are not lysed by these CTLs.
Figure 3.

Lytic ability of wt p53-specific CTL clone 1H11 on a panel of normal and tumor cells. (A) A panel of Eu3+-labeled C57BL/6-derived cell lines were tested for lysis by p53-specific CTL clone 1H11 in a cytotoxicity assay. The following targets were tested for recognition: MEC (filled circles); C3: HPV16 + EJras (open triangles); 5A (open diamonds) and 5D (filled diamonds): p53 + N-ras; 6J3: p53 + fos (open squares); 4J: p53 + H-ras (filled squares); and Con A–stimulated spleen cells (filled triangles). The p53 −/− Koko cell line (open circles) was taken along as the negative control. (B) Lack of recognition of freshly isolated thymocytes (open triangles) by the p53-specific CTL clone 1H11 in a 51Cr–cytotoxicicty assay (29). Thymocytes are recognized by the p53-specific CTL clone 1H11 when pulsed with the wt p53 peptide AIYKKSQHM (filled triangles). p53 −/− Koko cells, with (filled circles) and without (open circles) this peptide, were taken along as a positive control for recognition of the peptide.
Adoptive Transfer of wt p53-specific CTLs Eradicate Established Tumors in p53 +/+ B6 Nude Mice.
Since these wt p53-specific CTLs cross-reacted on nontransformed cells, we then assessed whether mice carrying a functional p53 gene would survive adoptive transfer of these potentially autoreactive CTLs and whether these CTLs could eradicate established 4J tumors in these mice without overt immunopathology. Adoptively transferred wt p53-specific CTLs were retrieved from the spleen of B6 p53 +/+ nude mice up to 3 mo after intravenous administration (Fig. 4, A and B), but not from splenocytes of untreated B6 nude mice (Fig. 4 C). No signs of autoimmune-induced damage were observed in mice that had recieved wt p53-specific CTLs. We subsequently tested the possibility of eradicating established 4J tumors by adoptive transfer of wt p53-specific CTLs. Tumors grew progressively in untreated mice (Fig. 5 A) and in mice injected with an irrelevant CTL clone recognizing an Ad5 E1B–derived epitope (5; Fig. 5 B). Adoptive transfer by intravenous infusion of the wt p53-specific clone 1H11 resulted in complete and permanent (>5 mo) tumor eradication in mice with small- (average size = 41 mm3; Fig. 5 C) and medium- (average size = 140 mm3; Fig. 5 D) sized tumors. Intratumoral injection of similar numbers of wt p53-specific CTLs in combination with rIL-2 also led to the complete eradication of medium-sized tumors (data not shown). Even large established 4J tumors (average size = 427 mm3) were eradicated in three out of six mice (Fig. 5 E) after intravenous infusion of the wt p53-specific clone 1H11. Mice that had rejected p53-induced tumors after treatment with wt p53-specific CTLs retained long-term tumor-specific CTL immunity, since wt p53-specific CTLs could be retrieved from spleens of these animals one month after CTL treatment (Fig. 6, A and B).
Figure 4.

Long-term survival of adoptively transferred wt p53-specific CTLs in B6 nude mice. Recovery of wt p53-specific CTLs (clone 1H11) 3 mo after adoptive transfer of these CTLs into C57BL/6 nude mice as tested in a Eu3+ release assay (46). Spleen cells of mice that had received wt p53-specific CTLs in the presence (A) or absence of 6 × 105 IU IL-2 (B), and naive (C) mice were restimulated in vitro with 4J and tested for their peptide specificity on Eu3+-labeled p53 −/− Koko cells (open circles), p53 −/− Koko cells pulsed with the wt p53 peptide AIYKKSQHM (filled circles), and recognition 4J (filled squares) cells after 7 d of culture.
Figure 5.

Tumor eradication by adoptive transfer of wt p53-specific CTLs. C57BL/6 nude mice were subcutaneously challenged with 107 4J tumor cells. After 3 wk, CTL treatment of established tumors was started. Treatment consisted of intravenous adoptive transfer of 2 × 107 CTL clone 1H11 in 300 μl PBS on day 0, and a subcutaneous injection of 6 × 105 IU rIL-2 (Cetus Corp., Emeryville, CA) in 50% incomplete Freund's adjuvant (IFA) on days 0 and 7. Tumor growth is given as a percentage relative to the size of the tumor when the treatment was started. Mice were given no treatment (A; n = 5), treatment with an irrelevant clone recognizing an Ad5E1B epitope (B; n = 4; reference 5), or treatment with the wt p53-specific CTL clone 1H11 (C–E). Mice treated with the wt p53-specific clone 1H11 consisted of three groups: mice bearing small tumors (C; 100% = 41 mm3; n = 6), medium-sized tumors (D; 100% = 140 mm3; n = 4), and large tumors (E; 100% = 427 mm3; n = 6) at the start of treatment.
Figure 6.

Recovery of wt p53-specific CTLs from nude mice after successful tumor eradication. wt p53-specific CTL clone 1H11 was retrieved from the spleens of C57BL/6 nude mice 1 mo after tumor eradication as tested in a Eu3+ release assay. CTLs were administered intravenously (A) or intratumorally (B). A nontreated naive mouse (C) was taken along as a negative control. Spleen cells were restimulated in vitro with 4J cells and tested for their peptide specificity on p53 −/− Koko cells (open circles), Koko cells pulsed with the p53 peptide AIYKKSQHM (filled circles), and recognition of 4J cells (filled squares) after 7 d of culture.
Prevention of Outgrowth of a p53-overexpressing Tumor in Immunocompetent C57BL/6 Mice.
To investigate whether the wt p53-specific CTLs would also persist in immunocompetent p53 +/+ C57BL/6 mice or would be deleted in the presence of a T cell compartment absent in C57BL/6 nude mice, p53-specific CTLs were transferred into p53 +/+ C57BL/6 immunocompetent mice and the spleens of these mice were assayed for p53-specific activity after 14 d. wt p53-specific CTLs, recognizing the peptide and 4J tumor cells, could be retrieved from spleens of C57BL/6 p53 +/+ immunocompetent mice at day 14 (Fig. 7 A) and up to at least 7 wk after adoptive transfer of these CTLs (data not shown). We also evaluated the ability of these wt p53-specific CTLs to prevent the outgrowth of the more aggressive tumor, 5D, transformed by mutant p53 and N-ras, in a Winn assay (Table 2). The 5D tumor, in contrast to the 4J tumor, is tumorigenic in immunocompetent syngeneic B6 mice. In the group of mice challenged intraperitoneally with 5D and simultaneously treated with control CTL clone 9.5, recognizing an HPV16 E7-derived epitope, 12 out of 12 animals developed a progressively growing tumor and died within 3 wk. In the group of mice challenged intraperitoneally with 5D and treated with the wt p53-specific CTL clone 1H11, only 1 out of 12 developed a progressively growing tumor, thus demonstrating in vivo activity against the tumor without demonstrable autoimmune pathology.
Figure 7.

Recovery of wt p53-specific CTL activity after adoptive transfer of CTLs into immunocompetent mice. wt p53-specific CTL clone 1H11 was recovered from the spleens of C57BL/6 p53 +/+ immunocompetent mice 14 d after the CTLs were administered intavenously (A), as tested in a Eu3+ release assay. A nontreated C57BL/6 p53 +/+ immunocompetent mouse (B) was taken along as a negative control. Spleen cells were restimulated in vitro with 4J and tested for their peptide specificity on p53 −/− Koko cells (open circles), Koko cells pulsed with the p53 peptide AIYKKSQHM (filled circles), and recognition of 4J cells (filled squares) after 7 d of culture.
Table 2.
Winn Type Tumor Neutralization by wt p53-specific CTL Clone 1H11
| Tumor incidence | ||||
|---|---|---|---|---|
| Tumor dose | Control clone 9.5 | wt p53-specific CTL clone 1H11 | ||
| 5 × 104 | 4/4 | 1/4 | ||
| 1 × 104 | 4/4 | 0/4 | ||
| 0.2 × 104 | 4/4 | 0/4 | ||
Immunocompetent B6 mice were simultaneously injected intraperitoneally with 5D (p53/N-ras) tumor cells at various doses and a control clone 9.5 (2 × 107) recognizing the HPV16 E7–derived epitope RAHYNIVTF or the wt p53-specific CTL clone 1H11 (2 × 107). On the day of treatment and 7 d later, a subcutaneous injection of 6 × 105 IU rIL-2 (Cetus Corp.) in 50% IFA was given.
Evaluation of Autoimmune Pathology.
3 mo after adoptive transfer of the p53-specific CTL clone 1H11 into nontumor-bearing nude animals and one month after successful eradication of small- and medium-sized tumors, tissues (liver, kidney, spleen, small intestine, large intestine, lung, stomach, heart, brain, skin, lymph nodes, and bone marrow) were collected and examined microscopically (hematoxylin and eosin stained). Examination of coded samples by two independent investigators showed no evidence of immunopathology in normal tissues of these nude mice (Fig. 8). An increased infiltrate of mononuclear cells was observed in normal tissue of cured mice. This is probably caused by the administration of rIL-2, since mice treated with an irrelevant clone also show this infiltrate (data not shown). Similarly, tissues from C57BL/6 p53 +/+ immunocompetent mice did not show evidence of immunopathology at times when, after adoptive transfer, the wt p53-specific CTLs could be recovered (Fig. 7 A). Staining of spleen and skin for CD4 and CD8 showed no difference between treated animals and nontreated control animals (data not shown).
Figure 8.
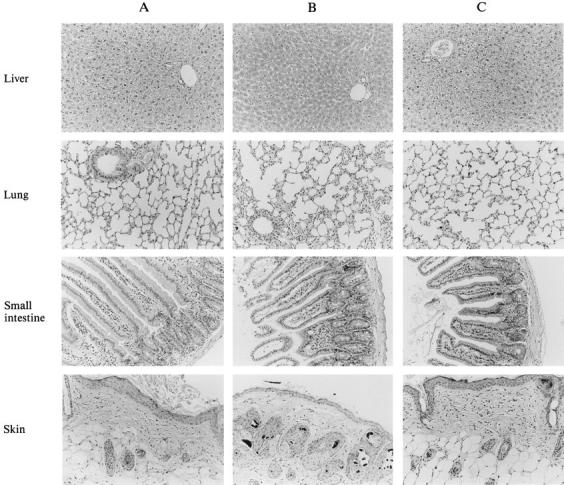
Histology of various tissues after adoptive transfer of wt p53-specific CTLs. Hematoxylin and eosin–stained sections of liver, small intestine, skin, and lung of a nontreated control mouse (A; tested for CTL activity as shown in Fig. 4 C); a mouse treated with an irrelevant clone directed against the Ad5E1B-derived epitope VNIRNCCYI (aa 192–200; reference 5) (B; from the group depicted in Fig. 5 B), and a mouse 1 mo after complete tumor eradication by the adoptive transfer of the wt p53-specific CTL clone 1H11 (C; tested for CTL activity as shown in Fig. 6 A).
Discussion
The search for widely expressed tumor antigens as targets for MHC class I–restricted CTLs is of great importance for the development of T cell–mediated immunotherapy of cancer. The tumor suppressor protein p53 is potentially such an antigen, with altered expression in up to 50% of all human tumors (19). In the present study we demonstrate that CTLs recognizing a murine wt p53-derived epitope were able to eradicate a p53-overexpressing tumor in p53 +/+ B6 nude mice in the absence of demonstrable immunopathology.
wt p53-specific CTLs were generated by immunizing C57BL/6 p53 −/− mice with syngeneic p53-overexpressing 4J tumor cells. As expected, a wt p53-derived sequence was found to serve as an excellent epitope, the mutant p53 area being devoid of MHC class I–binding motif carrying sequences (49). The wt p53-specific CTLs recognized the H-2Kb–binding epitope AIYKKSQHM (aa 158–166; Fig. 2 A) with high affinity (Fig. 2 B), comparable to previously published CTL clones against viral epitopes that were able to eradicate established tumors (2, 4, 5).
C57BL/6 nude and immunocompetent mice carrying the wt p53 gene survived the adoptive transfer of wt p53-specific CTLs. These potentially autoreactive CTLs persist normally in p53 +/+ mice (Fig. 4, A and B, and 7 A) without damage to normal tissues (Fig. 8 C). This persistence of large amounts of self-reactive T cells was elegantly shown in a study of Ohashi et al. (51). They demonstrated that CTLs expressing a transgenic TCR recognizing a lymphocytic choriomeningitis virus glycoprotein-derived epitope remained functionally unresponsive towards β islet cells expressing the transgene lymphocytic choriomeningitis virus glycoprotein. Similar observations were made by Goverman et al. (52), who showed that animals with large amounts of functionally, autoreactive T cells expressing the transgenic TCR specific for the naturally expressed myelin basic protein can be present without causing experimental allergic encephalomyelitis. However, in both models the transgenic TCR-carrying T cells can be activated to cause autoimmunity (51, 52). In contrast, the wt p53-specific CTLs were active in tumor-bearing C57BL/6 nude mice and eradicated established tumors (Fig. 5, C–E) without autoimmunity. In this respect, our study corroborates observations in Friend leukemia virus envelope (FLVenv) transgenic mice in which adoptively transferred FLVenv-autoreactive T cells can eradicate FLVenv expressing tumor cells without damage to normal tissue expressing this artificial autoantigen (53).
A simple explanation for the observed tumor selectivity can be the increased expression of the p53 protein resulting from the p53 mutation. Alternatively, the lack of “danger” signals delivered by normal tissues (54) might protect against the destruction by the potentially autoreactive wt p53-specific CTL. In fact, in normal tissues, homeostatic mechanisms apparently control tissue damage by potentially autoreactive T cells. Only powerful inflammatory stimuli can provoke autoaggression mediated by these otherwise dormant T cells, as illustrated by the examples of experimental allergic encephalomyelitis (55, 56) and adjuvant arthritis (57). The explanation why, in this particular instance, no autoimmune tissue damage occurs despite the infusion of a large number of activated cloned CTLs may lie in insufficient antigen display by the MHC class I molecules in combination with lack of proper costimulation (54) and downregulatory chemokine and cytokine conditions (55).
Recent reports show the induction of wt p53 peptide-specific responses with cross-reactivity on endogenously p53-expressing targets (28, 29, 31–34). However, the in vivo relevance of these responses remains to be demonstrated. In reports in which wt p53 was used as an immunogen (30, 58), therapeutic effects were found consisting of eradication of established tumors or protection against a subsequent tumor challenge. However, the mechanism of antitumor activity in these studies remains to be clarified. In our model, a well-defined high affinity CD8+ CTL clone recognizing a wt p53 epitope eradicated established tumors in B6 nude mice and prevented the outgrowth of a more aggressive tumor injected simultaneously in the peritoneal cavity in B6 immunocompetent mice.
To generate these CTLs, responding lymphocytes from p53 knockout mice were used for the obvious reason that these mice are not tolerant of p53. Since p53 is a crucial protein serving as a checkpoint in the cell cycle of stimulated T cells (59) which are open to attack by wt p53-specific CTLs (Fig. 3 A), one of the major challenges is to try to break tolerance of wt p53 and to generate high affinity wt p53-specifc CTLs in normal p53 +/+ individuals.
Activation of CTLs to autoantigens seems feasible in cancer patients as evidenced by the recent analyses of responses against melanoma-associated antigens and against p53 (11–14, 16–18, 31). From the blood of healthy donors, CTLs reactive against the autoantigen tyrosinase and against wt p53 can be aroused from their unresponsive state by appropriate in vitro stimulation (29, 31, 60). Our data support the idea to use widely expressed tumor-associated antigens such as p53 for CTL-mediated immunotherapy.
Acknowledgments
We would like to thank T. Ottenhof and B. Roep for critically reading the manuscript. G. Schijf and K. Goris were very helpful in technical assistance and maintaining the mice.
Footnotes
H.W. Nijman was supported by The Netherlands Organization for Scientific Research grant 900-716-075.
Abbreviations used in this paper: aa, amino acids; FLVenv, Friend leukemia virus envelope; MEC, mouse embryo cell; wt, wild type.
M.P.M. Vierboom and H.W. Nijman contributed equally to this paper.
References
- 1.Melief CJM. Tumor eradication by adoptive transfer of cytotoxic T lymphocytes. Adv Cancer Res. 1992;58:143–175. doi: 10.1016/s0065-230x(08)60294-8. [DOI] [PubMed] [Google Scholar]
- 2.Kast WM, Offringa R, Peters PJ, Voordouw AC, Meloen RH, Van der Eb AJ, Melief CJM. Eradication of adenovirus E1-induced tumors by E1A-specific cytotoxic T lymphocytes. Cell. 1989;59:603–614. doi: 10.1016/0092-8674(89)90006-8. [DOI] [PubMed] [Google Scholar]
- 3.Greenberg PD. Adoptive T cell therapy of tumors: mechanisms operative in the recognition and elimination of tumor cells. Adv Immunol. 1991;49:281–355. doi: 10.1016/s0065-2776(08)60778-6. [DOI] [PubMed] [Google Scholar]
- 4.Feltkamp MCW, Vreugdenhil GR, Vierboom MPM, Ras E, Van der Burg SH, Ter J, Schegget, Melief CJM, Kast WM. CTL raised against a subdominant epitope offered as a synthetic peptide eradicate human papillomavirus type 16–induced tumors. Eur J Immunol. 1995;25:2638–2642. doi: 10.1002/eji.1830250935. [DOI] [PubMed] [Google Scholar]
- 5.Toes REM, Offringa R, Blom HJJ, Brandt RMP, Van der Eb AJ, Melief CJM, Kast WM. An adenovirus type 5 early region 1B-encoded CTL epitope-mediating tumor eradication by CTL clones is down-modulated by an activated ras oncogene. J Immunol. 1995;154:3396–3405. [PubMed] [Google Scholar]
- 6.van der Bruggen P, Traversari C, Chomez P, Lurquin C, de Plaen E, van den Eynde B, Knuth A, Boon T. A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. Science (Wash DC) 1991;254:1643–1647. doi: 10.1126/science.1840703. [DOI] [PubMed] [Google Scholar]
- 7.van den Eynde B, Lethé B, van Pel A, de Plaen E, Boon T. The gene coding for a major tumor rejection antigen of tumor P815 is identical to the normal gene of syngeneic DBA/2 mice. J Exp Med. 1991;173:1373–1384. doi: 10.1084/jem.173.6.1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mandelboim O, Berke G, Fridkin M, Feldman M, Eisenstein M, Eisenbach L. CTL induction by a tumor-associated antigen octa-peptide derived from a murine lung carcinoma. Nature (Lond) 1994;369:67–71. doi: 10.1038/369067a0. [DOI] [PubMed] [Google Scholar]
- 9.Boon T, Cerottini J-C, Van den Eynde B, Van der Bruggen P, Van Pel A. Tumor antigens recognized by T lymphocytes. Annu Rev Immunol. 1994;12:337–365. doi: 10.1146/annurev.iy.12.040194.002005. [DOI] [PubMed] [Google Scholar]
- 10.Van Pel A, Van der Bruggen P, Coulie PG, Brichard VG, Lethé B, Van den Eynde B, Uyttenhove C, Renauld J-C, Boon T. Genes coding for tumor antigens recognized by cytolytic T lymphocytes. Immunol Rev. 1995;145:229–250. doi: 10.1111/j.1600-065x.1995.tb00084.x. [DOI] [PubMed] [Google Scholar]
- 11.Traversari C, Van der Bruggen P, Lüscher IF, Lurquin C, Chomez P, Van Pel A, De Plean E, Amar-Costesec A, Boon T. A nonapeptide encoded by human gene MAGE-1 is recognized on HLA-A1 by cytolytic T lymphocytes directed against tumor antigen MZ2-E. J Exp Med. 1992;176:1453–1457. doi: 10.1084/jem.176.5.1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brichard V, Van Pel A, Wolfel T, Wolfel C, De Plaen E, Lethe B, Coulie P, Boon T. The tyrosinase gene codes for an antigen recognized by autologous cytotoxic T lymphocytes on HLA-A2 melanomas. J Exp Med. 1993;178:489–495. doi: 10.1084/jem.178.2.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gaugler B, van den Eynde B, van der Bruggen P, Romero P, Gaforio JJ, de Plaen E, Lethé B, Brasseur F, Boon T. Human gene MAGE-3 codes for an antigen recognized on a melanoma by autologous cytolytic T lymphocytes. J Exp Med. 1994;179:921–930. doi: 10.1084/jem.179.3.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.De Plaen E, Arden K, Traversari C, Gaforio JJ, Szikora J-P, De Smet C, Brasseur F, Van der Bruggen P, Lethé B, Lurquin C, et al. Structure, chromosomal localization, and expression of 12 genes of the mage family. Immunogenetics. 1994;40:360–369. doi: 10.1007/BF01246677. [DOI] [PubMed] [Google Scholar]
- 15.Kawakami Y, Eliyahu S, Delgado CH, Robbins PF, Rivoltini L, Topalian SL, Miki T, Rosenberg SA. Cloning of the gene coding for a shared human melanoma antigen recognized by autologous T cells infiltrating into tumor. Proc Natl Acad Sci USA. 1994;91:3515–3519. doi: 10.1073/pnas.91.9.3515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kawakami Y, Eliyahu S, Sakaguchi K, Robbins PF, Rivoltini L, Yannelli JR, Appella E, Rosenberg SA. Identification of the immunodominant peptides of the MART-1 human melanoma antigen recognized by the majority of HLA-A2–restricted tumor infiltrating lymphocytes. J Exp Med. 1994;180:347–352. doi: 10.1084/jem.180.1.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kawakami Y, Eliyahu S, Delgado CH, Robbins PF, Sakaguchi K, Appella E, Yannelli JR, Adema GJ, Miki T, Rosenberg SA. Identification of a human melanoma antigen recognized by tumor-infiltrating lymphocytes associated with in vivo tumor rejection. Proc Natl Acad Sci USA. 1994;91:6458–6462. doi: 10.1073/pnas.91.14.6458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bakker ABH, Schreurs MWJ, De Boer AJ, Kawakami Y, Rosenberg SA, Adema GJ, Figdor CG. Melanocyte lineage-specific antigen gp100 is recognized by melanoma-derived tumor-infiltrating lymphocytes. J Exp Med. 1994;179:1005–1009. doi: 10.1084/jem.179.3.1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hollstein M, Sidransky D, Vogelstein B, Harris CC. p53 Mutation in human cancers. Science (Wash DC) 1991;253:49–53. doi: 10.1126/science.1905840. [DOI] [PubMed] [Google Scholar]
- 20.Denissenko MF, Pao A, Tang M-S, Pfeifer GP. Preferential formation of benzo[a]pyrene adducts at lung cancer mutational hotspots in p53. Science (Wash DC) 1996;274:430–432. doi: 10.1126/science.274.5286.430. [DOI] [PubMed] [Google Scholar]
- 21.Kuerbitz SJ, Plunkett BS, Walsh WV, Kastan MB. Wild-type p53 is a cell cycle checkpoint determinant following irradiation. Proc Natl Acad Sci USA. 1992;89:7491–7495. doi: 10.1073/pnas.89.16.7491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zambetti GP, Levine AJ. A comparison of the biological activities of wild-type and mutant p53. FASEB J. 1993;7:855–865. doi: 10.1096/fasebj.7.10.8344485. [DOI] [PubMed] [Google Scholar]
- 23.Ludlow JW. Interactions between SV 40 large-tumor antigen and the growth suppressor proteins pRB and p53. FASEB J. 1993;7:866–871. doi: 10.1096/fasebj.7.10.8344486. [DOI] [PubMed] [Google Scholar]
- 24.Moran E. Interaction of adenoviral proteins with Rb and p53. FASEB J. 1993;7:880–885. doi: 10.1096/fasebj.7.10.8344487. [DOI] [PubMed] [Google Scholar]
- 25.Momand M, Zambetti GP, Olson DC, George D, Levine AJ. The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell. 1992;69:1237–1245. doi: 10.1016/0092-8674(92)90644-r. [DOI] [PubMed] [Google Scholar]
- 26.Noguchi Y, Chen Y-T, Old LJ. A mouse mutant p53 product recognized by CD4+ and CD8+ T cells. Proc Natl Acad Sci USA. 1994;91:3171–3175. doi: 10.1073/pnas.91.8.3171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nijman HW, Van den Burg SH, Vierboom MPM, Houbiers JGA, Kast WM, Melief CJM. p53, a potential target for tumor-directed T cells. Immunol Lett. 1994;40:171–178. doi: 10.1016/0165-2478(94)90189-9. [DOI] [PubMed] [Google Scholar]
- 28.Theobald M, Biggs J, Dittmer D, Levine AJ, Sherman LA. Targeting p53 as a general tumor antigen. Proc Natl Acad Sci USA. 1995;92:11993–11997. doi: 10.1073/pnas.92.26.11993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Houbiers JGA, Nijman HW, Van den Burg SH, Drijfhout JW, Kenemans P, Van de Velde CJH, Brand A, Momburg F, Kast WM, Melief CJM. In vitro induction of human cytotoxic T lymphocyte responses against peptides of mutant and wild-type p53. Eur J Immunol. 1993;23:2072–2077. doi: 10.1002/eji.1830230905. [DOI] [PubMed] [Google Scholar]
- 30.Mayordomo JI, Loftus DJ, Sakamoto H, De Cesare CM, Appasamy PM, Lotze MT, Storkus WJ, Appella E, DeLeo AB. Therapy of murine tumors with p53 wild-type and mutant sequence peptide-based vaccines. J Exp Med. 1996;183:1357–1365. doi: 10.1084/jem.183.4.1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Röpke M, Hald J, Guldenberg P, Zeuthen J, Norgaard L, Fugger L, Svejgaard A, Van der Burg S, Nijman HW, Melief CJM, Claesson MH. Spontaneous human squamous cell carcinomas are killed by a human cytotoxic T lymphocyte clone recognizing a wild-type p53- derived peptide. Proc Natl Acad Sci USA. 1996;93:14704–14707. doi: 10.1073/pnas.93.25.14704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bertholet S, Iggo R, Corradin G. Cytotoxic T lymphocytes responses to wild-type and mutant mouse p53 peptides. Eur J Immunol. 1997;27:798–801. doi: 10.1002/eji.1830270332. [DOI] [PubMed] [Google Scholar]
- 33.Lacabanne V, Viguier M, Guillet J-G, Choppin J. A wild-type p53 cytotoxic T cell epitope is presented by mouse hepatocarcinoma cells. Eur J Immunol. 1996;26:2635–2639. doi: 10.1002/eji.1830261114. [DOI] [PubMed] [Google Scholar]
- 34.Dahl AM, Beverley PCL, Stauss HJ. A synthetic peptide derived from the tumor-associated protein mdm-2 can stimulate autoreactive, high avidity cytotoxic T lymphocytes that recognize naturally processed protein. J Immunol. 1996;157:239–246. [PubMed] [Google Scholar]
- 35.Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA, Butel JS, Bradley A. Mice deficient for p53 developmentally normal but susceptible to spontaneous tumors. Nature (Lond) 1992;356:215–221. doi: 10.1038/356215a0. [DOI] [PubMed] [Google Scholar]
- 36.Feltkamp MCW, Smits HL, Vierboom MPM, Minnaar RP, De Jongh BM, Drijfhout JW, Ter J, Schegget, Melief CJM, Kast WM. Vaccination with a cytotoxic T lymphocyte epitope-containing peptide protects against a tumor induced by human papillomavirus type 16– transformed cells. Eur J Immunol. 1993;23:2242–2249. doi: 10.1002/eji.1830230929. [DOI] [PubMed] [Google Scholar]
- 37.Kaczmarek L, Oren M, Baserga R. Co-operation between the p53 protein tumor antigen and platelet-poor plasma in the induction of cellular DNA synthesis. Exp Cell Res. 1986;62:268–272. doi: 10.1016/0014-4827(86)90445-3. [DOI] [PubMed] [Google Scholar]
- 38.Gorer PA. Studies in antibody response of mice to tumour inoculation. Br J Cancer. 1950;4:372–381. doi: 10.1038/bjc.1950.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sijts EJAM, Ossendorp F, Mengedé EAM, Van den Elsen PJ, Melief CJM. An immunodominant MCF murine leukemia virus–encoded CTL epitope, identified by its MHC class I–binding motif, explains MuLV type specificity of MCF-directed CTL. J Immunol. 1994;152:106–116. [PubMed] [Google Scholar]
- 40.Kast WM, De Waal LP, Melief CJM. Thymus dictates major histocompatibility complex (MHC) specificity and immune response gene phenotype of class II MHC-restricted T cells but not of class I MHC-restricted T cells. J Exp Med. 1984;160:1752–1760. doi: 10.1084/jem.160.6.1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Feltkamp MCW, Vierboom MPM, Toes REM, Ossendorp F, Ter J, Schegget, Melief CJM, Kast WM. Competition inhibition of cytotoxic T lymphocyte (CTL) lysis, a ore sensitive method to identify candidate CTL epitopes that antibody-detected MHC class I stabilization. Immunol Lett. 1995;47:1–8. doi: 10.1016/0165-2478(95)00052-7. [DOI] [PubMed] [Google Scholar]
- 42.Ljunggren H-G, Stam N, Öhlen JC, Neefjes JJ, Höglund P, Meemels M-T, Bastin J, Schumacher TNM, Townsend A, Kärre K, Ploegh HL. Empty MHC class I molecules come out in the cold. Nature (Lond) 1990;346:476–480. doi: 10.1038/346476a0. [DOI] [PubMed] [Google Scholar]
- 43.Köhler, G., K. Fisher-Lindahl, and C. Heusser. 1981. Characterization of a monoconal anti H-2k antibody. In The Immune System. C. Steinberg and I. Lefkovits, editors. Karger, Basel. 2:202–208.
- 44.Neefjes JJ, Momburg F, Hämmerling GJ. Selective and ATP-dependent translocation of peptides by the MHC-encoded transporter. Science (Wash DC) 1993;261:769–771. doi: 10.1126/science.8342042. [DOI] [PubMed] [Google Scholar]
- 45.Neisig A, Roelse J, Sijts EJAM, Ossendorp F, Feltkamp MCW, Kast WM, Melief CJM, Neefjes JJ. Major differences in TAP-dependent translocation of MHC class I presentable peptides and the effect of flanking sequences. J Immunol. 1995;154:1273–1279. [PubMed] [Google Scholar]
- 46.Bouma GJ, Van der Meer-Prins PMW, Van Bree FPMJ, Van Rood JJ, Claas FHJ. Determination of cytotoxic T-lymphocyte precursor frequencies using Europium labeling as a nonradioactive alternative to labeling with Chromium-51. Hum Immunol. 1992;35:85–92. doi: 10.1016/0198-8859(92)90015-f. [DOI] [PubMed] [Google Scholar]
- 47.Seed B, Aruffo A. Molecular cloning of the CD2 antigen, the T cell erythrocyte receptor, by a rapid immunoselection procedure. Proc Natl Acad Sci USA. 1987;84:3365–3369. doi: 10.1073/pnas.84.10.3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pennica D, Goeddel DV, Hayflick JS, Reich N, Anderson CW, Levine AJ. The amino acid sequence of murine p53 determined from a c-DNA clone. Virology. 1984;134:477–482. doi: 10.1016/0042-6822(84)90316-7. [DOI] [PubMed] [Google Scholar]
- 49.Falk K, Rötschke O, Stevanovic S, Jung G, Rammensee H-G. Allele-specific motifs revealed by sequencing of self-peptides eluted from MHC molecules. Nature (Lond) 1991;351:290–296. doi: 10.1038/351290a0. [DOI] [PubMed] [Google Scholar]
- 50.Kast, W.M., and C.J.M. Melief. 1991. Fine peptide specificity of cytotoxic T lymphocytes against adenovirus-induced tumours and peptide–MHC binding. Int. J. Cancer. 6(Suppl.): 90–94. [DOI] [PubMed]
- 51.Ohashi PS, Oehen S, Buerki K, Pircher H, Ohashi CT, Odermatt B, Malissen B, Zinkernagel RM, Hengartner H. Ablation of “tolerance” and induction of diabetes by virus infection in viral antigen transgenic mice. Cell. 1991;65:305–317. doi: 10.1016/0092-8674(91)90164-t. [DOI] [PubMed] [Google Scholar]
- 52.Goverman JA, Woods L, Larson LP, Weiner H, Hood L, Zaller DM. Transgenic mice that express a myelin basic protein-specific T cell receptor develop spontaneous autoimmunity. Cell. 1993;72:551–560. doi: 10.1016/0092-8674(93)90074-z. [DOI] [PubMed] [Google Scholar]
- 53.Hu J, Kindsvogel W, Busby S, Bailey MC, Shi Y, Greenberg PD. An evaluation of the potential to use tumor-associated antigens as targets for antitumor T cell therapy using transgenic mice expressing a retroviral tumor antigen in normal lymphoid tissues. J Exp Med. 1993;177:1681–1690. doi: 10.1084/jem.177.6.1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Matzinger P. Tolerance, danger, and the extended family. Annu Rev Immunol. 1994;12:991–1045. doi: 10.1146/annurev.iy.12.040194.005015. [DOI] [PubMed] [Google Scholar]
- 55.Olsson T. Critical influences of the cytokine orchestration on the outcome of myelin antigen-specific T-cell autoimmunity in experimental autoimmune encephalomyelitus and multiple sclerosis. Immunol Rev. 1995;144:245–268. doi: 10.1111/j.1600-065x.1995.tb00072.x. [DOI] [PubMed] [Google Scholar]
- 56.Ridgway WM, Weiner HL, Fathman CG. Regulation of autoimmune response. Curr Opin Immunol. 1994;6:946–955. doi: 10.1016/0952-7915(94)90018-3. [DOI] [PubMed] [Google Scholar]
- 57.van Eden W, Anderton SM, van der Zee R, Prakken BJ, Broeren CPM, Wauben MHM. (Altered) Self peptides and the regulation of self reactivity in the peripheral T cell pool. Immunol Rev. 1996;149:55–73. doi: 10.1111/j.1600-065x.1996.tb00899.x. [DOI] [PubMed] [Google Scholar]
- 58.Roth J, Dittmer D, Rea D, Tartaglia J, Paoletti E, Levine AJ. p53 as a target for cancer vaccines: recombinant canarypox virus vectors expressing p53 protect mice against a lethal tumor cell challenge. Proc Natl Acad Sci USA. 1996;93:4781–4786. doi: 10.1073/pnas.93.10.4781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Milner J. Different forms of p53 detected by monoclonal antibodies in non-dividing and dividing lymphocytes. Nature (Lond) 1984;310:143–145. doi: 10.1038/310143a0. [DOI] [PubMed] [Google Scholar]
- 60.Visseren MJW, Van Elsas A, Van der Voort EIH, Ressing ME, Kast WM, Schrier PI, Melief CJM. CTL specific for the tyrosinase autoantigen can be induced from healthy donor blood to lyse melanoma cells. J Immunol. 1995;154:3991–3998. [PubMed] [Google Scholar]
- 61.Kast WM, Roux L, Curren J, Blom HJJ, Voordouw AC, Meloen RH, Kolakofski D, Melief CJM. Protection against lethal Sendai virus infection by in vivo priming of virus-specific cytotoxic T lymphocytes with an unbound peptide. Proc Natl Acad Sci USA. 1991;88:2283–2287. doi: 10.1073/pnas.88.6.2283. [DOI] [PMC free article] [PubMed] [Google Scholar]