

Abstract
A method has been developed for de novo peptide sequencing using matrix-assisted laser desorption ionization mass spectrometry. This method will facilitate biological studies that require rapid determination of peptide or protein sequences, e.g., determination of posttranslational modifications, identification of active compounds isolated from combinatorial peptide libraries, and the selective identification of proteins as part of proteome studies. The method involves fast, one-step addition of a sulfonic acid group to the N terminus of tryptic peptides followed by acquisition of postsource decay (PSD) fragment ion spectra. The derivatives are designed to promote efficient charge site-initiated fragmentation of the backbone amide bonds and to selectively enhance the detection of a single fragment ion series that contains the C terminus of the molecule (y-ions). The overall method has been applied to pmol quantities of peptides. The resulting PSD fragment ion spectra often exhibit uninterrupted sequences of 20 or more amino acid residues. However, fragmentation efficiency decreases considerably at amide bonds on the C-terminal side of Pro. The spectra are simple enough that de novo sequence tagging is routine. The technique has been successfully applied to peptide mixtures, to high-mass peptides (up to 3,600 Da) and to the unambiguous identification of proteins isolated from two-dimensional gel electrophoresis. The PSD spectra of these derivatized peptides often allow far more selective protein sequence database searches than those obtained from the spectra of native peptides.
Knowledge of protein sequences is fundamentally important for understanding many physiological processes at the molecular level (1). Tandem mass spectrometry has become an increasingly essential tool for protein and peptide sequencing because of its speed, sensitivity, and applicability to complex mixtures (2). Recently, postsource decay (PSD) matrix-assisted laser desorption ionization (MALDI) mass spectrometry was developed for high-sensitivity peptide sequencing applications (3–8). Subpicomole limits of analysis were reported as a result of the high yield of fragment ions and the high ion transmission inherent with time-of-flight mass spectrometry (5). Kaufmann et al. (5) also noted several problems associated with PSD MALDI sequencing of peptides, including the complexity of the resulting fragmentation patterns and the lack of computer algorithms capable of interpreting the complex spectra. The recent incorporation of delayed extraction (DE) (9, 10), a technique designed to improve precursor-ion mass resolution and mass-measurement accuracy, reduced the rate of PSD fragmentation by at least an order of magnitude. As a result, many low-intensity precursor ions obtained by DE MALDI do not produce enough PSD fragmentation to allow derivation of even short sequence tags (7).
Positively charged derivatives have been used in desorption mass spectrometry for many years to improve sensitivity by enhancing ionization efficiencies (11, 12). Localization of a positive charge at a particular site on a peptide (13, 14) also profoundly affects the nature of the fragment ions observed in high-collision energy tandem mass spectrometry (15–26). A protonated Arg or Lys, or a cationic derivative located near the N terminus of a peptide, promotes the formation of fragment ions containing the N terminus (a, b, c ions; the nomenclature for peptide fragment ions has been discussed in ref. 27). A protonated Arg or Lys or a fixed-charge group near the C terminus enhances fragment ions containing the C terminus of the peptide (x, y, z ions). It is not surprising, therefore, that several researchers have used N-terminal fixed-charge derivatives (quaternary ammonium and phosphonium groups) to improve MALDI sensitivity and to try to direct and simplify peptide fragmentation patterns observed in PSD mass spectra (28–35). In our hands, generally poor-quality MALDI PSD mass spectra have been obtained with fixed positive-charge derivatives. An alternative strategy to increase the yield of sequence-specific fragment ions by PSD was developed by Dikler and coworkers (36), who modified the guanidine side chain of Arg-containing peptides. That modification was done to minimize charge localization on the basic Arg residue and to facilitate protonation of the backbone amide groups.
Several previous studies (7, 30, 33, 37) raise questions about the internal energy content of precursor ions derived from DE MALDI and whether there is enough energy to optimally produce sequence-specific products by the charge-remote fragmentation mechanisms that are required with fixed positive-charge derivatives. Wysocki and coworkers (37) showed that addition of a fixed positive charge at the N terminus of a peptide increased the internal energy needed to fragment the peptide. Those results are consistent with the electrospray ionization findings of Gaskell and coworkers (38), who showed that the formation of precharged derivatives was detrimental to the yield of sequence-specific fragment ions after low-energy collisional activation of singly charged species. On the other hand, experimental (37) and theoretical (39) studies reveal that protonation of backbone amide nitrogen atoms enhance peptide fragmentation because amide bond orders are lowered dramatically by protonation.
Questions about the internal energy content of precursor ions derived from DE MALDI (7, 30, 33) and the compelling arguments for the mobile proton model of peptide fragmentation (37, 39) led us to investigate an alternative approach to promote and direct fragmentation in PSD mass spectrometry. This method involves addition of a strong acid group to the N terminus of tryptic peptides before MALDI analyses. These derivatives facilitate low-energy charge site-initiated fragmentation, and they suppress the detection of N-terminal fragment ions in the positive ion mode of analysis. Dramatic increases in fragmentation efficiencies and fragment ion signal-to-noise ratios have been observed for derivatized peptides relative to nonderivatized peptides having the same sequences. This approach provides simple, easy-to-interpret fragment ion mass spectra that can be routinely interpreted de novo.
In this paper we demonstrate fast, one-step derivatization chemistry that can be applied generally to tryptic peptides. The role of the mobile proton in causing cleavage of backbone amide bonds was demonstrated by comparing the fragmentation behavior of MH+, MNa+, and (M-H)− molecular ions, and by comparing differences in the fragmentation patterns of tryptic peptides containing either one or two Arg residues. The acidity of the derivatizing group was shown to have a profound effect on the observed PSD fragment ion mass spectra. Peptides derivatized with sulfonic acid groups at the N terminus show y-type fragment ions almost exclusively, whereas peptides derivatized with carboxylic acids also show complementary N-terminal fragment ions (a- and b-type). The overall sensitivity of the method has been demonstrated by applying it to pmol quantities of peptides isolated from HPLC and by using it for the unambiguous identification of proteins separated by two-dimensional (2D) gel electrophoresis. The utility of the method for identification of phosphorylation sites has been established by analysis of a model phosphorylated peptide. Reliable sequence tags have been obtained from large peptides (up to 3,600 Da) having masses exceeding the range of conventional triple quadrupole instruments. The PSD spectra often exhibit uninterrupted sequences of 20 or more aa, particularly when the peptides lack Pro residues. This capability will increase the utility of PSD MALDI data for the design of specific oligonucleotide probes for gene cloning experiments and for protein sequence database searches used in proteome studies.
MATERIALS AND METHODS
Chemicals.
The model peptides used in this study include: CDPGYIGSR (C-0668), ASHLGLAR (A-8651), YGGFLR (E-8757), YGGFLRR (D-4524), PDVDHVFLRF-NH2 (P-2178), CLNRQLpSSGVSEIR (H-2654), and oxidized insulin B-chain (I-6383). They were obtained from Sigma, and they were used without further purification. Dithiodiglycolic acid (D-5392) was obtained from Sigma. DTSSP (3,3′-dithiobis[sulfosuccinimidylpropionate], 21578) was obtained from Pierce. Traut’s reagent (33,056–6), S-acetylmercaptosuccinic anhydride (19,732–7), 2-sulfobenzoic acid cyclic anhydride (19,169–8), chlorosulfonylacetyl chloride (25,977–2), phthalic anhydride (32,006–4), 3-nitrophthalic anhydride (15,688–4), 4-nitrophthalic anhydride (22,820–1), and succinic anhydride (23,969–0) were obtained from Aldrich.
N-Terminal Derivatization Procedures.
The two derivatization procedures most often used in this work involved coupling either 2-sulfobenzoic acid cyclic anhydride or chlorosulfonylacetyl chloride to the N terminus of peptides. 2-Sulfobenzoic acid cyclic anhydride was prepared at a concentration of 0.1 M in dry tetrahydrofuran (THF) just before use. High-level reactions involving 1 nM of peptide were carried out as follows: 1 nM of peptide was diluted into 20 μl of 0.05 M trimethylamine. Two microliters of the fresh 2-sulfobenzoic acid cyclic anhydride solution was added, and the reaction mixture was vortexed for 30 sec. The reaction proceeded for 1–2 min at room temperature before dilution and MALDI analyses. The concentration of the coupling reagent was decreased by a factor of as much as 100 when derivatizing smaller quantities of peptides. Reactions with chlorosulfonylacetyl chloride were carried out under nonaqueous conditions. Peptide (1 nM) was mixed with 2 μl of 0.02 M sulfoacetic acid, which had been formed by mixing 2 μl of neat chlorosulfonylacetyl chloride with 500 μl of H2O. The mixture was dried and then reconstituted in 20 μl of THF/diisopropylethyl amine 4:1. Two microliters of 0.1 M chlorosulfonylacetyl chloride in dry THF was added, and the mixture was votexed for 30 sec. The coupling reaction proceeded for 1–2 min at room temperature. The sample was then dried, reconstituted in 20 μl H2O, and further diluted before MALDI analyses. Derivatization of 2D gel isolates was accomplished in the same way except the concentrations of sulfoacetic acid and chlorosulfonylacetyl chloride were reduced 10- to 100-fold. These isolates were obtained from single 2D gels (not pooled samples), and they contained <10 pM per component.
Reactions involving phthalic anhydride, 3- and 4-nitrophthalic anhydride, and succinic anhydride were carried out in 0.05 M trimethylamine as described above for 2-sulfobenzoic acid cyclic anhydride.
Performic acid oxidation of CDPGYIGSR and some of the derivatized peptide intermediates was done by mixing 1–5 nM of peptide (in 5–20 μl H2O) with 10 μl of fresh formic acid (88%)/H2O2 (30%) prepared at a ratio of 19:1 (vol/vol). Oxidation was allowed to proceed for 30 min at room temperature, and the samples were dried before analysis.
Mass Spectrometry.
All mass spectrometry experiments were carried out on a PerSeptive Biosystems (Framingham, MA) Voyager DE-RP or Voyager DE-STR equipped with a N2 laser (337 nm, 3-nsec pulse width, 20-Hz repetition rate). The mass spectra were acquired in the reflectron mode with DE. External mass calibration was performed with low-mass peptide standards, and mass-measurement accuracy was typically ±0.3 Da. High levels of peptides and derivatized peptides were diluted to about 10 pM/μl in 0.1% trifluoroacetic acid (TFA). The samples then were diluted 5- to 10-fold further in α-cyano-4-hydroxycinnamic acid, which had been prepared by dissolving 10 mg in 1 ml of aqueous 50% acetonitrile containing 0.1% TFA (40). The 2D gel isolates were analyzed from thin film surfaces of α-cyano-4-hydroxycinnamic acid/nitrocellulose prepared by the fast evaporation method (41).
PSD fragment ion spectra were acquired for several peptides and peptide derivatives after isolation of the appropriate precursor ion by using timed ion selection. Fragment ions were refocused onto the final detector by stepping the voltage applied to the reflectron in the following ratios: 1.0000 (precursor ion segment), 0.9126, 0.6049, 0.4125, 0.2738, 0.1975, and 0.1213 (fragment segments). The individual segments were stitched together by using software provided by PerSeptive Biosystems. All precursor ion segments were acquired at low laser power (variable attenuator = 1,450) for <256 laser pulses to avoid saturating the detector. The laser power was increased (variable attenuator = 1,650) for all of the remaining segments of the PSD acquisitions. Typically, 256 laser pulses were acquired for each fragment-ion segment. The PSD data were acquired at a digitization rate of 20 MHz. Mass calibration was done with peptide standards only about once a month. Metastable decompositions were measured in all PSD mass spectrometry experiments.
Identification of Proteins Isolated From 2D Gel Electrophoresis.
2D-gel-separated protein spots were in-gel digested with trypsin following the method of Shevchenko and coworkers (42). Peptides were analyzed directly by MALDI after loading 5–10% aliquots of the digestion solutions. The measured tryptic peptide masses were used as inputs to search the NCBInr5.18.98 database containing 299,924 protein sequences. All searches were carried out by using the protein prospector software developed at the University of California, San Francisco (see P. R. Baker and K. R. Clauser, http://prospector.ucsf.edu). No restrictions were placed on the species of origin of the protein. The allowed protein molecular mass range was 1,000 to 150,000 Da. Isoelectric points were allowed to range from 3.0 to 10.0, and oxidation of Met was included as a side reaction. Up to one missed tryptic cleavage was considered, and a conservative mass accuracy of ± 0.6 Da was used for all tryptic-mass searches. The number of tryptic peptides required for identification varied as a function of the quality of the MALDI mass spectra. The protein prospector software also was used to search PSD fragment ion spectra either against the entire protein sequence database or against the saved set of proteins identified from the appropriate tryptic mass searches. Only y-type or (y-NH3) ions were allowed fragment-ion possibilities when searching the PSD spectra of derivatized peptides.
RESULTS AND DISCUSSION
Method to Promote Charge-Site-Initiated Fragmentation in PSD MALDI of Tryptic Peptides.
Gaskell and coworkers (43, 44) previously demonstrated that the oxidation of cysteine to cysteic acid in peptides containing a C-terminal Arg strongly increases the yield of y-type fragment ions observed by low-energy collisional activation of protonated molecules formed by electrospray ionization. These studies formed the starting point of our efforts to develop a general procedure for high-sensitivity tryptic peptide sequencing using PSD MALDI. We have developed several convenient routes to produce tryptic peptides containing an N-terminal sulfonic acid (a),
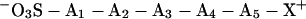 |
a |
where Ai are amino acid residues. This strategy recognizes that the basic C terminus (X = Lys or Arg) will be protonated after MALDI. The strong acid group at the N terminus will be deprotonated, exactly counterbalancing the C-terminal positive charge. One additional proton then is required to ionize the derivative for mass spectrometry analyses. That proton will be more or less free to randomly ionize the backbone amide groups because the most basic residue already is occupied. The resulting PSD spectra should show increased fragmentation by direct cleavage of protonated amide bonds. The major products of these fragmentation reactions have structures like b and c,
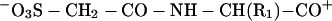 |
b |
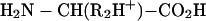 |
c |
which would be produced by fragmentation of a derivatized tryptic dipeptide (R2 is the basic side chain of a Lys or Arg residue). Fragments containing the N terminus will be suppressed in the positive ion mode because the negative charge on the sulfonate group neutralizes the C-terminal positive charge in b. Internal cleavage ions will not be enhanced because they lack the positively charged C-terminal Lys or Arg, and a single series of y-type ions (c) should be enhanced because they contain the protonated C terminus.
A commercially available peptide, CDPGYIGSR, was analyzed to determine whether derivatives like a exhibit the predicted effect on PSD MALDI fragmentation behavior. Fig. 1, Upper shows the PSD fragment ion spectrum of the native peptide, while Fig. 1, Lower shows the corresponding spectrum after performic acid oxidation of the N-terminal cysteine to cysteic acid. The spectrum of the native peptide shows an incomplete series of y-ions (y1, y2, y3, y7, and y8) dominated by the low-energy cleavage of the Asp-Pro amide bond. Prominent b4 and weak b5 ions are also evident. The spectrum was much improved after oxidation, Fig. 1, Lower. The N-terminal fragment ions were suppressed and several y-ions (y4, y5, y6, and y8) were enhanced considerably. The complete sequence of the peptide could be determined by PSD MALDI after oxidation. These promising results led us to investigate general methods to derivatize tryptic peptides.
Figure 1.

PSD fragment ion mass spectra of CDPGYIGSR before (Upper) and after (Lower) oxidation with performic acid.
N-Terminal Derivatization Methods.
A number of approaches for the introduction of sulfonic acid groups to the N terminus of peptides were evaluated in this study. The first method involved introduction of an N-terminal sulfur that subsequently could be oxidized. Dithiodiglycolic anhydride (I) and DTSSP (3,3′-dithiobis[sulfosuccinimidylpropionate]) (II), a commonly used protein crosslinking agent (45), enable the introduction of a disulfide bond at the N terminus of peptides. 2-Iminothiolane III (Traut’s reagent) (46) provides an N-terminal sulfhydryl group, whereas S-acetyl mercaptosuccinic anhydride (IV) provides an acetylated sulfhydryl group. However, all of these derivatives require performic acid oxidation to yield the desired sulfonic acid. Oxidation of labile amino acid residues made the resulting mass spectra more difficult to interpret. The second approach, which involved direct introduction of the sulfonic acid, was a more effective method. Sulfobenzoic acid cyclic anhydride (V) couples readily to free amines in aqueous solution. This reaction introduces an aromatic sulfonic acid group directly at the N terminus of the peptide. On the other hand, chlorosulfonylacetyl chloride (VI) did not provide high yields of derivatized peptides when the coupling reaction was carried out in aqueous solution. Presumably, it is too reactive with water. Coupling proceeded well in dry THF and subsequent addition of water converted the chlorosulfonyl group into the desired sulfonic acid.
Mass Spectrometry of Derivatized Peptides.
The MALDI mass spectra of the derivatized tryptic peptides often exhibit MH+ ions for the starting peptide as well as for the desired final product. The ratio of these two ions does not provide a good estimate of the overall product yield. The positive-ion response of the derivatized peptide is expected to be considerably lower than that of the unreacted starting material because of the added sulfonic acid group. In-source fragmentation of the derivative to regenerate starting material also alters the ratio of these ions. To investigate this effect, the ratio of starting peptide to derivatized product was examined as a function of laser power for the mixture formed by adding VI to ASHLGLAR in dry THF. The ratio of starting peptide to derivatized product increased from 0.25 at the lowest laser power used (variable attenuator set to 1,150) to 1.09 at the highest power (attenuator set to 1,650). Negative-ion MALDI is a more convenient method to monitor these reactions because the molecular anions are more stable and the MALDI spectra are simpler.
The PSD fragment ion spectra of the derivatives formed by reacting V and VI with various tryptic peptides are similar. This is true for all of the tryptic peptides we examined, whether they contain a C-terminal Arg or Lys. The spectra of both derivatives exhibit prominent y-ions, with few other ions observed. The elements of O3S-C6H4-CO are readily lost from products formed with V, and the most abundant product ion in those PSD spectra generally corresponds to the mass of the unreacted starting tryptic peptide. The relative abundance of the y-ions decreases toward lower mass. Fragmentation at amide bonds on the C-terminal side of Pro is less favorable than it is for other amino acid residues (for all derivatives examined). Derivatives formed with VI show losses of SO3 and O3S-CH2-CO to a small extent, in addition to the abundant y-series ions. The product ion formed by loss of the elements of O3S-CH2-CO generally is not the most abundant ion in the PSD spectra of these derivatives. Lower-mass y-series ions dominate, which suggests that peptide derivatives formed with VI are more stable than those formed with V under MALDI conditions. A larger fraction of the PSD product ion current will be directed into structurally significant y-ions with chlorosulfonylacetyl chloride derivatives than with derivatives formed with 2-sulfobenzoic acid cyclic anhydride.
Dramatic improvements often are observed in PSD MALDI fragment ion spectra after derivatization. Results obtained from a 24-aa peptide, formed by tryptic digestion of an enzyme isolated from a commercial laundry product, are given in Fig. 2. The peptide was isolated by reverse-phase HPLC. The PSD spectrum obtained from the native peptide (Fig. 2, Upper) demonstrates little structurally significant fragmentation. The spectrum of the derivative formed with VI was obtained under very similar experimental conditions. It shows a complete series of y-ions, enabling rapid verification of the amino acid sequence (NTATSLGSTNLYGSGLVNAEAATR) of this portion of the enzyme.
Figure 2.

PSD MALDI mass spectra of a tryptic peptide before (Upper) and after (Lower) derivatization.
The Role of the Mobile Ionizing Proton.
The role of the mobile ionizing proton was assessed by comparing the PSD fragment ion spectra of the MH+, MNa+, and (M-H)− ions of the derivative formed between ASHLGLAR and V. The spectrum of the protonated derivative showed a complete series of y-ions. It also showed an MH+ molecular ion that was of about half the abundance to the most prominent product ion, which corresponded to loss of 184 Da (O3S-C6H4-CO) as discussed above. Changing the ionizing agent from a proton to a sodium cation greatly reduced PSD fragmentation. The y-ion series was virtually absent from the spectrum of the MNa+ precursor ion. The most intense ion in the spectrum was the parent MNa+ ion. The main product ion corresponded to loss of 201 Da (the elements of HO3S-C6H4-CO-NH2). Weak signals also were observed for loss of 184 Da (O3S-C6H4-CO) and loss of (184 + 156 Da), where the 156-Da fragment comes from the Arg residue at the C terminus. Similarly poor results were observed for the (M-H)− ion, which also lacks the mobile ionizing proton. No y-ions were observed and the undissociated molecular ion was the largest ion observed in the PSD spectrum. The only other prominent fragment ions correspond to loss of the C-terminal R (−156 Da) and loss of 184 Da. It would not be possible to sequence this peptide by using the fragment ion mass spectra obtained from either the MNa+ or (M-H)− precursor ions.
The importance of the mobile proton in promoting fragmentation in these systems was explored further by obtaining the PSD spectra of the derivatives formed between V and YGGFLR and YGGFLRR. Wysocki’s group (37) previously examined the doubly charged ions formed from some bradykinin-related peptides that contained two Arg residues. Their experiments demonstrated that both protons were preferentially localized at the basic Arg residues and neither proton was mobile across the peptide backbone. We expected similar charge localization with these peptides and a corresponding reduction in PSD fragmentation. Derivatized YGGFLR showed typical behavior, i.e., a complete and abundant y-ion series dominated by y5 and MH+-(O3S-C6H4-CO). The nondissociated MH+ ion had a relative abundance of about 30% compared with that of y5. Derivatized YGGFLRR showed quite different PSD behavior when run under the same conditions as for YGGFLR. The MH+ precursor ion was the most abundant ion in this PSD spectrum and the fragment ions were reduced in relative abundance by at least a factor of 2 or 3. The y-ion series was evident in the spectrum, but all y-ions except y1 were accompanied with a (yi-17) ion that was at least twice as large as the corresponding yi ion. Tang and Boyd (47) observed similar (y-17) ions in the electrospray tandem mass spectra of singly charged peptides containing a C-terminal Arg.
It should be noted that YGGFLRR is an interesting model for peptides formed by incomplete tryptic digestion of proteins. The differences in fragmentation behavior between complete and incomplete tryptic peptides must be considered when searching protein sequence databases against the PSD spectra of these derivatives (see below).
Acidity of the Derivatizing Group.
A number of model peptides (ASHLGLAR, YGGFLRR, and PDVDHVFLRF-NH2) were derivatized with various anhydrides like phthalic anhydride, 3- and 4-nitrophthalic anhydride and succinic anhydride to test the effect of acid strength on the PSD MALDI spectra of derivatized tryptic-like peptides. The spectra of the peptides derivatized with the anhydrides showed extensive fragmentation, but they were much more complex than those obtained with any of the sulfonic acid derivatives. They showed a- and b-ions, both with and without the added anhydride, in addition to y-ions. None of the anhydride derivatives we investigated gave simple, single-series spectra like the ones obtained after derivatization with V or VI. They were unable to suppress detection of the N-terminal fragment ions, probably because the resulting acids are more basic (pKas ranging from 1.8 to 4.2, ref. 48) than protonated amide groups [pKa of protonated acetamide = −1 (nonaqueous)]. Strong acids, like sulfonic acids [pKas about −7 (nonaqueous)], appear to be required to suppress detection of the N-terminal fragment ions and produce easy-to-interpret spectra.
Phosphopeptide Analysis.
The utility of this method for determination of phosphorylation sites was demonstrated by analysis of a model phosphopeptide CLNRQLpSSGVSEIR obtained commercially. The PSD MALDI mass spectrum obtained from the native peptide showed poor sequence coverage. Only weak y1 and y7 ions were observed above background noise. PSD MALDI of the peptide derivatized with V showed much better fragment ion sensitivity and sequence coverage. A complete series of unmodified y-ions was observed from y1 to y7 confirming that Ser residues 8 and 11 (numbered from the N terminus) were not phosphorylated. The phosphate group was located on Ser-7 by the observation of an abundant (y8-H3PO4) ion. Facile loss of neutral phosphate is a characteristic of serine- and threonine-phosphorylated peptides (49). The y9, y10, y11, and y12 fragment ions also showed quantitative loss of H3PO4. The y11 and y12 ions showed further loss of NH3, as discussed above for tryptic peptides containing a second basic residue. No N-terminal fragment ions were observed in this spectrum even though the peptide contains an Arg at residue 4.
Variant Proteins.
The MALDI mass spectrum of a tryptic digest obtained from about 1 nmol of an enzyme isolated from a commercial product showed many masses identical to those of savinase, a widely used commercial protease. One of the expected tryptic peptides GVLVVAASGNSGAGSISYPAR (MH+ = 1,933 Da) was missing from the spectrum, and an unknown ion was observed at m/z 1963. This finding suggests that the isolate is a savinase variant. Eleven single amino acid changes could account for the observed +30-Da mass shift (four G → S, four A → T, and three V → E). Two or more amino acid changes are also possible, and a complete sequence is required to identify the variant. The PSD MALDI spectrum obtained after derivatization of the isolate (with VI) is given in Fig. 3. A complete series of y-ions is observed for this 21-aa peptide. The spectrum verifies that only a single amino acid has been changed. The Gly at residue 14 was converted to a Ser. Incidentally, the spectrum was automatically interpreted by using a sequencing program previously written by PerSeptive Biosystems. That program had been developed for automated interpretation of MALDI mass spectra of peptide ladders generated chemically (50) or by carboxypeptidase Y digestion (51). The computer-assigned amino acid residues are indicated in Fig. 3. One error was made in the automated interpretation of this spectrum, i.e., N (114 Da) was assigned as D (115 Da). Careful mass calibration should minimize this problem. The result illustrates that automated interpretation of these PSD fragment ion mass spectra is feasible.
Figure 3.

PSD MALDI mass spectrum of a derivatized tryptic peptide from a variant protein. The y1 ion is observed at m/z 175 establishing the C-terminal residue as R (not labeled by the software). Mass differences between adjacent y-ions establish the identities of the remaining amino acid residues (indicated on the figure).
High-Mass Peptides.
PSD mass spectrometry of high-mass peptides was demonstrated with oxidized insulin B-chain (MMcalc = 3,495.9 Da). The mass of this peptide far exceeds the upper limit of most conventional triple quadrupole instruments. The fragment ion mass spectrum of the native peptide showed a relatively abundant product-ion series consisting mainly of y-ions. All of the y-ions between y9 and y24 were clearly observed. This abundant fragment ion series probably results because the native peptide contains a basic Arg and Lys residue near the C terminus and it has sulfonic acid groups attached to the two Cys residues. The sequence-specific fragments defining the N terminus of the molecule, y25 to y29, were entirely absent from this spectrum. The PSD spectrum of this compound was improved by derivatization with V. The derivatized peptide showed significant enhancement of the y25, y26, y27, y28, and y29 ions. A complete series of y-ions was observed from y8 to y29 after derivatization and no prominent b-ions were detected. The spectrum is less complex than that obtained on a MALDI ion trap mass spectrometer (52), which showed significant b-type ions interspersed with the y-ions.
Identification of Proteins Isolated from 2D Gel Electrophoresis.
We use MALDI and PSD mass spectrometry extensively for the identification of proteins separated from 2D gel electrophoresis, and these derivatization methods have proven useful even with the low-pmol levels of tryptic peptides generally available from these experiments. In a recent study, an excellent MALDI mass map was obtained from a protein spot after in-gel tryptic digestion (42, 53). It showed several abundant MH+ signals including ions at m/z 1060.8, 1090.8, 1271.0, 1299.0, 1312.0, 1344.0, 1399.1, 1450.1, 1460.1, and 1794.4. Database searching against the 299,924 entries in the NCBInr5.18.98 protein sequence library produced a list of 41 candidate proteins that matched five or more of the input tryptic masses. The top molecular weight search (MOWSE) scores were quite low, only in the 560 range. The entire peptide mixture was then reacted with VI, and PSD mass spectrometry was carried out on the derivatized peptide weighing about 1,572 Da (1450 + 122 Da), Fig. 4. A sequence tag was obtained and it consisted of y-ions at m/z 574.5, 661.7, 875.9, 1003.8, 1103.9, 1204.6, 1304.1, and the (MH+ derivative) ion at m/z 1450.3. That spectrum was searched against the entire protein sequence database, and six candidate proteins (all mitochondrial aspartate aminotransferases) were returned. This level of specificity was obtained because the database search was constrained to consider y-ions as the only allowed fragments. This search constraint is possible because N-terminal and internal cleavage ions are not prominent in the PSD mass spectra of these derivatives. Much less specificity was obtained (239 candidate proteins returned) when this same fragment ion mass spectrum was searched against the full library and all fragment ion types [a, b, (b + H2O), (b-NH3), (b-H2O), internal, and y] were allowed.
Figure 4.

PSD MALDI fragment ion mass spectrum of a derivatized peptide obtained from an in-gel tryptic digestion of a protein separated by 2D gel electrophoresis.
Confirmation of the protein identification was sought by PSD MALDI of the derivative weighing 1,916 Da (1,794 + 122 Da). The PSD spectrum contained seven fragment ions at m/z 724.6, 1154.2, 1299.3, 1439.0, 1551.9, 1665.5, and 1779.2 in addition to the protonated precursor ion. This spectrum was searched against the full database assuming that the fragments were y-ions. The search did not return mitochondrial aspartate aminotransferase or any other candidate protein. Researching the database allowing both y and (y-NH3) ions returned 16 candidate proteins, 10 of which were mitochondrial aspartate aminotransferases. This latter search confirmed the protein identification. Inclusion of (y-NH3) ions as possible fragments was required for this particular search because the peptide turned out to be an incomplete tryptic product, ILIRPLYSNPPLNGAR, that contained a second Arg residue. In this case, the PSD fragment ions were exclusively (y-NH3) ions and not y-ions.
Acknowledgments
We thank Dr. Raymond A. Grant, Karen B. Begley, and Angela M. Fieno for the separations and sample preparations associated with the 2D gel electrophoresis experiments reported in this paper.
ABBREVIATIONS
- PSD
postsource decay
- MALDI
matrix-assisted laser desorption ionization
- DE
delayed extraction
- 2D
two-dimensional
- THF
tetrahydrofuran
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
References
- 1.Allen G. Sequencing of Proteins and Peptides. New York: Elsevier/North–Holland; 1981. [Google Scholar]
- 2.Carr S A, Hemling M E, Bean M F, Roberts G D. Anal Chem. 1991;63:2802–2824. doi: 10.1021/ac00024a003. [DOI] [PubMed] [Google Scholar]
- 3.Spengler B, Kirsch D, Kaufmann R, Jaeger E. Rapid Commun Mass Spectrom. 1992;6:105–108. doi: 10.1002/rcm.1290060207. [DOI] [PubMed] [Google Scholar]
- 4.Spengler B, Kirsch D, Kaufmann R. J Phys Chem. 1992;96:9678–9684. [Google Scholar]
- 5.Kaufmann R, Spengler B, Lutzenkirchen F. Rapid Commun Mass Spectrom. 1993;7:902–910. doi: 10.1002/rcm.1290071010. [DOI] [PubMed] [Google Scholar]
- 6.Kaufmann R, Kirsch D, Spengler B. Int J Mass Spectrom Ion Processes. 1994;131:355–385. [Google Scholar]
- 7.Kaufmann R, Chaurand P, Kirsch D, Spengler B. Rapid Commun Mass Spectrom. 1996;10:1199–1208. doi: 10.1002/(SICI)1097-0231(19960731)10:10<1199::AID-RCM643>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 8.Spengler B. J Mass Spectrom. 1997;32:1019–1036. [Google Scholar]
- 9.Brown R S, Lennon J J. Anal Chem. 1995;67:1998–2003. doi: 10.1021/ac00109a015. [DOI] [PubMed] [Google Scholar]
- 10.Vestal M, Juhasz P. J Am Soc Mass Spectrom. 1998;9:892–911. [Google Scholar]
- 11.Keough T, DeStefano A J. Anal Chem. 1981;53:25–29. [Google Scholar]
- 12.Busch K L, Unger S E, Vincze A, Cooks R G, Keough T. J Am Chem Soc. 1982;104:1507–1511. [Google Scholar]
- 13.Johnson R S, Martin S A, Biemann K. Int J Mass Spectrom Ion Processes. 1988;86:137–154. [Google Scholar]
- 14.Vath J E, Biemann K. Int J Mass Spectrom Ion Processes. 1990;100:287–299. [Google Scholar]
- 15.Kidwell D A, Ross M M, Colton R J. J Am Chem Soc. 1984;106:2219–2220. [Google Scholar]
- 16.Renner D, Spiteller G. Biomed Environ Mass Spectrom. 1986;13:405–410. doi: 10.1002/bms.1200150204. [DOI] [PubMed] [Google Scholar]
- 17.Wagner D S, Salari A, Gage D A, Leykam J, Fetter J, Hollingsworth R, Watson J T. Biol Mass Spectrom. 1991;20:419–425. doi: 10.1002/bms.1200200705. [DOI] [PubMed] [Google Scholar]
- 18.Zhao Y-F, Zhang D-Q, Xue C-B. Int J Peptide Protein Res. 1991;37:457–461. doi: 10.1111/j.1399-3011.1991.tb00761.x. [DOI] [PubMed] [Google Scholar]
- 19.Watson J T, Wagner D S, Chang Y-S, Strahler J R, Hanash S M, Gage D A. Int J Mass Spectrom Ion Processes. 1991;111:191–209. [Google Scholar]
- 20.Yang H-J, He M-Y, Ye Y-H, Zhao Y-F. Org Mass Spectrom. 1992;27:746–749. [Google Scholar]
- 21.Kiplinger J P, Contillo L, Hendrick W L, Grodski A. Rapid Commun Mass Spectrom. 1992;6:747–752. doi: 10.1002/rcm.1290061207. [DOI] [PubMed] [Google Scholar]
- 22.Chang Y-S, Gage D A, Watson J T. Biol Mass Spectrom. 1993;22:176–180. doi: 10.1002/bms.1200220306. [DOI] [PubMed] [Google Scholar]
- 23.Bunk D M, Macfarlane R D. Int J Mass Spectrom Ion Processes. 1993;126:123–136. [Google Scholar]
- 24.Stults J T, Shannon J L, McCune S, Wetzel R. Anal Chem. 1993;65:1703–1708. doi: 10.1021/ac00061a012. [DOI] [PubMed] [Google Scholar]
- 25.Downard K M, Biemann K. J Am Soc Mass Spectrom. 1994;5:966–975. doi: 10.1016/1044-0305(94)80015-4. [DOI] [PubMed] [Google Scholar]
- 26.Zaia J, Biemann K. J Am Soc Mass Spectrom. 1995;6:428–436. doi: 10.1016/1044-0305(95)00018-9. [DOI] [PubMed] [Google Scholar]
- 27.Biemann K. Biomed Environ Mass Spectrom. 1988;16:99–111. doi: 10.1002/bms.1200160119. [DOI] [PubMed] [Google Scholar]
- 28.Bartlet-Jones M, Jeffery W A, Hansen H F, Pappin D J C. Rapid Commun Mass Spectrom. 1994;8:737–742. doi: 10.1002/rcm.1290080916. [DOI] [PubMed] [Google Scholar]
- 29.Liao P-C, Allison J. J Mass Spectrom. 1995;30:511–512. [Google Scholar]
- 30.Liao P-C, Huang Z-H, Allison J. J Am Soc Mass Spectrom. 1997;8:501–509. [Google Scholar]
- 31.Schurch S, Scott J R, Wilkins C L. Int J Mass Spectrom Ion Processes. 1997;169/170:141–152. [Google Scholar]
- 32.Stimson E, Truong O, Richter W J, Waterfield M D, Burlingame A L. Int J Mass Spectrom Ion Processes. 1997;169/170:231–240. [Google Scholar]
- 33.Spengler B, Luetzenkirchen F, Metzger S, Chaurand P, Kaufmann R, Jeffery W, Bartlet-Jones M, Pappin D J C. Int J Mass Spectrom Ion Processes. 1997;169/170:127–140. [Google Scholar]
- 34.Huang Z-H, Wu J, Roth K D W, Yang Y, Gage D A, Watson J T. Anal Chem. 1997;69:137–144. doi: 10.1021/ac9608578. [DOI] [PubMed] [Google Scholar]
- 35.Strahler J R, Smelyanskiy Y, Lavine G, Allison J. Int J Mass Spectrom Ion Processes. 1997;169/170:111–126. [Google Scholar]
- 36.Dikler S, Kelly J W, Russell D H. J Mass Spectrom. 1997;32:1337–1349. doi: 10.1002/(SICI)1096-9888(199712)32:12<1337::AID-JMS599>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 37.Dongre A R, Jones J L, Somogyi A, Wysocki V H. J Am Chem Soc. 1996;118:8365–8374. [Google Scholar]
- 38.Burlet O, Orkiszewski R S, Ballard K D, Gaskell S J. Rapid Commun Mass Spectrom. 1992;6:658–662. doi: 10.1002/rcm.1290061106. [DOI] [PubMed] [Google Scholar]
- 39.McCormack A L, Somogyi A, Dongre A R, Wysocki V H. Anal Chem. 1993;65:2859–2872. doi: 10.1021/ac00068a024. [DOI] [PubMed] [Google Scholar]
- 40.Beavis R C, Chaudhary T, Chait B T. Org Mass Spectrom. 1992;27:156–158. [Google Scholar]
- 41.Arnott D, O’Connell K L, King K L, Stults J T. Anal Biochem. 1998;258:1–18. doi: 10.1006/abio.1998.2566. [DOI] [PubMed] [Google Scholar]
- 42.Shevchenko A, Wilm M, Vorm O, Mann M. Anal Chem. 1996;68:850–858. doi: 10.1021/ac950914h. [DOI] [PubMed] [Google Scholar]
- 43.Burlet O, Yang C-Y, Gaskell S J. J Am Soc Mass Spectrom. 1992;3:337–344. doi: 10.1016/1044-0305(92)87061-3. [DOI] [PubMed] [Google Scholar]
- 44.Cox K A, Gaskell S J, Morris M, Whiting A. J Am Soc Mass Spectrom. 1996;7:522–531. doi: 10.1016/1044-0305(96)00019-0. [DOI] [PubMed] [Google Scholar]
- 45.Lomant A J, Fairbanks G. J Mol Biol. 1976;104:243–261. doi: 10.1016/0022-2836(76)90011-5. [DOI] [PubMed] [Google Scholar]
- 46.Traut R J, Bollen A, Sun T T, Hershey J W B, Sunberg J, Pierce L R. Biochemistry. 1973;12:3266–3273. doi: 10.1021/bi00741a019. [DOI] [PubMed] [Google Scholar]
- 47.Tang X-J, Boyd R K. Rapid Commun Mass Spectrom. 1992;6:651–657. doi: 10.1002/rcm.1290061105. [DOI] [PubMed] [Google Scholar]
- 48.Dean J A. Lange’s Handbook of Chemistry. 14th Ed. New York: McGraw Hill; 1992. [Google Scholar]
- 49.Annan R S, Carr S A. Anal Chem. 1996;68:3413–3421. doi: 10.1021/ac960221g. [DOI] [PubMed] [Google Scholar]
- 50.Chait B T, Wang R, Beavis R C, Kent S B H. Science. 1993;262:89–92. doi: 10.1126/science.8211132. [DOI] [PubMed] [Google Scholar]
- 51.Patterson D H, Tarr G E, Regnier F E, Martin S A. Anal Chem. 1995;67:3971–3978. doi: 10.1021/ac00117a024. [DOI] [PubMed] [Google Scholar]
- 52.Quin J, Steenvoorden R J J M, Chait B T. Anal Chem. 1996;68:1784–1791. doi: 10.1021/ac9511612. [DOI] [PubMed] [Google Scholar]
- 53.Shevchenko A, Jensen O N, Podtelejnikov A V, Sagliocco F, Wilm M, Vorm O, Mortensen P, Shevchenko A, Boucherie H, Mann M. Proc Natl Acad Sci USA. 1996;93:14440–14445. doi: 10.1073/pnas.93.25.14440. [DOI] [PMC free article] [PubMed] [Google Scholar]