

Abstract
Endoglin is a transforming growth factor β (TGFβ) superfamily auxiliary receptor. We had previously shown that it suppressed prostate cancer (PCa) cell motility, and that its expression was lost during PCa progression. The mechanism by which endoglin inhibits PCa cell motility is unknown. Here we demonstrate that endoglin abrogates TGFβ-mediated cell motility, but does not alter cell surface binding of TGFβ. By measuring Smad-specific phosphorylation and Smad-responsive promoter activity, endoglin was shown to constitutively activate Smad1, with little-to-no effect upon Smad3. Knockdown of Smad1 increased motility and abrogated endoglin's effects. As type I activin receptor-like kinases (ALKs) are necessary for Smad activation, we went on to show that knockdown of ALK2, but not TGFβRI (ALK5), abrogated endoglin-mediated decreases in cell motility and constitutively active ALK2 was sufficient to restore a low-motility phenotype in endoglin deficient cells. These findings provide the first evidence that endoglin decreases PCa cell motility through activation of the ALK2-Smad1 pathway.
Keywords: transforming growth factor β, prostate cancer, motility, endoglin, smad1, ALK2
Introduction
Death from prostate cancer (PCa) is due to the formation of metastases (Carroll et al., 2001). The metastatic behavior of cells is heavily dependent upon the cellular functions of migration and invasion (Poste and Fidler, 1980; Bissell and Radisky, 2001). Using gene arrays to identify regulators of PCa cell motility, we uniquely identified endoglin (ENG) (Jovanovic et al., 2001). ENG is a 180 kDa homodimeric type I transmembrane transforming growth factor β (TGFβ) binding protein (Cheifetz et al., 1992). We demonstrated that ENG expression was lost during PCa cell progression, and that this led to increased cell invasion and migration (Liu et al., 2002). The mechanism by which ENG regulates PCa cell motility is not known.
ENG has been shown to bind TGFβ through interaction with other TGFβ superfamily receptors (Barbara et al., 1999). Canonical TGFβ signaling minimally involves a heteromeric complex involving type II (RII) and type I (RI) serine/threonine kinase receptors, resulting in RI activation, which in turn phosphorylates/activates Smad transcription factors (Shi and Massague, 2003). ENG is considered an accessory TGFβ superfamily receptor, and studies in endothelial cells suggest that it plays a vital role in regulating whether activin receptor-like kinase 1 (ALK1) versus ALK5 RI subtypes are activated in response to TGFβ (Lebrin et al., 2004; Blanco et al., 2005; Pece-Barbara et al., 2005).
The current study was undertaken to elucidate the mechanism by which ENG regulated PCa cell motility. In addition to inhibiting PCa cell motility, we show that ENG abrogates TGFβ-induced cell motility. Further, ENG was shown to activate Smad1, and Smad1 was shown to inhibit PCa cell motility. Finally, Smad1 and ALK2, an RI subtype, were shown to be necessary for ENG's anti-motility effects.
Results
ENG inhibits TGFβ-induced prostate cell motility
Human PC3-M PCa cells possess nearly undetectable levels of ENG and exhibit a high degree of cell motility (migration and invasion) (Liu et al., 2002). In PC3-M cells transfected with either wild-type ENG or empty-control vector (VC), TGFβ1 significantly increased both cell migration and invasion through gelatin in VC cells (Figure 1a and b). However, in ENG cells, migration and invasion were significantly reduced compared to VC cells, and there was complete abrogation of TGFβ-mediated increases.
Figure 1.
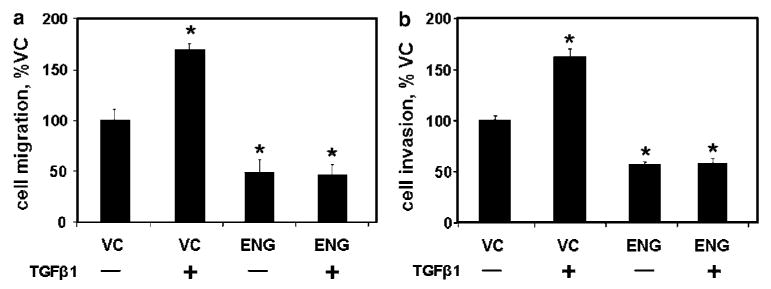
Endoglin (ENG)-mediated inhibition of cell motility is not altered by transforming growth factor β1 (TGFβ1). PC3-M cells were transfected with either ENG or with empty vector (VC) and β-galactosidase (β-gal) then treated with TGFβ1 (or not) for 24 h, as indicated. Cell migration (a) and invasion (b) were measured as described in the Materials and methods. Data are the mean±s.e.m. (N = 4) percent of VC cells from a single experiment; similar results were obtained in replicate experiments. (* denotes two-sided t-test P-value <0.05).
To determine if ENG-associated abrogation of TGFβ-mediated cell motility was a consequence of altered TGFβ1 binding, we utilized flow cytometry to quantify cell surface bound biotinylated TGFβ1 in the presence or absence of ENG (Figure 2). While a fivefold increase in cell surface type II TGFβ receptor (TGFβRII) expression was associated with an increase in TGFβ1 binding, a 13-fold increase in ENG did not alter TGFβ1 binding, compared to VC cells (Figure 2a and b). The specificity of biotinylated TGFβ1 binding was demonstrated by showing that it's binding was much greater than that of either biotinylated negative control protein, or of biotinylated TGFβ1 in the presence of TGFβ1 blocking antibody, in VC, ENG or TGFβRII cells (Figure 2c–e).
Figure 2.
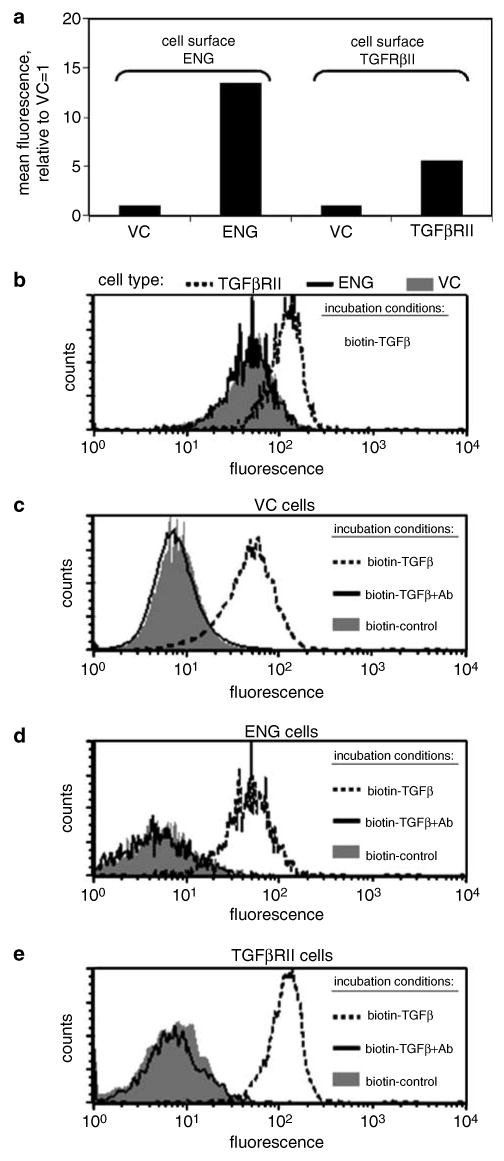
Endoglin (ENG) does not alter cell surface TGFβ1 binding. PC3-M cells were transfected with ENG, type II transforming growth factor β receptor (TGFβRII) or with empty vector (VC), as indicated. (a) Cell surface expression of ENG or TGFβRII was measured by FACS. Values represent the mean fluorescent intensity, normalized to the isotype control, from a single experiment. In (b), TGFβRII (broken line), ENG (solid line) or VC cells (solid gray) were incubated with biotinylated TGFβ1 (biotin-TGFβ), and, after washing, cell-associated biotin was measured by FACS analysis. Next, VC (c), ENG (d) or TGFβRII (e) cells were incubated with biotin-TGFβ (broken line), biotinylated negative control protein (biotin-control; solid gray) or with biotin-TGFβ pre-incubated with TGFβ-blocking antibody (biotin-TGFβ+Ab; solid line), and cell-associated biotin was measured by FACS analysis. Histograms from a single experiment are depicted. Cell transfection and FACS analysis were performed as described in the Materials and methods. All experiments were repeated with similar results.
ENG promotes Smad1 signaling
We next evaluated the ability of ENG to activate Smads. In the absence of TGFβ1, phosphorylated Smad1, -2 and -3 (pSmad1, -2, -3) levels were all low in VC cells, while in ENG cells, pSmad1 levels were elevated (Figure 3a). In the presence of TGFβ1, levels of pSmad1, -2 and -3 all increased in both VC and ENG cells. However, comparison of VC and ENG cells treated with TGFβ1 reveals that in ENG cells pSmad1 levels were elevated, whereas there was little-to-no change in pSmad2 and -3 levels. Importantly, the levels of total Smad1, -2 and -3 protein were unaltered by TGFβ1 treatment or ENG status.
Figure 3.
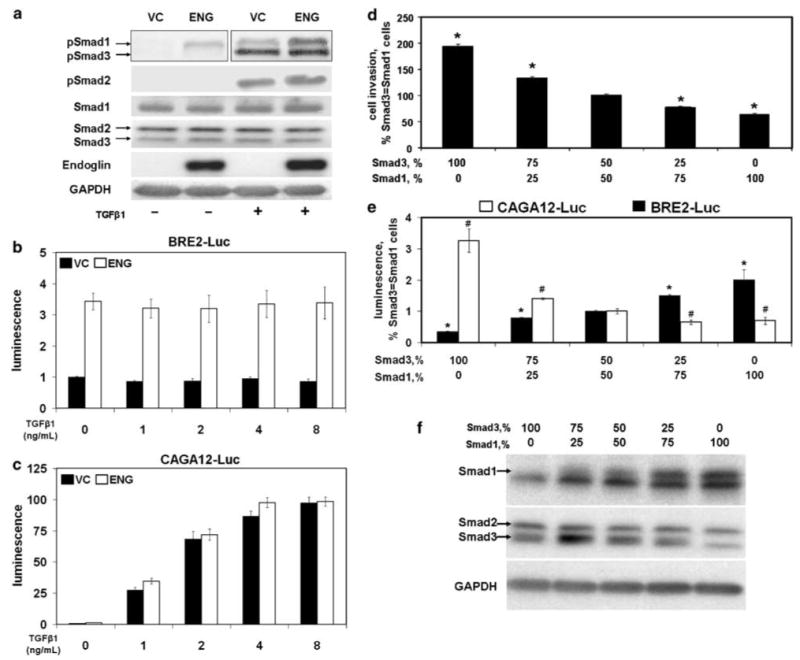
Smad1 is activated by endoglin (ENG) and inhibits cell invasion. (a) ENG enhances phosphorylation of Smad1. PC3-M cells were transfected with ENG or VC, as in Figure 1. After treatment of cells with TGFβ1 (or not) for 30 min, resultant cell lysates were then probed by western blot for phosphorylated-Smad1, -2 and -3 (pSmad1, -2 and -3); total Smad1, -2 and -3 (Smad1, -2 and -3) and GAPDH, as described in the Materials and methods. Note: the pSmad1 and -3 blot for cells not treated with TGFβ1 was exposed longer than the TGFβ1 treated group in an attempt to detect pSmad3. (b and c) ENG enhances Smad1 promoter activity. Cells were co-transfected with ENG or VC, β-gal and BRE2-luciferase (BRE2-Luc) (b) or CAGA12-luciferase (CAGA12-Luc) (c). Six hours after changing to serum-free media, cells were treated with the indicated concentration of TGFβ1 for 18 h, and luciferase activity measured. VC cells not treated with TGFβ1 were normalized to 1.0 for each reporter construct. (d) Smad3 promotes cell invasion and Smad1 decreases it. Cells were transfected with the indicated ratios of Smad3 and -1, along with β-gal, and cell invasion measured. Data are the mean±s.e.m. (N = 4) of a single experiment, with similar results seen in a separate experiment (also N = 4). Compared to cells transfected with equal amounts of Smad3 and -1, * denotes values that differ by P<0.05 (two-sided t-test). The effects of different ratios of transfected Smad3 and -1, upon promoter activity (e; luciferase assay) and protein expression (f; western blot) are depicted. For promoter activity, cells were co-transfected with BRE2-Luc or CAGA12-Luc, as indicated. * and # denote values for BRE2-Luc and CAGA12-Luc, respectively, which differ from cells transfected with equal amounts of Smad3 and -1 with a P-value of <0.05. For all promoter assays, luciferase activity was normalized to β-gal; depicted values represent the mean±s.e.m. of two experiments performed on different days (N = 2 for each experiment).
The functional significance of ENG-mediated activation of Smad1 was evaluated next by measuring Smad1 and Smad3 promoter activity. As shown in Figure 3b, ENG increased Smad1-responsive BRE2-luciferase (BRE2) activity over threefold relative to VC. Importantly, exogenous TGFβ1 did not affect BRE2 promoter activity, nor did it affect ENG-mediated activation of BRE2. In contrast, ENG had little-to-no effect on Smad3-responsive CAGA12-luciferase (CAGA12) activity (Figure 3c). Further, TGFβ1-mediated activation of CAGA12 was concentration dependent, and was essentially unaffected by ENG status. These findings demonstrate that ENG can selectively enhance Smad1 signaling, and that was not affected by exogenous TGFβ1 concentrations, which activate Smad3 signaling.
The above findings supported the notion that Smad1 and Smad3 have opposing effects upon cell motility. If this were true, then relative increases in Smad3 should promote motility, while relative increases in Smad1 should inhibit it. This was shown to be the situation by transfecting cells with different ratios of Smad3 and Smad1 plasmid, and measuring the effect upon cell invasion (Figure 3d). Compared to the situation where equal amounts of Smad3 and Smad1 were used, increases in Smad3 increased invasion by nearly two-fold, while increases in Smad1 decreased invasion to 64%. By performing Smad1 and -3 promoter assays (Figure 3e), and by measuring protein expression by western blot (Figure 3f), the functional activity and protein expression level of individual Smad constructs was shown to vary in accordance with the amount of construct used during transfection. These findings further support the notion that Smad3 promotes cell motility, while Smad1 inhibits it. They also suggest that the ratio of Smad3 to Smad1 is an important determinant of motility.
Smad1 is necessary for endoglin-mediated regulation of cell motility
A series of experiments were performed to determine if Smad1 was necessary for ENG-mediated inhibition of PCa cell motility. Cell migration (Figure 4a) and invasion (Figure 4b) were decreased by either ENG or Smad1, and further decreased by the combination of ENG and Smad1. Importantly, with ENG, Smad1 or the combination of ENG and Smad1, similar levels of cell motility were observed irrespective of TGFβ1 treatment status. In contrast, in VC cells, TGFβ1 increased motility. These findings demonstrate that Smad1 and ENG have similar effects upon cell motility, and raise the notion that Smad1 may be necessary for ENG function.
Figure 4.
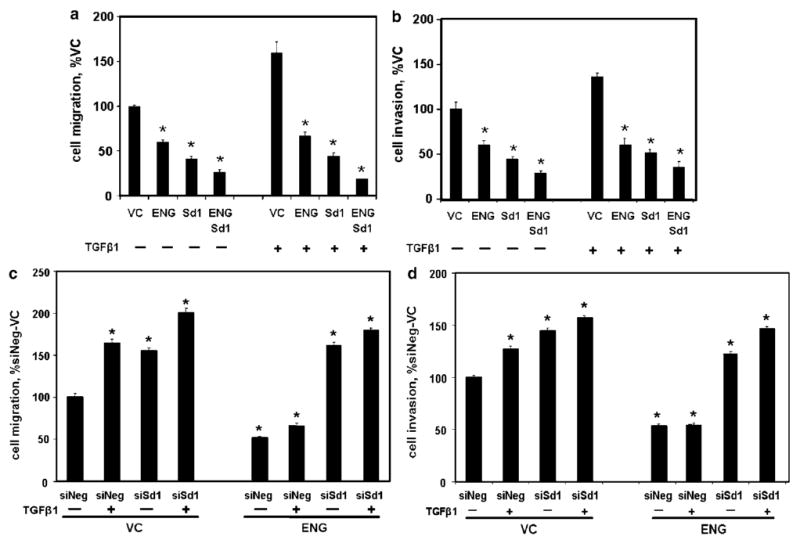
Smad1 is necessary for endoglin (ENG)-mediated inhibition of cell motility. (a and b) ENG and Smad1 inhibit cell motility. PC3-M cells were transfected with VC, ENG, Smad1 (Sd1) or ENG and Sd1, then treated with TGFβ1 for 24 h, as indicated, and cell migration (a) and invasion (b) measured, as in Figure 1, and expressed as the percent of the corresponding VC cells. (c and d) Smad1 is necessary for ENG function. Cells were transfected with ENG or VC followed by either siRNA to Smad1 (siSd1) or nontargeting siRNA (siNeg), treated with TGFβ1 and cell migration (c) and invasion (d) measured, and expressed as the percent of non-TGFβ1-treated control cells (siNeg/VC). All values are the mean±s.e.m. (N = 4) of a single experiment, and repeated (also N = 4), with similar results. *P-value of <0.05, compared to control.
If Smad1 was necessary for ENG function, then its knockdown by small-interfering RNA (siRNA) should abrogate ENG-mediated effects upon cell motility. The efficacy and specificity of siRNA to Smad1 (siSd1) was first confirmed (Supplementary Figure 1). Knockdown of Smad1 was then shown to significantly increase cell migration (Figure 4c) and invasion (Figure 4d) in the presence or absence of TGFβ1 in VC cells, compared to cells treated with nontargeting siRNA (siNeg). Importantly, while ENG decreased both migration and invasion, siSd1 completely abrogated ENG's effects, in the presence or absence of TGFβ1. These findings demonstrate that Smad1 is necessary for ENG-mediated inhibition of PCa cell motility.
ENG-mediated regulation of cell motility requires the ALK2 receptor
ENG lacks a consensus kinase domain (Gougos and Letarte, 1990) making it unlikely that it directly activates Smad1. However, the TGFβ superfamily RI subtypes, ALK1 and ALK5, have kinase activity, and in endothelial cells have been shown to interact with ENG, resulting in augmented Smad signaling (Lebrin et al., 2004; Blanco et al., 2005). Utilizing quantitative reverse transcription (qRT)-PCR, ALK1 was undetectable in PC3 and PC3-M PCa cells (Figure 5a). ALK2 has the highest sequence homology with ALK1 (ten Dijke et al., 1993). Further, ALK1 and ALK2 bind common ligands, interact with common RII subtypes, and mediate TGFβ-associated function (Attisano et al., 1993; Ebner et al., 1993a, b; Miettinen et al., 1994; Ward et al., 2002; Goumans et al., 2003; Lebrin et al., 2004; Blanco et al., 2005; Desgrosellier et al., 2005). Using qRT-PCR, we found that ALK2 was expressed at high levels in PCa cells, and was over fourfold higher than ALK5 in both PC3 and PC3-M cells (Figure 5a).
Figure 5.
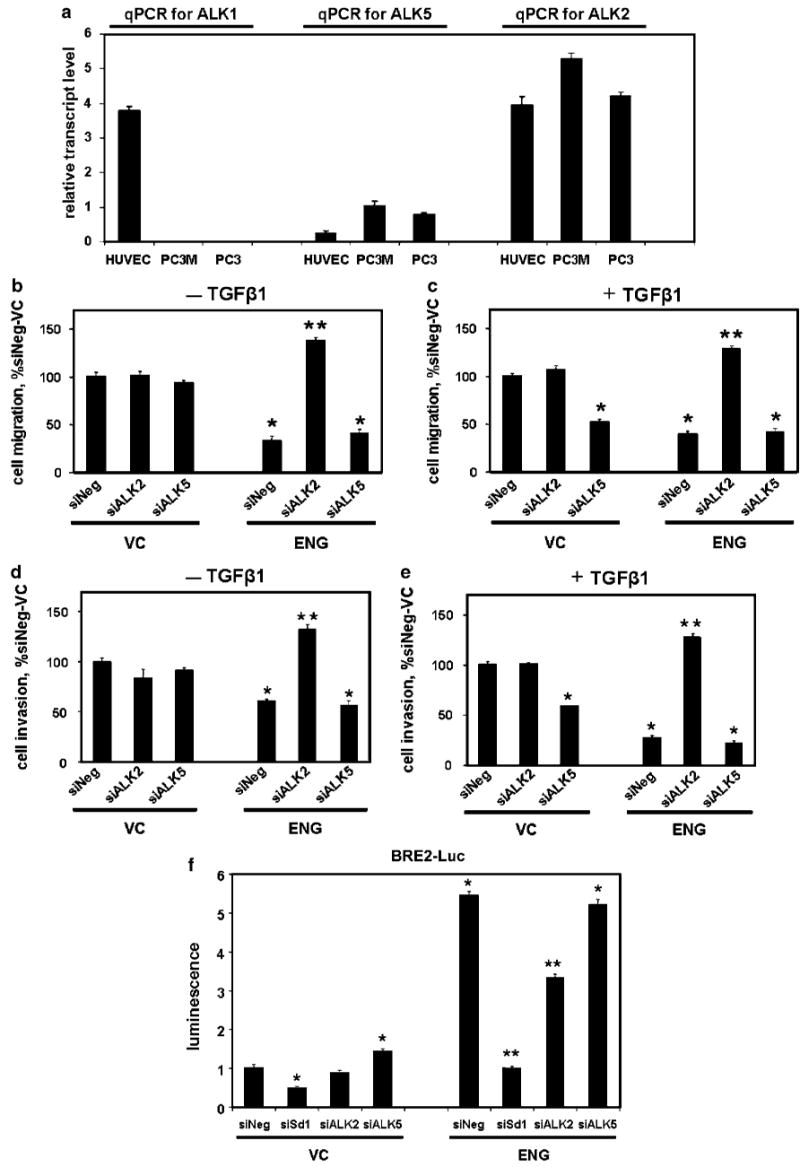
Activin receptor-like kinase 2 (ALK2) is necessary for endoglin (ENG) function. (a) Human prostate cancer cells express ALK2, but not ALK1. ALK1, ALK2 and ALK5 transcript levels were measured by quantitative reverse transcription (qRT)-PCR in PC3, PC3-M and human umbilical vein endothelial cells (HUVEC) cells (positive control), as described in the Materials and methods. Mean±s.d. relative transcript levels (normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH)) are depicted. (b–e) ALK2 is necessary for ENG-mediated decreases in cell motility. PC3-M cells were transfected with ENG or empty-control vector (VC), along with β-gal, then with siALK2, siALK5 or siNeg, treated with TGFβ1 (c and e), or not (b and d) and cell migration (b and c) and invasion (d and e) measured. Data are the mean±s.e.m., expressed as percent siNeg/VC. (f) ALK2 supports ENG-mediated activation of Smad1. Cells were transfected with ENG or VC, along with β-gal and BRE2-Luc, and then with siSd1, siALK2, siALK5 or siNeg. Luciferase activity was measured (normalized to β-gal) and expressed as the mean±s.e.m. relative to VC-siNeg. (g–i) Constitutively active ALK2 induces the ENG phenotype. Cells were transfected with HA-ALK2, HA-caALK2, ENG or VC, and cell invasion (g), luciferase activity (h) and HA-, ENG and GAPDH protein expression, by western blot (i), were measured. In (h), cells were co-transfected with β-gal and BRE2-Luc. Values are from a single experiment (replicates of N = 2 for qRT-PCR and luciferase; N = 4 for motility assays), and all experiments were repeated with similar findings. *P-value of <0.05, compared to siNeg/VC or to VC/VC (for G and H); **P-value of <0.05, compared to siNeg/ENG.
The importance of ALK2 and ALK5 in ENG-mediated effects upon cell motility was evaluated next. ALK2 (siALK2) and ALK5 (siALK5) siRNA was first shown to effectively and specifically suppress ALK2 and ALK5 transcript levels, respectively (Supplementary Figure 2). As can be seen in Figure 5b–e, ENG decreased cell migration and invasion in siNeg (nontargeting siRNA) cells in either the presence or absence of TGFβ1. Importantly, siALK2 abrogated ENG's anti-motility effects, and in fact increased motility to levels above those observed with VC cells. Interestingly, siALK2 had little-to-no effect upon cell motility in VC cells, consistent with ALK2-mediated regulation of cell motility being dependent upon ENG. In contrast, siALK5 had no impact upon ENG function in the presence or absence of TGFβ1. However, in VC cells, siALK5 decreased cell motility only in the presence of TGFβ1, consistent with the pro-motility role of TGFβ-ALK5. These findings demonstrate that ALK2 is necessary for ENG-mediated suppression of cell motility in human PCa.
Studies next evaluated the dependency of ENG upon ALK2, for activation of the Smad1-responsive BRE2 promoter. In VC cells, BRE2 activity was low overall, was decreased by siSd1, was unaffected by siALK2, and was slightly increased by siALK5 (Figure 5f). In ENG cells, promoter activity increased by more than fivefold compared to VC-siNeg cells. Importantly, in ENG cells, siALK2 and siSd1 both reduced Smad1 promoter activity, as compared to siNeg, and siALK5 had no appreciable effect. These findings demonstrate that ALK2 supports ENG-mediated Smad1 activation.
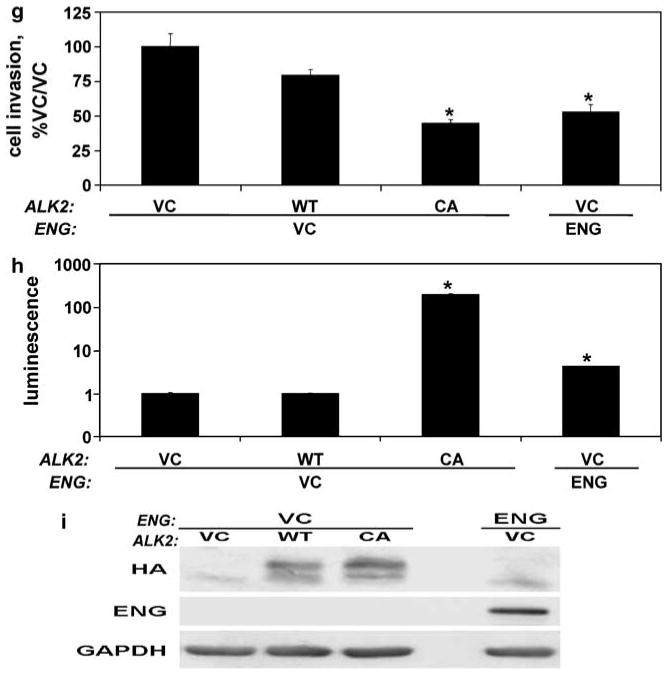
To determine whether constitutively active ALK2 could induce an ENG phenotype in the absence of ENG, cells were transfected with wild-type ALK2 (HA-ALK2), constitutive active ALK2 (HA-caALK2) or with ENG, and cell invasion and Smad1 promoter activation were measured (Figure 5g–i). HA-caALK2 decreased the invasion of ENG-deficient cells, and did so to the same degree as was seen by ENG replacement (Figure 5g). Similarly, both HA-caALK2 and ENG significantly increased Smad1 promoter activity (Figure 5h).
Effects in other prostate cells
To rule out the possibility that the above studies were limited to PC3-M cells, a series of investigations were performed with PC3 PCa cells. Compared to PC3-M cells, PC3 cells express higher levels of ENG (Liu et al., 2002), are less invasive (Huang et al., 2005), have a lower propensity to detach (Bergan et al., 1996b), are less metastatic (Kozlowski et al., 1984) and are ∼10-fold less sensitive to TGFβ (as assessed by growth inhibition) (Liu et al., 2001).
ENG decreased invasion, and abrogated TGFβ-mediated increases in invasion, in PC3 cells (Figure 6a). Smad1 phosphorylation was enhanced by ENG, in the presence or absence of TGFβ1, with little-to-no effect upon Smad2 or Smad3 phosphorylation (Figure 6b). Using siRNA to knockdown ALK2, ALK5 or Smad1, as indicated, we then confirmed that Smad1 and ALK2 were necessary for ENG-mediated decreases in cell invasion, in the presence or absence of TGFβ1 (Figure 6c and d). Also, similar to PC3-M cells, knockdown of ALK5 inhibited TGFβ1-mediated cell invasion in VC cells while having little-to-no effect in ENG expressing cells (Figure 6d). Finally, constitutive active ALK2 decreased invasion and increased Smad1 promoter activity in ENG deficient PC3 cells, thus giving them the same phenotype as ENG replete cells (Figure 6e–g). In total, findings in PC3 cells corroborate those found in PC3-M cells.
Figure 6.
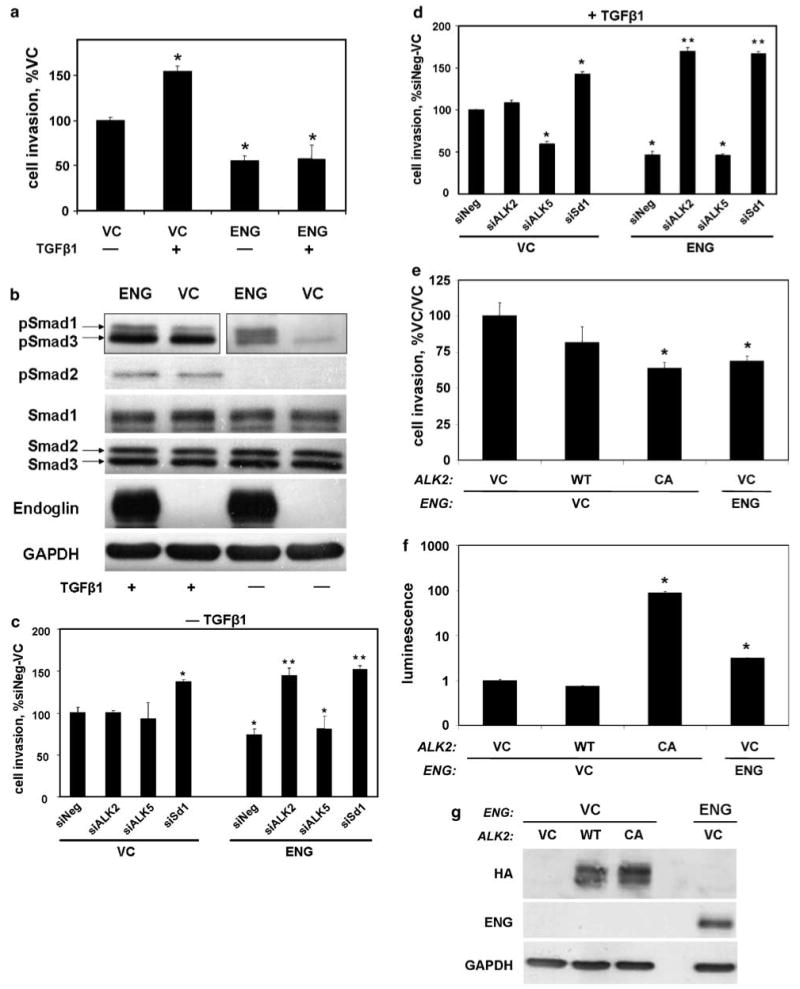
The activin receptor-like kinase 2 (ALK2)-Smad1 pathway is important for endoglin (ENG) (a) ENG inhibits cell invasion. PC3 cells were transfected with either ENG or with empty vector (VC), along with β-gal, and treated with transforming growth factor β1 (TGFβ1) (or not) for 24 h. Depicted are the mean±s.e.m. number of invading cells as a percent of no TGFβ1 vector control. (b) ENG increases Smad1 phosphorylation. Cells were transfected with either ENG or VC and treated with TGFβ (or not) for 30 min, and the resultant cell lysates were probed for the indicated phospho- and total protein species by western blot. Note the blot for pSmad1 and -3, for cells not treated with TGFβ1, was exposed longer than the TGFβ1 treated group in an attempt to detect pSmad3. (c and d) ENG-mediated inhibition of cell invasion is dependent upon ALK2 and Smad1. Cells were transfected as indicated, as in Figure 5, and treated with TGFβ (d) for 24 h, or not (c). Depicted are the mean±s.e.m. number of invading cells as a percent of siNeg/VC for a single experiment. (e–g) Constitutively active ALK2 induces the ENG phenotype. Cells were transfected with HA-ALK2, HA-caALK2, ENG or VC, and cell invasion (e), luciferase activity; normalized to β-gal (f), and HA-, ENG and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) protein expression, by western blot (g), were measured. In (f), cells were co-transfected with β-gal and BRE2-Luc. All experiments (N = 4 replicates for invasion) were repeated with similar results. *P-value <0.05, compared to VC (a), siNeg/VC (c and d) or VC/VC (e and f); **P-value of <0.05, compared to siNeg/ENG.
Discussion
We demonstrate for the first time that ENG abrogates TGFβ-mediated increases in PCa cell motility. Both Smad1 and ALK2 were shown to be necessary for ENG-mediated suppression of cell motility. Though Smad1 activity has previously been linked to the presence of ENG (Lebrin et al., 2004; Blanco et al., 2005; Pece-Barbara et al., 2005), this was the first time that it had been shown to be necessary for ENG function. Further, it is also the first time that ALK2 has been linked to ENG function. Finally, to our knowledge this is the first time Smad1 and ALK2 have been shown to suppress PCa cell motility. From these findings, we propose a model for ENG-mediated regulation of PCa cell motility (Figure 7). In this model, ENG expression facilitates ALK2-mediated Smad1 activation, thereby suppressing cell motility. During PCa progression, ENG expression is lost (Liu et al., 2002). Under conditions of low ENG expression, TGFβ-mediated activation of the ALK5-Smad3 pathway predominates, thereby increasing cell motility.
Figure 7.

Model of endoglin (ENG)-mediated regulation of prostate cell motility.
The current findings neither prove nor refute independent activation of the TGFβ-ALK5-Smad3-pro-motility and the ENG-ALK2-Smad1-anti-motility pathways. In particular, ENG was shown to selectively activate Smad1, as compared to Smad3. Conversely, TGFβ was shown to activate Smad3 promoter activity, but not Smad1 promoter activity. These findings suggest that Smad1 and Smad3 are independently activated. However, TGFβ did in fact increase Smad1 phosphorylation, thus providing evidence of cross-talk between the pathways. Also, the ratio of Smad3 to Smad1 appeared to be an important determinant of cell motility, thus suggesting interaction between the pathways. Given that TGFβ-mediated phosphorylation of Smad1 did not translate into increased Smad1 promoter activity, additional regulatory mechanisms are implicated. Ongoing investigations are seeking to elucidate these co-regulatory mechanisms.
In the current system, ENG did not alter TGFβ binding to the cell surface. Lastres et al. (1996) also found that the long isoform of ENG (used here) did not alter TGFβ binding, while Letamendia et al. (1998) found that ENG slightly increased TGFβ binding. Although we have shown that ENG does not affect overall TGFβ surface binding, and that TGFβ does not alter ENG's regulation of Smad1 promoter activation or cell motility, we cannot completely rule out the possibility that ENG's effects do not require TGFβ, or that of another ligand.
In accordance with our findings in PCa cells, others have shown in endothelial cells that ENG promotes Smad1 activity (Lebrin et al., 2004; Blanco et al., 2005). However, in contrast to our findings, those groups found that ENG-mediated Smad1 promoter activation was enhanced by TGFβ. Importantly, though TGFβ-mediated activation of Smad1 promoter activity differs between endothelial and PCa cells, within a given cell type, responses are consistent with the corresponding ENG function studies. Specifically, in endothelial cells, both ENG function and Smad1 promoter activation are responsive to TGFβ (Lebrin et al., 2004). In contrast, in PCa cells we show that both ENG function and Smad1 promoter activation are unaffected by exogenous TGFβ. Finally, Lebrin et al. (2004) reported that in endothelial cells, TGFβ-mediated Smad1 phosphorylation is associated with Smad1 promoter activation. In contrast, in PCa cells we show that TGFβ-mediated Smad1 phosphorylation is not associated with Smad1 promoter activation. These findings support the notion that similar pathways may have important differences in different cell types. Thus, they highlight the importance of evaluating pathway-specific function in a given cell type, and the potential pitfalls associated with generalization of function.
In light of these cell type-specific differences, we thought it important to elucidate a mechanism by which ENG promoted Smad1 signaling. We found that the mechanism in PCa cells was, in fact, different from that described in endothelial cells. Specifically, the RI subtype, ALK2, was shown to facilitate ENG-mediated activation of Smad1 promoter activity and function in PCa cells. In endothelial cells, it is ALK1 that facilitates ENG function (Lebrin et al., 2004; Blanco et al., 2005). We were unable to detect ALK1 expression in PCa cells, but found that ALK2 is highly expressed. As ALK1 and ALK2 are highly homologous (ten Dijke et al., 1993), it is not surprising that they perform related functions. It is likely that differences between ALK1 and ALK2 contribute, at least in part, to differences in ENG function observed between endothelial and PCa cells.
ALK5 represents the canonical TGFβ superfamily RI subtype which activates Smad3 (Massague, 1998). Thus, it was thus not surprising that we found that Smad3 enhanced cell motility and that loss of ALK5 abrogated TGFβ-induced cell motility. Our data suggest that ENG functions independently of ALK5 in PCa cells. This finding was surprising because in endothelial cells, ALK5 appears to be important for ENG-ALK1-mediated function (Lebrin et al., 2004). Though we have not proven that ENG interacts with a TGFβ family receptor complex in human prostate cells, a potential explanation is that ENG may facilitate ALK2-homodimer interaction with an RII. It will be important to decipher the mechanism by which ENG, ALK2 and Smad1 functionally interact to inhibit PCa cell motility. These studies are currently under way.
In summary, ENG-mediated decreases in PCa cell motility were shown to be facilitated by ALK2 and Smad1. Loss of ENG expression seen with PCa progression would thus minimize opposition to the TGFβ-ALK5-Smad3-pro-motility pathway, thereby tipping the balance in favor of increased cell motility. Additional studies will be necessary in order to understand interactions between these two pathways likely taking place at the level of cell surface receptors, as well as at the level of Smad subtype activation.
Materials and methods
Antibodies
Anti-ENG-phycoerythrin (PE) (R&D Systems, Minneapolis, MN, USA), anti-ENG (clone 35; BD Biosciences, San Jose, CA, USA), anti-phosphorylated-Smad3,-1 (detects Smad3 Ser423/425/Smad1 Ser463/46) and anti-Smad2,-3 (Cell Signaling Technology, Danvers, MA, USA), anti-phosphorylated Smad2 (detects Ser465/467); Chemicon, Temecula, CA, USA), anti-Smad1 (Upstate Biotechnology, Lake Placid, NY, USA), anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH, clone: CSA-335E; Stressgen, Victoria, CA, USA), anti-HA (Santa Cruz Biotechnology, Santa Cruz, CA, USA). Anti-mouse, anti-rabbit IgG-horseradish peroxidase from Amersham Pharmacia Biotech (Piscataway, NJ, USA). Recombinant human TGFβ1 from R&D Systems. Vectors: β-galactosidase (pCMV-β-gal; Stratagene, La Jolla, CA, USA); BRE2-luciferase pGL3 and CAGA12-luciferase pGL3 (described and provided by Peter ten Dijke, Netherlands Cancer Institute (Dennler et al., 1998; Monteiro et al., 2004)); Smad1 pCDNA3.1 (described and provided by Mark de Caestecker, Vanderbilt-Ingram Cancer Center (de Caestecker et al., 1997)); Smad3 pCS2, HA-tagged ALK5 pCMV5 and TGFβRII pCMV5 (described and provided by Joan Massague, Howard Hughes Medical Institute (Wrana et al., 1992; Wieser et al., 1995; Ulloa et al., 1999)); ENG-long isoform pcDNA3 (described by us (Liu et al., 2002)); HA-ALK2 pCMV5 (provided by Andreas Lux, University of Applied Sciences Mannheim, Germany and described by Jeff Wrana (Attisano et al., 1993); constitutively active (Q207D) ALK2 pCMV5 (caALK2) was engineered by site-specific mutagenesis using the Quickchange II (Stratagene, La Jolla, CA, USA) system. Constructs were confirmed by sequencing.
Cell culture and transfection
The origin and culture conditions for PC3 and PC3-M established cell lines have been described (Bergan et al., 1996b). Transient transfection of plasmids was performed with Mirus LT1 transfection reagent (Mirus, Madison, WI, USA) as per the manufacturer's instructions. Transfection of Smad1 (catalog#: M-012732-00-0005), ALK2 (M-004924-01-0005), ALK5 (M-003929-01-0005) and negative control (D-001210-01-05) SMARTpool siRNA-utilized DharmaFECT (all from Dharmacon, Lafayette, CO, USA) was performed 5h post-plasmid transfection as per the manufacturer's instructions.
Cell migration and invasion assays
Cell invasion and migration assays were performed as described, with modifications (Huang et al., 2005). Cells were co-transfected with β-gal and expression vector. Cells invaded through a gelatin-coated Nuclepore Track-Etch Membrane, with 8 μm pores (Whatman, Clifton, NJ, USA), toward serum-free NIH-3T3 conditioned medium. Migration assays utilized uncoated membranes. Transfected cells were visualized with a β-gal staining kit (Stratagene), and the percent invaded-transfected cells counted.
Flow cytometry
Flow cytometric analysis (FACS) was performed as described, with modifications (Bergan et al., 1996a). Cell surface TGFβ1, ENG and TGFβRII were detected using a TGFβ1 flurokine assay kit (containing biotinylated-TGFβ1, TGFβ1-blocking antibody and biotinylated-negative control protein), anti-ENG-PE-conjugated IgG and anti-TGFßRII-PE IgG, respectively (all from R&D Systems) as per the manufacturer's instructions. Median fluorescent intensity was determined on a Beckman Coulter (Fullerton, CA, USA) Epics-XL-MCL flow cytometry machine.
Western blot
24 h after plating, cells were serum starved for 8 h, then treated with 2 ng/ml TGFβ1 (or not) for 30 min before cell lysis. Western blotting of equal amounts of resultant protein was performed as described (Xu et al., 2006).
Smad promoter luciferase reporter assays
Cells were co-transfected with the indicated luciferase reporter and β-gal. Luciferase and β-gal activity were measured as described (Hayes et al., 2003), using luciferase and β-galactosidase Assay Systems (Promega, San Luis Obispo, CA, USA) as per the manufacturer's instructions.
qRT-PCR
RNA isolation and real time qRT-PCR were performed as described (Ding et al., 2006). Reactions were run in duplicate on a single Applied Biosystems 7500 Real Time PCR workstation, using a TaqMan universal PCR kit and validated gene-specific exon spanning primers and probe sets (all from Applied Biosystems, Foster City, CA, USA). Gene expression was normalized to GAPDH.
Supplementary Material
Supplementary Information accompanies the paper on the Oncogene Web site (http://www.nature.com/onc).
Acknowledgments
We express our gratitude to Dr Shan Chen for her many helpful suggestions throughout the manuscript's preparation. This work was funded by the following grants to Raymond C Bergan: a merit review award from the Veterans Administration and a Specialized Program of Research Excellence (SPORE) grant CA90386, from the National Cancer Institute, National Institutes of Health, Department of Health and Human Services. Clarissa S Craft was funded by a training grant, T32CA09560, from the National Institutes of Health.
References
- Attisano L, Carcamo J, Ventura F, Weis FM, Massague J, Wrana JL. Identification of human activin and TGF beta type I receptors that form heteromeric kinase complexes with type II receptors. Cell. 1993;75:671–680. doi: 10.1016/0092-8674(93)90488-c. [DOI] [PubMed] [Google Scholar]
- Barbara NP, Wrana JL, Letarte M. Endoglin is an accessory protein that interacts with the signaling receptor complex of multiple members of the transforming growth factor-beta superfamily. J Biol Chem. 1999;274:584–594. doi: 10.1074/jbc.274.2.584. [DOI] [PubMed] [Google Scholar]
- Bergan R, Hakim F, Schwartz GN, Kyle E, Cepada R, Szabo JM, et al. Electroporation of synthetic oligodeoxy-nucleotides: a novel technique for ex vivo bone marrow purging. Blood. 1996a;88:731–741. [PubMed] [Google Scholar]
- Bergan R, Kyle E, Nguyen P, Trepel J, Ingui C, Neckers L. Genistein-stimulated adherence of prostate cancer cells is associated with the binding of focal adhesion kinase to beta-1-integrin. Clin Exp Metastasis. 1996b;14:389–398. doi: 10.1007/BF00123398. [DOI] [PubMed] [Google Scholar]
- Bissell MJ, Radisky D. Putting tumours in context. Nat Rev Cancer. 2001;1:46–54. doi: 10.1038/35094059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco FJ, Santibanez JF, Guerrero-Esteo M, Langa C, Vary CP, Bernabeu C. Interaction and functional interplay between endoglin and ALK-1, two components of the endothelial transforming growth factor-beta receptor complex. J Cell Physiol. 2005;204:574–584. doi: 10.1002/jcp.20311. [DOI] [PubMed] [Google Scholar]
- Carroll PR, Lee KL, Fuks ZY, Kantoff PW. In: CANCER: Principals and Practices of Oncology. Devita Vt, Hellman S, Rosenberg Sa., editors. Lippincott-Raven; New York: 2001. pp. 1418–1479. [Google Scholar]
- Cheifetz S, Bellon T, Cales C, Vera S, Bernabeu C, Massague J, et al. Endoglin is a component of the transforming growth factor-beta receptor system in human endothelial cells. J Biol Chem. 1992;267:19027–19030. [PubMed] [Google Scholar]
- de Caestecker MP, Hemmati P, Larisch-Bloch S, Ajmera R, Roberts AB, Lechleider RJ. Characterization of functional domains within Smad4/DPC4. J Biol Chem. 1997;272:13690–13696. doi: 10.1074/jbc.272.21.13690. [DOI] [PubMed] [Google Scholar]
- Dennler S, Itoh S, Vivien D, ten Dijke P, Huet S, Gauthier JM. Direct binding of Smad3 and Smad4 to critical TGF beta-inducible elements in the promoter of human plasminogen activator inhibitor-type 1 gene. EMBO J. 1998;17:3091–3100. doi: 10.1093/emboj/17.11.3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desgrosellier JS, Mundell NA, McDonnell MA, Moses HL, Barnett JV. Activin receptor-like kinase 2 and Smad6 regulate epithelial-mesenchymal transformation during cardiac valve formation. Dev Biol. 2005;280:201–210. doi: 10.1016/j.ydbio.2004.12.037. [DOI] [PubMed] [Google Scholar]
- Ding Y, Xu L, Chen S, Jovanovic BD, Helenowski IB, Kelly DL, et al. Characterization of a method for profiling gene expression in cells recovered from intact human prostate tissue using RNA linear amplification. Prostate Cancer Prostatic Dis. 2006;9:379–391. doi: 10.1038/sj.pcan.4500888. [DOI] [PubMed] [Google Scholar]
- Ebner R, Chen RH, Lawler S, Zioncheck T, Derynck R. Determination of type I receptor specificity by the type II receptors for TGF-beta or activin. Science. 1993a;262:900–902. doi: 10.1126/science.8235612. [DOI] [PubMed] [Google Scholar]
- Ebner R, Chen RH, Shum L, Lawler S, Zioncheck TF, Lee A, et al. Cloning of a type I TGF-beta receptor and its effect on TGF-beta binding to the type II receptor. Science. 1993b;260:1344–1348. doi: 10.1126/science.8388127. [DOI] [PubMed] [Google Scholar]
- Gougos A, Letarte M. Primary structure of endoglin, an RGD-containing glycoprotein of human endothelial cells. J Biol Chem. 1990;265:8361–8364. [PubMed] [Google Scholar]
- Goumans MJ, Valdimarsdottir G, Itoh S, Lebrin F, Larsson J, Mummery C, et al. Activin receptor-like kinase (ALK)1 is an antagonistic mediator of lateral TGFbeta/ALK5 signaling. Mol Cell. 2003;12:817–828. doi: 10.1016/s1097-2765(03)00386-1. [DOI] [PubMed] [Google Scholar]
- Hayes SA, Huang X, Kambhampati S, Platanias LC, Bergan RC. p38 MAP kinase modulates Smad-dependent changes in human prostate cell adhesion. Oncogene. 2003;22:4841–4850. doi: 10.1038/sj.onc.1206730. [DOI] [PubMed] [Google Scholar]
- Huang X, Chen S, Xu L, Liu YQ, Deb DK, Platanias LC, et al. Genistein inhibits p38 MAP kinase activation, MMP-2, and cell invasion in human prostate epithelial cells. Cancer Res. 2005;65:3470–3478. doi: 10.1158/0008-5472.CAN-04-2807. [DOI] [PubMed] [Google Scholar]
- Jovanovic BD, Huang S, Liu Y, Naguib KN, Bergan RC. A simple analysis of gene expression and variability in gene arrays based on repeated observations. Am J Pharmacogenomics. 2001;1:145–152. doi: 10.2165/00129785-200101020-00007. [DOI] [PubMed] [Google Scholar]
- Kozlowski JM, Fidler IJ, Campbell D, Xu ZL, Kaighn ME, Hart IR. Metastatic behavior of human tumor cell lines grown in the nude mouse. Cancer Res. 1984;44:3522–3529. [PubMed] [Google Scholar]
- Lastres P, Letamendia A, Zhang H, Rius C, Almendro N, Raab U, et al. Endoglin modulates cellular responses to TGF-beta 1. J Cell Biol. 1996;133:1109–1121. doi: 10.1083/jcb.133.5.1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebrin F, Goumans MJ, Jonker L, Carvalho RL, Valdimarsdottir G, Thorikay M, et al. Endoglin promotes endothelial cell proliferation and TGF-beta/ALK1 signal transduction. EMBO J. 2004;23:4018–4028. doi: 10.1038/sj.emboj.7600386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letamendia A, Lastres P, Botella LM, Raab U, Langa C, Velasco B, et al. Role of endoglin in cellular responses to transforming growth factor-beta. A comparative study with betaglycan. J Biol Chem. 1998;273:33011–33019. doi: 10.1074/jbc.273.49.33011. [DOI] [PubMed] [Google Scholar]
- Liu Y, Jovanovic B, Pins M, Lee C, Bergan RC. Over expression of endoglin in human prostate cancer suppresses cell detachment, migration and invasion. Oncogene. 2002;21:8272–8281. doi: 10.1038/sj.onc.1206117. [DOI] [PubMed] [Google Scholar]
- Liu YQ, Kyle E, Patel S, Housseau F, Hakim F, Lieberman R, et al. Prostate cancer chemoprevention agents exhibit selective activity against early stage prostate cancer cells. Prostate Cancer Prostatic Dis. 2001;4:81–91. doi: 10.1038/sj.pcan.4500506. [DOI] [PubMed] [Google Scholar]
- Massague J. TGF-beta signal transduction. Annu Rev Biochem. 1998;67:753–791. doi: 10.1146/annurev.biochem.67.1.753. [DOI] [PubMed] [Google Scholar]
- Miettinen PJ, Ebner R, Lopez AR, Derynck R. TGF-beta induced transdifferentiation of mammary epithelial cells to mesenchymal cells: involvement of type I receptors. J Cell Biol. 1994;127:2021–2036. doi: 10.1083/jcb.127.6.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteiro RM, de Sousa Lopes SM, Korchynskyi O, ten Dijke P, Mummery CL. Spatio-temporal activation of Smad1 and Smad5 in vivo: monitoring transcriptional activity of Smad proteins. J Cell Sci. 2004;117:4653–4663. doi: 10.1242/jcs.01337. [DOI] [PubMed] [Google Scholar]
- Pece-Barbara N, Vera S, Kathirkamathamby K, Liebner S, Di Guglielmo GM, Dejana E, et al. Endoglin null endothelial cells proliferate faster and are more responsive to transforming growth factor beta1 with higher affinity receptors and an activated Alk1 pathway. J Biol Chem. 2005;280:27800–27808. doi: 10.1074/jbc.M503471200. [DOI] [PubMed] [Google Scholar]
- Poste G, Fidler IJ. The pathogenesis of cancer metastasis. Nature. 1980;283:139–146. doi: 10.1038/283139a0. [DOI] [PubMed] [Google Scholar]
- Shi Y, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- ten Dijke P, Ichijo H, Franzen P, Schulz P, Saras J, Toyoshima H, et al. Activin receptor-like kinases: a novel subclass of cell-surface receptors with predicted serine/threonine kinase activity. Oncogene. 1993;8:2879–2887. [PubMed] [Google Scholar]
- Ulloa L, Doody J, Massague J. Inhibition of transforming growth factor-beta/SMAD signalling by the interferon-gamma/STAT pathway. Nature. 1999;397:710–713. doi: 10.1038/17826. [DOI] [PubMed] [Google Scholar]
- Ward SM, Desgrosellier JS, Zhuang X, Barnett JV, Galper JB. Transforming growth factor beta (TGFbeta) signaling via differential activation of activin receptor-like kinases 2 and 5 during cardiac development. Role in regulating parasympathetic responsiveness. J Biol Chem. 2002;277:50183–50189. doi: 10.1074/jbc.M209668200. [DOI] [PubMed] [Google Scholar]
- Wieser R, Wrana JL, Massague J. GS domain mutations that constitutively activate T beta R-I, the downstream signaling component in the TGF-beta receptor complex. EMBO J. 1995;14:2199–2208. doi: 10.1002/j.1460-2075.1995.tb07214.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrana JL, Attisano L, Carcamo J, Zentella A, Doody J, Laiho M, et al. TGF beta signals through a heteromeric protein kinase receptor complex. Cell. 1992;71:1003–1014. doi: 10.1016/0092-8674(92)90395-s. [DOI] [PubMed] [Google Scholar]
- Xu L, Chen S, Bergan RC. MAPKAPK2 and HSP27 are downstream effectors of p38 MAP kinase-mediated matrix metalloproteinase type 2 activation and cell invasion in human prostate cancer. Oncogene. 2006;25:2987–2998. doi: 10.1038/sj.onc.1209337. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information accompanies the paper on the Oncogene Web site (http://www.nature.com/onc).