

Abstract
Facile “writing” of DNA fragments that encode entire gene sequences potentially has widespread applications in biological analysis and engineering. Rapid writing of open reading frames (ORFs) for expressed proteins could transform protein engineering and production for protein design, synthetic biology, and structural analysis. Here we present a process, protein fabrication automation (PFA), which facilitates the rapid de novo construction of any desired ORF from oligonucleotides with low effort, high speed, and little human interaction. PFA comprises software for sequence design, data management, and the generation of instruction sets for liquid-handling robotics, a liquid-handling robot, a robust PCR scheme for gene assembly from synthetic oligonucleotides, and a genetic selection system to enrich correctly assembled full-length synthetic ORFs. The process is robust and scalable.
Keywords: gene assembly, automation, synthetic ORF, protein expression, fabrication
The potential of synthetic gene construction has long been recognized, starting in 1979 with the enzymatic assembly of a single suppressor tRNA gene from short oligonucleotides (Sekiya et al. 1979). Subsequently, many enzymatic assembly methods have been explored, using simple ligation (Heyneker et al. 1976; Goeddel et al. 1979), the FokI enzyme (Mandecki and Bolling 1988), serial cloning (Hayden and Mandecki 1988), and the polymerase chain reaction (Dillon and Rosen 1990). The construction of a single gene has been extended to the construction of gene clusters (Kodumal et al. 2004), plasmids (Stemmer et al. 1995), and viral genomes (Cello et al. 2002; Smith et al. 2003).
Most synthetic gene approaches have been limited to the construction of single genetic elements. For protein engineering projects such as the redesign of binding specificity (de Lorimier et al. 2002; Shifman and Mayo 2002; Kortemme et al. 2004), introduction of novel activities (Dwyer et al. 2004), design of new protein folds (Kuhlman et al. 2003), or construction of fragments for protein crystallization and NMR structural studies (Zheng et al. 2005), it is desirable to synthesize in parallel many closely related alleles of defined sequence for a single protein. Synthesis of oligonucleotides on arrayed microchips has been used to multiplex gene construction. Such applications have produced a 180-base-pair (bp) DNA sequence (Richmond et al. 2004), a dozen genes (Zhou et al. 2004), and a 14.6-kb operon (Tian et al. 2004). In this method, an oligonucleotide array synthesized on a spatially addressable glass chip is harvested as a large mixed pool; individual, full-length genes are subsequently constructed by a PCR-mediated assembly of their pool. Although a collection of nonhomologous genes can be constructed in this way, the approach is not suitable for constructing multiple, closely related alleles with uniquely predefined sequences, because the assembly step will combinatorially scramble near-identical sequences by cross-hybridization. Here we present a method, protein fabrication automation (PFA), that multiplexes the synthesis of closely related open reading frames (ORFs) from oligonucleotides by maintaining addressability of each individual gene during the assembly process. This method has utility in protein design and engineering for the simultaneous construction of relatively large numbers of variants of predetermined sequence and for the construction of protein fragments for structural studies.
Results
Automation scheme
The PFA scheme assembles complete ORFs from synthetic oligonucleotides (Fig. 1). Addressability is maintained by confining each gene to a separate well in a 96-well microplate. Sequences of arbitrary length and (dis)similarity can be assembled in this way. Successfully assembled sequences are enriched using a genetic selection scheme that takes advantage of the fact that sequences encode ORFs: the full-length PCR product is cloned in a vector that places a genetic marker (chloramphenicol acetyltransferase) in frame with the encoded synthetic ORF. This arrangement suppresses frameshift errors in the ORF, which are the most common errors using synthetic oligonucleotides, due to the synthesis errors that introduce single base deletions (Temsamani et al. 1995). Specialized software is used to guide design of a suitable assembly scheme and maintain flow of information and materials. Five components are therefore integrated in the resulting PFA process pipeline:
Figure 1.
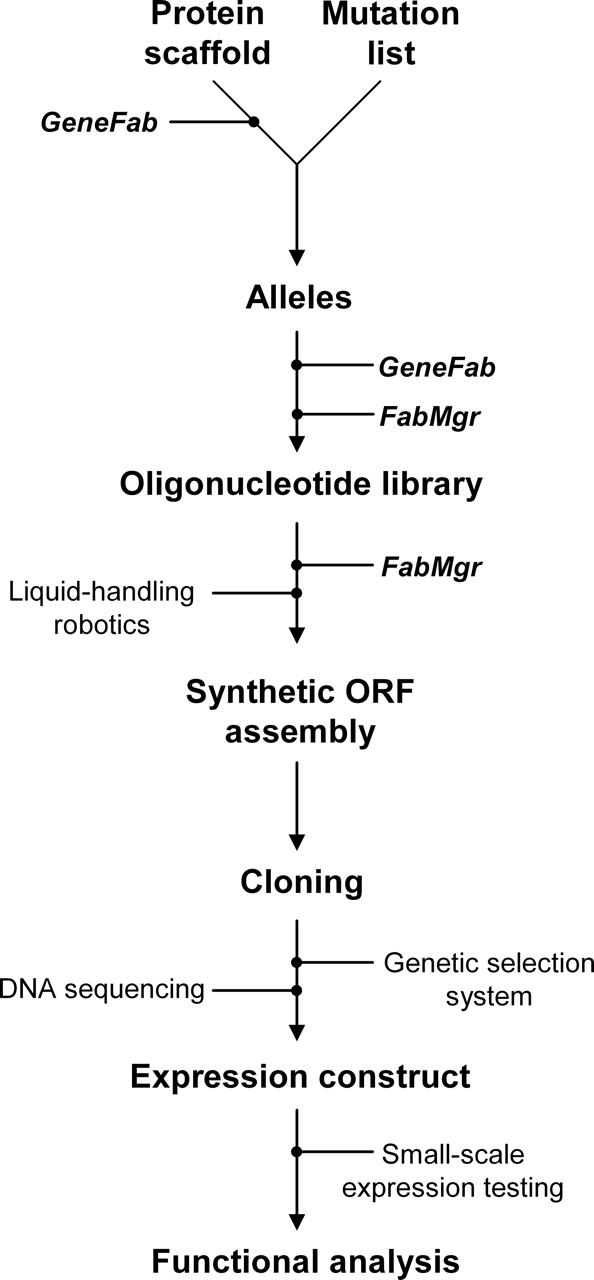
The protein fabrication automation (PFA) process.
Software to design the oligonucleotide topology (length, location, orientation) for ORF assembly, to generate DNA sequences for multiple alleles, to maintain a database of oligonucleotides for minimization of synthesis cost and effort, to generate an automated instruction set (program) for liquid-handling robots, and to maintain the flow of information and material.
Liquid-handling robotics to mix the required subset of synthetic oligonucleotides for each individual ORF in separate microplate wells.
A robust PCR gene assembly scheme.
A genetic selection scheme to suppress frameshift mutations in the final cloned synthetic ORF product.
DNA sequencing to confirm the synthetic ORF sequence.
The PCR scheme to assemble full-length synthetic ORFs
All synthetic gene construction technologies produce final, full-length products by hybridization-mediated assembly of a collection of individual oligonucleotides with overlapping ends (Heyneker et al. 1976; Goeddel et al. 1979; Sekiya et al. 1979). Recently developed techniques couple the assembly step with gene amplification (Dillon and Rosen 1990; Stemmer et al. 1995). The critical factor in selecting a suitable assembly scheme for automation is not maximum length that can be achieved in a one-step reaction nor minimization of iterative rounds of assembly (although this is desirable to avoid introduction of polymerase-mediated replication errors), but its robustness: The reactions need to work always under one set of defined conditions for all assemblies.
We found coupled PCR/ligation-based gene assembly (Strizhov et al. 1996; Seyfang and Jin 2004) to be more costly, less efficient, and much less robust than typical PCR-only-based methods. Classical PCR-based assembly schemes (Dillon and Rosen 1990; Stemmer et al. 1995) are often sensitive to small changes in reaction conditions and can show an unpredictable dependence on sequence (Lin et al. 2002; Gao et al. 2003; Young and Dong 2004). We found that the inside-out nucleation (ION) scheme (Gao et al. 2003) is highly robust. In this scheme DNA fragments are assembled from nested pairs of oligonucleotides, the inner one of which nucleates the reaction (Fig. 2A). In our implementation of the ION-PCR scheme, we use individual oligonucleotides 50–60 bases in length with 25-bp overlaps. The 60-bp limit is imposed by constraints in the current exigencies of commercial manufacturing (cost per base of that length; the avoidance of oligonucleotide purification). We anticipate that these will change over time. In general, it is advantageous to use the longest oligonucleotides available, which still maintain cost effectiveness, because increasing oligonucleotide length diminishes the complexity of the PCR assembly scheme.
Figure 2.

Synthetic ORF assembly scheme. Lengths, locations, and orientations of oligonucleotides (half arrows) and fragments (double arrows) are shown to scale for (A) the ION-PCR assembly and (B) the SOE-PCR to assemble a full-length gene in this three-fragment scheme. Letters “s” and “a” indicate sense or antisense oligonucleotides, respectively; “A,” “B,” and “C” indicate subfragment identity.
Under these conditions ION-PCR robustly assembles fragments up to 375 or 445 bp in length, using five or six nested pairs, respectively. Although longer fragments can be assembled by ION-PCR using more nested pairs, we find that the schemes become sensitive to DNA sequence at ≥6 oligonucleotide pairs (see Electronic Supplemental Material), and require “thermal balancing” (approximate matching of the stabilities of the oligonucleotide overlap regions; Hoover and Lubkowski 2002). Assembly of long fragments that exceed six nested pairs (450-bp limit, using our currently selected oligonucleotide and overlap lengths) decreases process robustness and is therefore not suitable for automation. Based on these observations, we opted to develop a two-step assembly scheme, in which subfragments are generated by ION-PCR in a primary round of reactions, which are then combined in a second round of PCR reactions using splice overlap extension (SOE, Fig. 2B) to assemble full-length synthetic ORFs. Although this approach increases the number of steps, which is not desirable for handcrafted gene construction, it fulfills the robustness criterion essential for automation. Furthermore, we found that it is not necessary to purify the primary reaction products of the ION-PCR: mixing 100-fold dilutions of unpurified primary reaction fragments with flanking primers permits robust assembly of full-length fragments in a secondary SOE-PCR assembly. We have been able to routinely assemble full-length ORFs up to 1.3 kb from four fragments (Fig. 3).
Figure 3.

Full-length assembly for genes of varying lengths. A, ankyrin binding protein (Binz et al. 2004), 519-bp ORF (two primary ION-PCR fragments are combined into the full-length gene by SOE-PCR). G, glucose binding protein from T. maritima, 927-bp ORF (three primary fragments). M, maltose binding protein from E. coli, 1125-bp ORF (four primary fragments).
The ION-PCR and SOE-PCR reaction conditions were optimized to further increase the robustness of the assembly method. Factors that were tested include choice of DNA polymerase and buffer, total and individual oligonucleotide concentrations, oligonucleotide purity, annealing temperature, and number of thermocycling steps (Electronic Supplemental Material). We, and others, have found that the DNA polymerase choice is a critical determinant for robust assembly (Wu et al. 2006), with KOD polymerase derived from Thermococcus kodakaraensis being the most successful (Takagi et al. 1997). Use of a concentration gradient for the ION pair oligonucleotides is also critical. Best results are obtained with a low inner pair concentration, and the others increasing in a geometric progression:
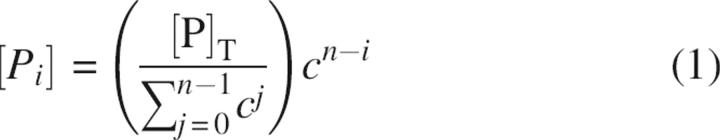 |
where [Pi] is the oligonucleotide concentration of the combined ith pair of primers (sense and anti-sense), [P]T is the chosen total oligonucleotide concentration, n is the total number of primer pairs, and c is a constant. Robust assemblies are observed for 0.65 ≤ c ≤ 0.75 and [P]T = 600 nM. Under these conditions, the reactions are not very sensitive to the annealing temperature (56°C < T m < 64°C) or number of thermal cycling steps (N > 12).
Automated setup of the assembly scheme and selection of oligonucleotides
Two programs have been developed for PFA (Fig. 4): GeneFab, to set up ORF topology and convert mutation lists into oligonucleotide sequences, and FabMgr, to maintain the flow of information, physical materials, and operations. GeneFab first generates an oligonucleotide scaffold from user-specified constraints. Given the length of a synthetic ORF (lorf), the maximum length of each oligonucleotide (lo), the maximum number of sense–anti-sense pairs in an ION primary fragment (np), splice-overlap length (of), and ION pair overlap length (oo), a sequence-independent primer topology is designed, specifying length, position, and orientation of all oligonucleotides. If these constraints can be satisfied, the algorithm determines SOE fragment end points and numbers of ION pairs per fragment by simple arithmetic; thus the number of fragments (nf) and the ION primary fragment size (lfrag) are determined by
Figure 4.

Software control for the flow of information, material, and operations. (1) User-specified amino acid sequence, sequence restrictions (e.g., forbidden restriction endonuclease sites, G:C content) and oligonucleotide and PCR fragment length limits are used by GeneFab to generate a gene scaffold assembly topology (positions and orientations of oligonucleotides). (2) User-specified reaction conditions for the ION- and SOE-PCR assemblies. (3) Allele formation is driven by mutation lists provided either by a user or by computational protein design algorithms. These are converted by GeneFab into specific oligonucleotide sequences using the scaffold topology parameters. (4) FabMgr queries the oligonucleotide database to identify duplicate oligonucleotides and generate synthesis orders for new oligonucleotide sequences. (5) New oligonucleotide microplates are added to physical and virtual inventory by FabMgr. (6) The Materials list contains all the information (reagents, oligonucleotides, source and destination locators, plasticware) required to assemble the alleles. (7) Liquid-handling robot requires specification of the robotic deck geometry (position of plates, source of water, enzyme, tips). (8) Human action is required to load the robotic deck with the correct oligonucleotides plates from the collection, plasticware (reaction plates, tips), and reagents (water, enzyme, and oligonucleotides as specified in the Materials list).
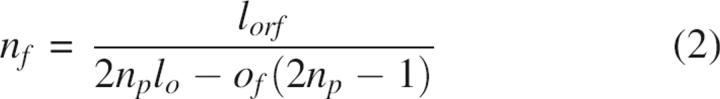 |
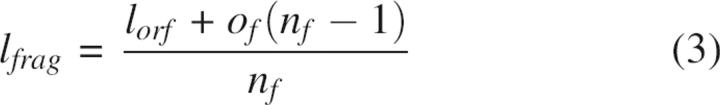 |
Oligonucleotide ION start and end points within fragments are decided in a two-pass scan. In the first pass, a maximum fragment length is calculated by using the user-specified limits for oligonucleotide length and primer pair constraints. The difference between this maximum length and the actual required fragment length (lfrag) is the excess length (Δl); if Δl < 0 in the first pass, the user-specified constraints cannot be satisfied, the process fails, and GeneFab prompts the user for a new set of constraints. In the second pass, Δl is used to calculate the final oligonucleotide lengths (lfinal), which are adjusted to give Δl = 0. Fragments are often not perfectly divisible by the total number of primers, resulting in the assignment of the 3′-most oligonucleotides being assigned one less base in length than the 5′-most oligonucleotides. This maximum value and final length calculation is repeated for each primary fragment in the ORF:
 |
The resulting assembly schemes are subsequently annotated with oligonucleotide concentrations to be used in ION-PCR gene amplification (see above). A starting, wild-type DNA sequence (“gene scaffold”) can either be imported or is generated by reverse translation of an amino acid sequence, using a codon table that is annotated to preferentially use codons associated with high levels of protein expression (Ikemura 1985; Kane 1995; Kurland and Gallant 1996; Baca and Hol 2000). Reverse translation is further constrained by a set of forbidden DNA sequences that prevent introduction of out-of-place sites within the ORF, including restriction sites used for cloning (see below) or regulatory elements such as ribosome-binding sites. GeneFab also adds appropriate flanking sequences for cloning and gene expression (Supplemental material). Once a scaffold topology has been designed, it remains constant for the design of alleles with point mutations.
Alleles are generated either by hand, using a graphical user interface to enter the desired mutations, or in batch mode from mutation lists. The latter mode in particular exploits the power of PFA by enabling facile generation of many variants that may have been specified by computational design (Hellinga and Richards 1991; Pinto et al. 1997; Benson et al. 1998; Looger et al. 2003) or fragment identification algorithms (Brezellec et al. 2006; Lubovac et al. 2006). The resulting list of oligonucleotides and their assembly relationships is passed to the FabMgr program, which manages the flow of material and operations in the PFA pipeline.
For each gene scaffold, FabMgr (Fig. 4) maintains a relational database (the oligonucleotide database) recording the DNA sequences and well locators of all oligonucleotides that have been used to engineer the gene, assembly combinations, and reaction conditions for generating each full-length construct. This database is used in three ways: first, it specifies the oligonucleotides needed for setting up both ION- and SOE-PCR gene assembly reactions (see below); second, it is used to generate orders for the synthesis of new oligonucleotides that are not present in the current collection for the gene scaffold; third, it manages the link between the virtual and physical oligonucleotide collection by maintaining microplate and well-locator information. The elimination of duplicates is essential when designing variants of single sequences, as many alleles will have regions of identical sequence, sharing oligonucleotides (we observe that the need for new syntheses diminishes during the course of a project with the reuse of oligonucleotides).
It is critical to maintain synchrony between the virtual (in the oligonucleotide database) and physical (in freezers) collection of oligonucleotides. Oligonucleotide orders are therefore generated automatically by FabMgr and synthesized in bar-coded microplates. Additionally, contamination of the collection is prevented by replicating stock plates into working plates that are used by the liquid-handling robot (see below).
Automation of the gene assembly reactions
Mixing of the oligonucleotides from the collection, buffers, and enzymes for the PCR-mediated assembly reactions, temperature cycling, dilutions, and materials handling are all carried out by conventional liquid-handling robots using 96-well microplates. To support PFA, liquid-handling robots need to have a multipipetting arm suitable for 96-well microplates, pipetting range of 1 μL to 180 μL per reaction well, aspirate and dispense functions that are individually addressable for each pipette, disposable pipette tips, and script-driven programmability.
Genes are assembled in two steps: generation of the primary fragments by ION-PCR (Fig. 2A), which are then combined appropriately and diluted 100-fold into a second reaction that assembles the full-length product by SOE-PCR (Fig. 2B). For a typical full-length 1-kb assembly, using three fragments of five ION pairs each, about 3300 pipetting steps (oligonucleotide cherry picking, reagents dispensing) are required to set up a 96-well primary reaction plate (32 alleles) followed by ∼230 steps (dilutions and reagent dispensing) to set up the secondary reaction: The need for automation is manifest.
Pipetting algorithms need to satisfy three criteria: avoidance of cross-contamination between reaction wells, minimization of disposable tips use to contain plasticware cost, and optimization of mechanical arm travel time (reagent master mix or oligonucleotide stock to reaction vessel; tip pickup and discard). Two dispense methods are used (Fig. 5): submersion of the pipette tip into the reaction well followed by content expulsion and rinsing with the reactants (submerged mixing) or expulsion of a drop above the surface of the reaction well (aerial expulsion). The former is limited to a single aspiration/dispense cycle bracketed by tip pickup and discard steps; the latter allows a pipette to be used multiple times in successive dispensals for a single aspiration step before the discard, minimizing tip usage and travel time. Figure 5 illustrates how the tip pickup, aspiration, dispense, and discard operations are combined for the dispensing of water (Fig. 5A) or enzyme (Fig. 5B) and cherry picking of oligonucleotides (Fig. 5C).
Figure 5.

Robotic pipetting operations. (A) Dispensing of water does not require prevention of cross-contamination or mixing. A simple multitip, multidispense fills all wells. (B) Submerged mixing for the addition of reagents to wells containing oligonucleotides requires tip discards between mixing tips. (C) Cherry picks of individual oligonucleotides requires aerial expulsion, single-tip addressability, and arbitrarily complex travel paths of the dispensing arm. Some destinations are shown for different sources (oligonucleotide libraries) and destinations (ION-PCR reaction plates). Note the use of single tips for the same oligonucleotide.
Water is dispensed first; the use of a single row of tips (Fig. 5A) minimizes both tip use and travel time. The master mix is dispensed last and requires a mixing step; therefore, only travel time can be minimized, as each well requires a separate tip (Fig. 5B). Above a critical volume (typically 3 μL), oligonucleotides can be dispensed with aerial expulsion. A single tip can therefore serve multiple wells in a multidispense operation, and several tips can be filled simultaneously to minimize travel time (Fig. 5C). To further limit travel time, plates are processed in order, one at a time; occasionally, therefore, not all eight tips are used (as shown in Fig. 5C). For dispensal of very small volumes (<3 μL), submerged mixing has to be used, and neither tip usage nor travel time can be optimized.
Table 1 summarizes the operations for setting up a plate of primary and secondary reactions. The FabMgr program produces a machine-independent (object) code that describes the assembly of the full-length gene products in generic operations (tip pick up, discard, individual tip aspirations, individual tip dispenses, arm moves from reagent or oligonucleotide stock plate positions to reaction wells). The object code is then converted into instrument-specific scripts. In the limit, about 1200 pipette tips would be needed to set up the example given above without optimization. We find that assembly reactions for a typical set of assemblies requiring 10–14 mutations per allele requires about 900 tips and takes 5 h including two thermal cycling steps of 45 min each. Figure 6 shows a set of 32 independent, full-length products generated fully automatically from 95 oligonucleotides.
Table 1.
Operations, machine use, and time for a typical PFA run

Figure 6.

Automated full-length gene assembly. Thirty-two alleles of Thermatoga maritima glucose binding protein (three primary ION-PCR fragments per allele) were assembled fully automatically from 96 primary ION-PCR fragments (top two panels) to yield full-length 927-bp ORFs by SOE-PCR (bottom panel).
Selection of full-length, in-frame synthetic ORFs
Oligonucleotides synthesized using phosphoramidite chemistry (Caruthers et al. 1983) are contaminated with single-base deletions that cause frameshift mutations that are the primary source of error in gene assembly reactions (Temsamani et al. 1995; Tian et al. 2004). To suppress such frameshifts, we clone the full-length synthetic ORFs in-frame with a C-terminal selectable marker, chloramphenicol acetyl transferase (Maxwell et al. 1999), in a specially constructed synthetic ORF selection (SOS) vector (Fig. 7A). Chloramphenicol-resistant colonies are then picked and the DNA sequence of the synthetic ORF determined. Similar strategies have been used by others also to select for full-length ORFs by antibiotic resistance (Seehaus et al. 1992; Daugelat and Jacobs 1999; Lutz et al. 2002) or to screen for or improve protein expression levels (Nixon and Benkovic 2000; Mossner et al. 2001; Cabantous et al. 2004). To test the efficiency of the selection step, we constructed a green fluorescent protein (GFPuv) gene (Crameri et al. 1996) by PFA and assessed the fraction of successful assemblies by counting green colonies. Active and inactive colonies were sequenced. Without selection 28% of the clones are functional, using unpurified oligonucleotides; with selection, 82% of the clones are functional. With this success rate, an average of only 1.2 clones needs to be sequenced to identify a correctly assembled synthetic ORF. Sequence analysis demonstrates that the genetic selection system removes all frameshifts present in an assembly reaction when assaying only functional clones (or 50-fold reduction in frameshifts overall; Table 2) while greatly enriching mutation-free clones (Fig. 8). Furthermore, the use of the SOS vectors obviates the need for PAGE-purified oligonucleotides, providing a significant savings in both cost and effort. Other gene assembly methods require PAGE-purified oligonucleotides to facilitate gene synthesis (Stemmer et al. 1995; Strizhov et al. 1996; Hoover and Lubkowski 2002; Gao et al. 2003; Carr et al. 2004; Seyfang and Jin 2004; Tian et al. 2004; Xiong et al. 2004; Young and Dong 2004).
Figure 7.

PFA vectors. (A) synthetic ORF selection plasmid (pSOS), used to select correctly assembled full-length ORFs from the SOE-PCR products. (B) synthetic ORF expression plasmid (pSOX), into which ORFs from pSOS are recloned for protein expression. (C) The synthetic ORF selection and expression plasmid (pSOS-X) combines the functionalities of pSOS and pSOX, obviating a recloning step. plac, lactose promoter; ptac, lactose/tryptophan fusion promoter; SD, Shine–Dalgarno sequence; His6, polyhistidine purification tag; UGA, amber stop codon; CS, cloning site; ori, origin of replication.
Table 2.
Efficacy of full-length ORF selection

Figure 8.

Total mutation frequency per clone. Insertions, deletions, point mutations (nonsense, missense, silent) are counted as mutational events. (White bars) no genetic selection; (black bars) SOS genetic selection.
Protein expression
Full-length synthetic ORFs are recloned into either pSOX (Fig. 7B) or standard protein expression vectors such as the pET series that utilize the T7 promoter (Studier et al. 1990). Expression is induced using autoinduction medium (Studier 2005) and tested either by gel electrophoresis or MALDI-TOF mass spectrometry on crude lysates.
Combined ORF selection and expression.
The genetic selection system and protein overexpression elements have been combined into one vector, pSOS-X (Fig. 7C). A suppressible stop codon facilitates genetic selection in permissive strains while prohibiting the fusion of chloramphenicol resistance in expression strains. This obviates the need for subcloning steps after genetic selection.
The integrated PFA pipeline
Pipelines can be characterized by their length (time taken to complete), width (number of products per run), and scalability (number of pipes that can be run in parallel). The minimum length of the PFA pipeline is 4 or 7 d using pSOS-X or pSOS, respectively (Fig. 9). Its width is 96/n or 384/n, where n is the number of SOE fragments per assembly scaffold and the numerator is the microplate format. There are multiple stagger points that allow pipes to be run in parallel without overlap of the same resource (not shown).
Figure 9.

PFA pipeline timeline: (A) Using the pSOS genetic selection plasmid, followed by recloning into the pSOX or a pET expression vector; (B) using the combined genetic selection and expression plasmid pSOS-X.
Discussion
The protein fabrication automation (PFA) method described here multiplexes the assembly of closely related synthetic open reading frames from oligonucleotides using a combination of a robust PCR gene assembly scheme, liquid-handling robotics, genetic selection to enrich correctly assembled ORFs, and software to maintain the flow of operations and materials. We have been able to routinely assemble 24–48 ORFs per run (up to 1.3 kb in length) in one pipeline cycle (∼1–2 wk), and can parallelize 2–3 pipelines at a given time. In this mode, we have constructed over 600 unique synthetic ORFs in nine protein scaffolds. This method is invaluable for protein engineering and design experiments in which large numbers of designs of explicitly defined sequence need to be constructed. One advantage that PFA has over general schemes for assembling large DNA segments (Kodumal et al. 2004; Shevchuk et al. 2004) is that function is inherently encoded in the genes of interest, enabling the use of simple selection schemes that greatly enhance the yield of correctly assembled segments. The PFA method presented here can be further optimized using coupled in vitro transcription and translation (Littlefield et al. 1955; Matthaei and Nirenberg 1961) of the full-length ORFs, obviating the need for molecular cloning, and can be applied to high-throughput assay systems (Aharoni et al. 2005; Miller et al. 2006).
Materials and methods
Software
GeneFab is written in Java and supports both a graphical user interface (GUI) and batch mode; FabMgr is written in C and driven by a text-based interface (a Java-based GUI is under development). The oligonucleotide relational database is maintained as a human-readable flat file and queried/updated with functions specialized to FabMgr. Software and documentation is freely available for download at http://www.biochem.duke.edu/hellinga/pfa.html.
Oligonucleotide synthesis
Oligonucleotides were purchased from Integrated DNA Technologies in 96-well format (100 μM concentration, 10 nmol scale) and used without further purification.
PCR reaction conditions
Primary ION-PCR reactions were carried out in a 50-μL total reaction volume containing 600 nM total oligonucleotide concentration, with individual oligonucleotide concentrations varying according to Equation 1 (c = 0.7), one unit Thermococcus kodakaraensis (KOD) Hot-Start DNA polymerase (EMD Biosciences), 40 μM each dNTP, 0.5 mM MgSO4, and 1/10 reaction volume of 10× KOD Hot-Start Buffer (EMD Biosciences). Reactions were set up by the liquid-handling robot and assembled in 16 thermocycling steps (94°C, 15 sec; 60°C, 30 sec; 68°C, 30 sec) after an initial 2-min preincubation at 95°C to denature the inhibitory antibody in the Hot-Start preparation. Secondary SOE-PCR reactions were set up automatically by diluting the primary reactions threefold with water (100 μL to 50 μL reaction). Primary reactions were combined (1.5 μL per diluted fragment reaction) into a new microplate for splice-overlap extension (Horton et al. 1989). Primer encoding for the beginning of the sense and antisense strand (200 nM; see Fig. 2B) were added, and the full-length ORF was concatenated in 16 thermocycling steps (94°C, 15 sec; 60°C, 30 sec; 72°C, 30 sec), after another 2-min preincubation at 95°C for antibody denaturation.
Liquid-handling robot
A 2-m Genesis Freedom (Tecan) liquid-handling platform was used for pipetting and PCR automation. The instrument included one gripping robotic arm, one eight-channel liquid-handling arm, a fast-wash module, low-volume tubing, pinch valves, disposable tip adaptors, an integrated PTC-200 PCR machine (Bio-Rad Laboratories), and carbon-impregnated disposable tips for liquid sensing during aspiration (to minimize reagent waster and verify reagent presence) but not dispensal. The various liquid classes that were used are available for download at http://www.biochem.duke.edu/hellinga/pfa.html.
SOS vector
The synthetic ORF selection (pSOS; GenBank EF030424; Electronic Supplemental Material Fig. 1a) vector combines an origin of replication from pUC18, a kanamycin resistance marker from Tn903, and a cloning site for synthetic ORFs consisting of a plac promoter with HpaI and PacI cloning sites. In this vector, the start codon and chloramphenicol resistance gene are out of frame, resulting in antibiotic sensitivity without a synthetic ORF inserted between the PacI and HpaI sites. pSOS is propagated in medium containing kanamycin.
Expression vectors
The selected ORF expression (pSOX; GenBank EF030425; Electronic Supplemental Material Fig. 1b) vector was assembled using standard enzyme-based cloning methods (Sambrook and Russell 2001). The vector contains a pMB1 origin of replication and an ampicillin resistance gene (from Tn3) for propagation. A synthetic cloning site was constructed consisting of a ptac promoter (Russell and Bennett 1982), a highly optimized Shine–Dalgarno site (Curry and Tomich 1988; Chen et al. 1994; Shultzaberger et al. 2001; Ma et al. 2002), and PacI and PmeI cloning sites. Additionally, a hexahistidine purification sequence tag (Smith et al. 1998) is placed to create a C-terminal fusion to the ORF. The cloning site is followed by the Escherichia coli rRNA transcriptional terminator sequence rrnB T1T2 (Glaser et al. 1983; Sarmientos et al. 1983). Finally, pSOX also possesses the lactose operon repressor, lacIq, to minimize basal transcription. The synthetic ORF selection and expression (pSOS-X; GenBank EF030426; Electronic Supplemental Material Fig. 1c) vector is nearly identical to pSOX with the addition of a chloramphenicol resistance gene inserted into the cloning site (with NaeI and PacI sites). An amber stop codon (TGA) is placed after the polyhistidine tag and before the chloramphenicol resistance gene. The origin of the genetic elements used to construct these vectors is provided as Electronic Supplemental Material. Alternatively, sequenced ORFs can be expressed under control of the T7 promoter (Studier et al. 1990) in a pET vector (EMD Biosciences).
Protein expression
Verified ORFs placed in pSOX or pET vectors were transformed into BL21 or BL21 (DE3) cells (Stratagene), respectively. When utilizing the pSOS-X vector, a ligated vector was first transformed into E. coli tRNA suppressive strain LE392 (ATCC #33572) for the purpose of genetic selection, then sequence verified and retransformed into BL21 cells for expression. Cultures were grown either in small scale (1–5 mL) or large scale (250–1000 mL) in ZYM-5052 autoinducing media (Studier 2005) at 30°C or 37°C overnight.
After incubation, cultures were pelleted and lysed by the addition of 5 mL BugBuster Master Mix (EMD Biosciences) per gram of wet cell paste at room temperature for 20 min, and the insoluble fraction was pelleted via centrifugation at 10, 000g for 15 min. At this point, the soluble fraction can be purified with nickel NTA sepharose (Smith et al. 1998) or assayed. Using standard methods, expression fractions can be analyzed with SDS-PAGE analysis (Sambrook and Russell 2001). Alternatively, expression can be verified using MALDI-TOF mass spectrometry. The soluble fraction (1 μL) was quickly mixed with 9 μL freshly prepared matrix (saturated sinapinic acid [Sigma-Aldrich] in 50% acetonitrile, 49.9% water, 0.1% trifluoroacetic acid) and 1–2 μL of the resulting solution were spotted on a MALDI stainless steel plate, air dried for 5–10 min, and analyzed in a Voyager-DE mass spectrometer (Applied Biosystems).
Electronic supplemental material (ESM)
Annotated sequence maps of the pSOS, pSOX, and pSOS-X vectors (ESM-Figure 1a.pdf, ESM-Figure 1b.pdf, and ESM-Figure 1c.pdf).
A spreadsheet listing the genetic elements of the above vectors (ESM-Figure 2.pdf).
Several tables indicating the outcome of our various optimization efforts (ESM-Figure 3.pdf).
Expanded Materials and Methods covering those optimizations and other routine molecular biology steps (ESM-Figure 4.pdf).
In addition to the ESM, we host the following components at http://www.biochem.duke.edu/hellinga/pfa.html:
5. The GeneFab and FabMgr software suite and documentation.
6. The liquid class definitions file we have compiled for use on a Tecan robotic platform running Gemini software for PFA.
7. Tecan-specific scripts and software for PCR machine loading and thermal cycling automation.
Acknowledgments
This work was funded by grants from the NIH Director's Pioneer Award (5 DP1 OD000122) and the Department of Homeland Security HSARPA (W81XWH-05-C-0161).
Footnotes
Reprint requests to: Homme W. Hellinga, Nanaline Building, Room #413D, Research Drive, DUMC 3711, Duke University Medical Center, Durham, NC 27710, USA; e-mail: hwh@biochem.duke.edu; fax: (919) 684-8885.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.062591607.
Abbreviations: PFA, protein fabrication automation; ORF, open reading frame; PSD, protein scaffold database; SOE, splice-overlap extension; PAGE, poly-acrylamide gel electrophoresis; SOS, synthetic ORF selection; SOX, selected ORF expression; SOS-X, synthetic ORF selection and expression.
Supplemental material: see www.proteinscience.org
References
- Aharoni A., Griffiths, A.D., and Tawfik, D.S. 2005. High-throughput screens and selections of enzyme-encoding genes. Curr. Opin. Chem. Biol. 9: 210–216. [DOI] [PubMed] [Google Scholar]
- Baca A.M. and Hol, W.G. 2000. Overcoming codon bias: A method for high-level overexpression of Plasmodium and other AT-rich parasite genes in Escherichia coli . Int. J. Parasitol. 30: 113–118. [DOI] [PubMed] [Google Scholar]
- Benson D.E., Wisz, M.S., Liu, W., and Hellinga, H.W. 1998. Construction of a novel redox protein by rational design: Conversion of a disulfide bridge into a mononuclear iron–sulfur center. Biochemistry 37: 7070–7076. [DOI] [PubMed] [Google Scholar]
- Binz H.K., Amstutz, P., Kohl, A., Stumpp, M.T., Briand, C., Forrer, P., Grutter, M.G., and Pluckthun, A. 2004. High-affinity binders selected from designed ankyrin repeat protein libraries. Nat. Biotechnol. 22: 575–582. [DOI] [PubMed] [Google Scholar]
- Brezellec P., Hoebeke, M., Hiet, M.S., Pasek, S., and Ferat, J.L. 2006. DomainSieve: A protein domain-based screen that led to the identification of dam-associated genes with potential link to DNA maintenance. Bioinformatics 22: 1935–1941. [DOI] [PubMed] [Google Scholar]
- Cabantous S., Terwilliger, T.C., and Waldo, G.S. 2004. Protein tagging and detection with engineered self-assembling fragments of green fluorescent protein. Nat. Biotechnol. 23: 102–107. [DOI] [PubMed] [Google Scholar]
- Carr P.A., Park, J.S., Lee, Y.J., Yu, T., Zhang, S., and Jacobson, J.M. 2004. Protein-mediated error correction for de novo DNA synthesis. Nucleic Acids Res. 32: e162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caruthers M.H., Beaucage, S.L., Becker, C., Efcavitch, J.W., Fisher, E.F., Galluppi, G., Goldman, R., deHaseth, P., Matteucci, M., and McBride, L., et al. 1983. Deoxyoligonucleotide synthesis via the phosphoramidite method. Gene Amplif. Anal. 3: 1–26. [PubMed] [Google Scholar]
- Cello J., Paul, A.V., and Wimmer, E. 2002. Chemical synthesis of poliovirus cDNA: Generation of infectious virus in the absence of natural template. Science 297: 1016–1018. [DOI] [PubMed] [Google Scholar]
- Chen H., Bjerknes, M., Kumar, R., and Jay, E. 1994. Determination of the optimal aligned spacing between the Shine–Dalgarno sequence and the translation initiation codon of Escherichia coli mRNAs. Nucleic Acids Res. 22: 4953–4957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crameri A., Whitehorn, E.A., Tate, E., and Stemmer, W.P. 1996. Improved green fluorescent protein by molecular evolution using DNA shuffling. Nat. Biotechnol. 14: 315–319. [DOI] [PubMed] [Google Scholar]
- Curry K.A. and Tomich, C.S. 1988. Effect of ribosome binding site on gene expression in Escherichia coli . DNA 7: 173–179. [DOI] [PubMed] [Google Scholar]
- Daugelat S. and Jacobs Jr, W.R. 1999. The Mycobacterium tuberculosis recA intein can be used in an ORFTRAP to select for open reading frames. Protein Sci. 8: 644–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lorimier R.M., Smith, J.J., Dwyer, M.A., Looger, L.L., Sali, K.M., Paavola, C.D., Rizk, S.S., Sadigov, S., Conrad, D.W., and Loew, L., et al. 2002. Construction of a fluorescent biosensor family. Protein Sci. 11: 2655–2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillon P.J. and Rosen, C.A. 1990. A rapid method for the construction of synthetic genes using the polymerase chain reaction. Biotechniques 9: 298–300. [PubMed] [Google Scholar]
- Dwyer M.A., Looger, L.L., and Hellinga, H.W. 2004. Computational design of a biologically active enzyme. Science 304: 1967–1971. [DOI] [PubMed] [Google Scholar]
- Gao X., Yo, P., Keith, A., Ragan, T.J., and Harris, T.K. 2003. Thermodynamically balanced inside-out (TBIO) PCR-based gene synthesis: A novel method of primer design for high-fidelity assembly of longer gene sequences. Nucleic Acids Res. 31: e143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaser G., Sarmientos, P., and Cashel, M. 1983. Functional interrelationship between two tandem E. coli ribosomal RNA promoters. Nature 302: 74–76. [DOI] [PubMed] [Google Scholar]
- Goeddel D.V., Kleid, D.G., Bolivar, F., Heyneker, H.L., Yansura, D.G., Crea, R., Hirose, T., Kraszewski, A., Itakura, K., and Riggs, A.D. 1979. Expression in Escherichia coli of chemically synthesized genes for human insulin. Proc. Natl. Acad. Sci. 76: 106–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayden M.A. and Mandecki, W. 1988. Gene synthesis by serial cloning of oligonucleotides. DNA 7: 571–577. [DOI] [PubMed] [Google Scholar]
- Hellinga H.W. and Richards, F.M. 1991. Construction of new ligand binding sites in proteins of known structure. I. Computer-aided modeling of sites with pre-defined geometry. J. Mol. Biol. 222: 763–785. [DOI] [PubMed] [Google Scholar]
- Heyneker H.L., Shine, J., Goodman, H.M., Boyer, H.W., Rosenberg, J., Dickerson, R.E., Narang, S.A., Itakura, K., Lin, S., and Riggs, A.D. 1976. Synthetic lac operator DNA is functional in vivo. Nature 263: 748–752. [DOI] [PubMed] [Google Scholar]
- Hoover D.M. and Lubkowski, J. 2002. DNAWorks: An automated method for designing oligonucleotides for PCR-based gene synthesis. Nucleic Acids Res. 30: e43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton R.M., Hunt, H.D., Ho, S.N., Pullen, J.K., and Pease, L.R. 1989. Engineering hybrid genes without the use of restriction enzymes: Gene splicing by overlap extension. Gene 77: 61–68. [DOI] [PubMed] [Google Scholar]
- Ikemura T.. 1985. Codon usage and tRNA content in unicellular and multicellular organisms. Mol. Biol. Evol. 2: 13–34. [DOI] [PubMed] [Google Scholar]
- Kane J.F.. 1995. Effects of rare codon clusters on high-level expression of heterologous proteins in Escherichia coli . Curr. Opin. Biotechnol. 6: 494–500. [DOI] [PubMed] [Google Scholar]
- Kodumal S.J., Patel, K.G., Reid, R., Menzella, H.G., Welch, M., and Santi, D.V. 2004. Total synthesis of long DNA sequences: Synthesis of a contiguous 32-kb polyketide synthase gene cluster. Proc. Natl. Acad. Sci. 101: 15573–15578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kortemme T., Joachimiak, L.A., Bullock, A.N., Schuler, A.D., Stoddard, B.L., and Baker, D. 2004. Computational redesign of protein–protein interaction specificity. Nat. Struct. Mol. Biol. 11: 371–379. [DOI] [PubMed] [Google Scholar]
- Kuhlman B., Dantas, G., Ireton, G.C., Varani, G., Stoddard, B.L., and Baker, D. 2003. Design of a novel globular protein fold with atomic-level accuracy. Science 302: 1364–1368. [DOI] [PubMed] [Google Scholar]
- Kurland C. and Gallant, J. 1996. Errors of heterologous protein expression. Curr. Opin. Biotechnol. 7: 489–493. [DOI] [PubMed] [Google Scholar]
- Lin Y., Cheng, G., Wang, X., and Clark, T.G. 2002. The use of synthetic genes for the expression of ciliate proteins in heterologous systems. Gene 288: 85–94. [DOI] [PubMed] [Google Scholar]
- Littlefield J.W., Keller, E.B., Gross, J., and Zamecnik, P.C. 1955. Studies on cytoplasmic ribonucleoprotein particles from the liver of the rat. J. Biol. Chem. 217: 111–123. [PubMed] [Google Scholar]
- Looger L.L., Dwyer, M.A., Smith, J.J., and Hellinga, H.W. 2003. Computational design of receptor and sensor proteins with novel functions. Nature 423: 185–190. [DOI] [PubMed] [Google Scholar]
- Lubovac Z., Gamalielsson, J., and Olsson, B. 2006. Combining functional and topological properties to identify core modules in protein interaction networks. Proteins 64: 948–959. [DOI] [PubMed] [Google Scholar]
- Lutz S., Fast, W., and Benkovic, S.J. 2002. A universal, vector-based system for nucleic acid reading-frame selection. Protein Eng. 15: 1025–1030. [DOI] [PubMed] [Google Scholar]
- Ma J., Campbell, A., and Karlin, S. 2002. Correlations between Shine–Dalgarno sequences and gene features such as predicted expression levels and operon structures. J. Bacteriol. 184: 5733–5745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandecki W. and Bolling, T.J. 1988. FokI method of gene synthesis. Gene 68: 101–107. [DOI] [PubMed] [Google Scholar]
- Matthaei H. and Nirenberg, M.W. 1961. The dependence of cell-free protein synthesis in E. coli upon RNA prepared from ribosomes. Biochem. Biophys. Res. Commun. 4: 404–408. [DOI] [PubMed] [Google Scholar]
- Maxwell K.L., Mittermaier, A.K., Forman-Kay, J.D., and Davidson, A.R. 1999. A simple in vivo assay for increased protein solubility. Protein Sci. 8: 1908–1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller O.J., Bernath, K., Agresti, J.J., Amitai, G., Kelly, B.T., Mastrobattista, E., Taly, V., Magdassi, S., Tawfik, D.S., and Griffiths, A.D. 2006. Directed evolution by in vitro compartmentalization. Nat. Methods 3: 561–570. [DOI] [PubMed] [Google Scholar]
- Mossner E., Koch, H., and Pluckthun, A. 2001. Fast selection of antibodies without antigen purification: Adaptation of the protein fragment complementation assay to select antigen-antibody pairs. J. Mol. Biol. 308: 115–122. [DOI] [PubMed] [Google Scholar]
- Nixon A.E. and Benkovic, S.J. 2000. Improvement in the efficiency of formyl transfer of a GAR transformylase hybrid enzyme. Protein Eng. 13: 323–327. [DOI] [PubMed] [Google Scholar]
- Pinto A.L., Hellinga, H.W., and Caradonna, J.P. 1997. Construction of a catalytically active iron superoxide dismutase by rational protein design. Proc. Natl. Acad. Sci. 94: 5562–5567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richmond K.E., Li, M.H., Rodesch, M.J., Patel, M., Lowe, A.M., Kim, C., Chu, L.L., Venkataramaian, N., Flickinger, S.F., and Kaysen, J., et al. 2004. Amplification and assembly of chip-eluted DNA (AACED): A method for high-throughput gene synthesis. Nucleic Acids Res. 32: 5011–5018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell D.R. and Bennett, G.N. 1982. Construction and analysis of in vivo activity of E. coli promoter hybrids and promoter mutants that alter the −35 to −10 spacing. Gene 20: 231–243. [DOI] [PubMed] [Google Scholar]
- Sambrook J. and Russell, D.W. 2001. Molecular cloning: A laboratory manual, 3rd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Sarmientos P., Sylvester, J.E., Contente, S., and Cashel, M. 1983. Differential stringent control of the tandem E. coli ribosomal RNA promoters from the rrnA operon expressed in vivo in multicopy plasmids. Cell 32: 1337–1346. [DOI] [PubMed] [Google Scholar]
- Seehaus T., Breitling, F., Dubel, S., Klewinghaus, I., and Little, M. 1992. A vector for the removal of deletion mutants from antibody libraries. Gene 114: 235–237. [DOI] [PubMed] [Google Scholar]
- Sekiya T., Takeya, T., Brown, E.L., Belagaje, R., Contreras, R., Fritz, H.J., Gait, M.J., Lees, R.G., Ryan, M.J., and Khorana, H.G., et al. 1979. Total synthesis of a tyrosine suppressor transfer RNA gene. XVI. Enzymatic joinings to form the total 207-base pair-long DNA. J. Biol. Chem. 254: 5787–5801. [PubMed] [Google Scholar]
- Seyfang A. and Jin, J.H. 2004. Multiple site-directed mutagenesis of more than 10 sites simultaneously and in a single round. Anal. Biochem. 324: 285–291. [DOI] [PubMed] [Google Scholar]
- Shevchuk N.A., Bryksin, A.V., Nusinovich, Y.A., Cabello, F.C., Sutherland, M., and Ladisch, S. 2004. Construction of long DNA molecules using long PCR-based fusion of several fragments simultaneously. Nucleic Acids Res. 32: e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shifman J.M. and Mayo, S.L. 2002. Modulating calmodulin binding specificity through computational protein design. J. Mol. Biol. 323: 417–423. [DOI] [PubMed] [Google Scholar]
- Shultzaberger R.K., Bucheimer, R.E., Rudd, K.E., and Schneider, T.D. 2001. Anatomy of Escherichia coli ribosome binding sites. J. Mol. Biol. 313: 215–228. [DOI] [PubMed] [Google Scholar]
- Smith D.L., Struck, D.K., Scholtz, J.M., and Young, R. 1998. Purification and biochemical characterization of the λ holin. J. Bacteriol. 180: 2531–2540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith H.O., Hutchison 3rd, C.A., Pfannkoch, C., and Venter, J.C. 2003. Generating a synthetic genome by whole genome assembly: phiX174 bacteriophage from synthetic oligonucleotides. Proc. Natl. Acad. Sci. 100: 15440–15445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stemmer W.P., Crameri, A., Ha, K.D., Brennan, T.M., and Heyneker, H.L. 1995. Single-step assembly of a gene and entire plasmid from large numbers of oligodeoxyribonucleotides. Gene 164: 49–53. [DOI] [PubMed] [Google Scholar]
- Strizhov N., Keller, M., Mathur, J., Koncz-Kalman, Z., Bosch, D., Prudovsky, E., Schell, J., Sneh, B., Koncz, C., and Zilberstein, A. 1996. A synthetic cryIC gene, encoding a Bacillus thuringiensis δ-endotoxin, confers Spodoptera resistance in alfalfa and tobacco. Proc. Natl. Acad. Sci. 93: 15012–15017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studier F.W.. 2005. Protein production by auto-induction in high density shaking cultures. Protein Expr. Purif. 41: 207–234. [DOI] [PubMed] [Google Scholar]
- Studier F.W., Rosenberg, A.H., Dunn, J.J., and Dubendorff, J.W. 1990. Use of T7 RNA polymerase to direct expression of cloned genes. Methods Enzymol. 185: 60–89. [DOI] [PubMed] [Google Scholar]
- Takagi M., Nishioka, M., Kakihara, H., Kitabayashi, M., Inoue, H., Kawakami, B., Oka, M., and Imanaka, T. 1997. Characterization of DNA polymerase from Pyrococcus sp. strain KOD1 and its application to PCR. Appl. Environ. Microbiol. 63: 4504–4510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temsamani J., Kubert, M., and Agrawal, S. 1995. Sequence identity of the n − 1 product of a synthetic oligonucleotide. Nucleic Acids Res. 23: 1841–1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian J., Gong, H., Sheng, N., Zhou, X., Gulari, E., Gao, X., and Church, G. 2004. Accurate multiplex gene synthesis from programmable DNA microchips. Nature 432: 1050–1054. [DOI] [PubMed] [Google Scholar]
- Wu G., Wolf, J.B., Ibrahim, A.F., Vadasz, S., Gunasinghe, M., and Freeland, S.J. 2006. Simplified gene synthesis: A one-step approach to PCR-based gene construction. J. Biotechnol. 124: 496–503. [DOI] [PubMed] [Google Scholar]
- Xiong A.S., Yao, Q.H., Peng, R.H., Li, X., Fan, H.Q., Cheng, Z.M., and Li, Y. 2004. A simple, rapid, high-fidelity and cost-effective PCR-based two-step DNA synthesis method for long gene sequences. Nucleic Acids Res. 32: e98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young L. and Dong, Q. 2004. Two-step total gene synthesis method. Nucleic Acids Res. 32: e59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H., Zhao, J., Wang, S., Lin, C.M., Chen, T., Jones, D.H., Ma, C., Opella, S., and Xie, X.Q. 2005. Biosynthesis and purification of a hydrophobic peptide from transmembrane domains of G-protein-coupled CB2 receptor. J. Pept. Res. 65: 450–458. [DOI] [PubMed] [Google Scholar]
- Zhou X., Cai, S., Hong, A., You, Q., Yu, P., Sheng, N., Srivannavit, O., Muranjan, S., Rouillard, J.M., and Xia, Y., et al. 2004. Microfluidic PicoArray synthesis of oligodeoxynucleotides and simultaneous assembling of multiple DNA sequences. Nucleic Acids Res. 32: 5409–5417. [DOI] [PMC free article] [PubMed] [Google Scholar]