

Abstract
FtsZ has two domains, the amino GTPase domain with a Rossmann fold, and the carboxyl domain that resembles the chorismate mutase fold. Bioinformatics analyses suggest that the interdomain interaction is stronger than the interaction of the protofilament longitudinal interfaces. Crystal B factor analysis of FtsZ and detected conformational changes suggest a connection between these domains. The unfolding/folding characteristics of each domain of FtsZ were tested by introducing tryptophans into the flexible region of the amino (F135W) and the carboxyl (F275W and I294W) domains. As a control, the mutation F40W was introduced in a more rigid part of the amino domain. These mutants showed a native-like structure with denaturation and renaturation curves similar to wild type. However, the I294W mutant showed a strong loss of functionality, both in vivo and in vitro when compared to the other mutants. The functionality was recovered with the double mutant I294W/F275A, which showed full in vivo complementation with a slight increment of in vitro GTPase activity with respect to the single mutant. The formation of a stabilizing aromatic interaction involving a stacking between the tryptophan introduced at position 294 and phenylalanine 275 could account for these results. Folding/unfolding of these mutants induced by guanidinium chloride was compatible with a mechanism in which both domains within the protein show the same stability during FtsZ denaturation and renaturation, probably because of strong interface interactions.
Keywords: FtsZ, folding, protein flexibility, GTPase
FtsZ is a GTPase that forms the Z-ring, the first step in bacterial cell division. This ring is located around the middle of the cell on the cytosolic side of the inner membrane and is essential for recruiting the rest of the proteins that form the divisome (Errington et al. 2003; Vicente et al. 2006). The 3D crystal structure of FtsZ has been resolved for four microorganisms: the extremophilic archaebacteria Methanococcus jannaschii (MjFtsZ) in GDP and GTP conformations and an engineered conformation of Thermotoga maritima (TmFtsZ); and the mesophilic Pseudomonas aeruginosa (PaFtsZ) in GDP state and Mycobacterium tuberculosis (MtbFtsZ) in both forms (Lowe 1998; Cordell et al. 2003; Leung et al. 2004; Oliva et al. 2004). In all cases, the 3D structures show the presence of an N-terminal domain, with a Rossmann fold and a GTP-binding pocket, and a C-terminal domain with a chorismate mutase fold that interacts with other proteins of the divisome.
FtsZ folding is a spontaneous and reversible process that occurs in the absence of chaperones, suggesting cooperative folding between the two domains (Andreu et al. 2002; Santra and Panda 2003). Recently, the amino and carboxyl domains of TmFtsZ were expressed separately, and it was determined that each domain folded independently, and that the GTPase activity was recovered only on mixing both domains (Oliva et al. 2004). The crystal structure highlighted that both FtsZ domains have a well-defined surface of interaction. However, the functional consequences of this interaction have not been analyzed. Protein–protein interactions have been characterized from crystallized complexes with respect to the nature of the contacts between each protein, such as the formation of interprotein hydrogen bonds, salt bridges, and the area involved (ΔASA), identifying real protein–protein interfaces (Jones and Thornton 1996; Henrick and Thornton 1998; Nooren and Thornton 2003). Protein interactions can be classified in two types according to the association constant values: “permanent,” which only dissociated by denaturation, and “non-obligate,” which can dissociate (Jones and Thornton 1996). From now on, this nomenclature will be used throughout this study. These approaches provide tools to analyze the interactions between the FtsZ domains, using their crystallographic structural features.
Two conformations, straight (GTP) and curved (GDP), have been postulated for FtsZ in relation to its dynamic assembly (Mukherjee and Lutkenhaus 1998; Lu et al. 2000), where the switch between these conformations would be given principally by the movement of the T3 loop (Diaz et al. 2001; Leung et al. 2004). Other movements associated with the nucleotide-binding site have been described for the calcium-induced association of filaments in bundling (Marrington et al. 2004). Inspection of the crystal structures in GTP and GDP conformations indicate minor displacements between the two states (Oliva et al. 2004). The free energy for the transition from the inactive state (GDP) to active (straight conformation) is lowered by GTP, allowing polymerization (Buey et al. 2006). Two models have been proposed to explain how the polymer remains stable after GTP hydrolysis. In the first model, GDP is exchanged for GTP in the polymer (Mingorance et al. 2001; Oliva et al. 2004), whereas in the second model, the monomers are released from the filaments, prior to the exchange of the nucleotides (Chen and Erickson 2005). The latter model has been supported by kinetic evidence that showed that the rate-limiting step for GTP hydrolysis was nucleotide turnover, similar to the exchange rate of FtsZ monomers from the polymers (Romberg and Mitchison 2004; Chen et al. 2005). Once hydrolysis has occurred, conformational changes should occur, weakening the longitudinal interactions. Interestingly, GTPase activity located in the amino domain, and polymerization, are affected by several mutations within the carboxyl domain of FtsZ (Lutkenhaus 1990; Lu et al. 2001; Huecas and Andreu 2004; Chen et al. 2005). To date, the only study to determine the effect of a mutation on the secondary structure of FtsZ, using UV circular dichroism, analyzed the importance of a residue located in the carboxyl zone (Y319W) of MjFtsZ (Oliva et al. 2003).
The aim of this study was to determine the unfolding/folding characteristics and functional relationship between the amino and carboxyl domains of Escherichia coli FtsZ (EcFtsZ). To follow the structural changes, one tryptophan was introduced in the flexible zones, either into the amino or the carboxyl domains. Selected on the basis of bioinformatics, F40W, F135W, F275W, and I294W mutants and the double mutant F275A/I294W were constructed. The structural properties of these mutants were determined following the equilibrium unfolding using circular dichroism and the intrinsic fluorescence of tryptophan. The effect of these mutations on the function of EcFtsZ was evaluated by in vivo complementation, GTPase activity, and in vitro polymerization. Using a bioinformatics approach that involved structural evaluation of GDP and GTP conformations, the results were analyzed considering the flexibility of FtsZ, the interactions between both domains, and the longitudinal interaction in the protofilament.
Results
In silico characterization of the interface between the amino and carboxyl domains of FtsZ
To determine the nature of the interaction between these domains, a 3D model of EcFtsZ was constructed by homology using FtsZ from P. aeruginosa, which shares 58% sequence identity with EcFtsZ, as a structural template (1OFU.pdb) (Cordell et al. 2003). Analysis of the model showed a ΔASA value for the interdomain interface of 1547 Å2, similar to the characteristic values for interfaces of permanent interactions (Jones and Thornton 1996), where 75% of the residues involved in the interaction surface correspond to hydrophobic residues (Table 1). EcFtsZ possessed eight interdomain H-bonds within the interface, with 0.5 H-bonds per 100 Å2 of ΔASA, a typical value for the permanent complex interface (Table 1). As shown in Figure 1A, secondary structural elements belonging to the interaction surface of the carboxyl domain were mainly the central α-helix (H7), which interacts with the H1-helix; and the β-sheet formed by the S4, S5, and S6 strands within the Rossmann fold of the amino domain. These results suggest the existence of a strong interaction between the amino and carboxyl domains of EcFtsZ, indicating a permanent association of both domains, which would maintain a global structural communication within the protein. Table 1 shows the same analysis for the crystal structure of FtsZ from P. aeruginosa (1OFU.pdb), M. tuberculosis FtsZ (1RQ7.pdb), and M. jannaschii (1FSZ.pdb), confirming the permanent character of the interaction. This behavior is conserved in all the FtsZ structures analyzed, from both extremophilic and mesophilic organisms. The longitudinal interaction surface of a MjFtsZ homodimer is 1038 Å2 for ΔASA, close to the value reported by Oliva et al. (2004) and typical of a non-obligate interaction, and values of 0.2 for H-bonds and 62% of hydrophobic amino acid residues, which are frequent in permanent interactions (Table 1).
Table 1.
Parameters for permanent and non-obligate interdomain and longitudinal interactions of EcFtsZ protofilaments

Figure 1.

Mutant derivatives of EcFtsZ and thermal factor analysis of MtbFtsZ and MjFtsZ structures. (A) Ribbon representation of EcFtsZ, showing the amino (light gray) the carboxyl (blue) domains. (Red balls and sticks) Point mutations; (yellow sticks) nucleotides. (B) Bar plot of the average normalized B-factor versus secondary structural element in MjFtsZ (black bars) and MtbFtsZ (white bars). The normalized B-factor was calculated by the equation B norm = (Bi − B min)/(B max − Bmin), where Bi is the B-factor for Cα of the ith amino acid, and B min and B max are the minimum and maximum value of Cα B-factor in the protein, respectively. Secondary structural elements were labeled according to Oliva et al. (2004). Protein representation was made using the DS Modeling v1.1 package (Accelrys) and PovRay 3.6 (Persistence of Vision Raytracer Pty. Ltd.).
Thermal factor analysis
To study the implicit flexibility of MjFtsZ and MtbFtsZ, an analysis of the B-factors included in the PDB files was carried out (Fig. 1B). In both cases, the structures (1FSZ.pdb and 1RQ7.pdb) possess a rather rigid nucleotide binding site—including T4, S4-strand, and H4-helix—and similar flexibility in the C-terminal domain (T7, S7–H9, H9–S8, S8–H10, H10–S9, and S9–S10 loops; S7, S9, and S10 strands; and H9- and H10-helices). However, MtbFtsZ contains more flexible regions in the N-terminal domain (H1–S2, S2–H2, T3, and H6–H7 loops and H2-helix). These results show that the amino domain, including the GTP-binding site, is more rigid than the carboxyl domain.
Based on the flexibility of the protein and the conserved phenylalanine residues, one tryptophan was introduced in each mutant as an intrinsic fluorescent probe at different positions in each domain of the protein. Tryptophan is the least abundant residue in proteins (1.3%) (Creighton 1993), and this residue is absent in native EcFtsZ. To rule out a secondary effect of the mutation on the global stability of EcFtsZ structure, protein unfolding and refolding at equilibrium were analyzed through circular dichroism and fluorescence spectroscopy.
Mutant design
To choose the sites for the introduction of tryptophan, the residue frequency on a multiple alignment of FtsZ sequences from different organisms was calculated. Position F135 in the EcFtsZ sequence has a high level of conservation of aromatic residues (99%). The flexibility analysis of MjFtsZ and MtbFtsZ suggests that this residue in EcFtsZ is located in a flexible zone close to the nucleotide binding site (Fig. 1). Therefore, F135 located in the H5-S5 loop of the amino domain was replaced by tryptophan. In the carboxyl domain, the frequency analysis showed that at position I294 the aromatic residues are 50% conserved among all species analyzed. This residue, which is located in the S9 strand, has inherent flexibility that could avoid the effect of a bulky group in these regions over the function and the stability of these constructs (Taniuchi and Anfinsen 1969). To test this hypothesis, an F40W mutation located in the S2 strand, a more rigid zone of FtsZ was constructed. The locations of the tryptophans in the mutants are shown in Figure 1A.
Structural characterization of wild-type and mutant forms of EcFtsZ
The normalized fluorescence spectra of the F40W, F135W, and I294W mutants were measured in native conditions (Fig. 2A). These spectra of the F135W, I294W, and F40W mutants showed maxima emission wavelengths at 343, 339, and 340 nm, respectively. The far-UV CD spectra in native conditions of wild-type (wt) and F40W, F135W, and I294W mutants of EcFtsZ are shown in Figure 2B. The spectra are typical of α/β protein structures and the minimum at 222 nm associated with α-helix absorption is similar between wt and mutant EcFtsZ. The small differences observed at 208 nm for the I294W mutant did not affect the secondary structure content of this mutant. The change at 222 nm for the F40W mutant corresponded only to a 2% difference in the secondary structure content with respect to wt FtsZ. This change is compatible with the introduction of a bulky amino acid in a more rigid region of the protein.
Figure 2.

Fluorescence normalized spectra and circular dichroism (CD) of tryptophan mutants of EcFtsZ. (A) Uncorrected fluorescence emission spectra of F135W (dashed line), I294W (solid line), and F40W (dotted line) mutants, recorded in the same day in buffer 50 mM Tris-HCl (pH 7.5) at 25°C, at the same protein concentration (12 μM). λexcitation = 295 nm. Maximum emission fluorescence occurred at 343, 340, and 339 nm for F135W, F40W, and I294W mutants, respectively. (B) Far-UV circular dichroism spectra of (♦) EcFtsZ wt, (▪) F40W, (○) F135W, and (△) I294W mutants (7.5 μM), recorded under the same experimental conditions.
Functional characterization of wt EcFtsZ and the mutant derivatives
The GTPase activity and polymerization capacity of the F40W, F135W, and I294W mutants of EcFtsZ, with respect to wt EcFtsZ (initial velocity of 1.8 ± 0.1 μM GTP/μM protein·min) were determined. Figure 3A shows that the GTPase activity of F40W and F135W mutants diminished to 80% and 71%, respectively. Figure 3B shows that the turbidity of polymerization of the F40W and F135W mutants compared to wt FtsZ fell to 70% and 32%, respectively. Owing to the dependence of the turbidity on the size and shape of the polymers, the critical concentration (Cr) and the slope were determined plotting protein concentration versus the maximal polymerization turbidity value (Table 2).
Figure 3.

Functional characterization of wt and F40W and F135W mutants of EcFtsZ. (A) GTP hydrolysis activity was initiated by the addition of 1 mM GTP and stopped by adding 10% perchloric acid. Each protein (25 μM) sample was incubated in buffer containing 50 mM Mes (pH 6.5), 10 mM MgCl2, and 50 mM KCl for 5 min at 30°C. Percentages of activity are with respect to wt EcFtsZ. The errors were obtained by linear regression analysis. (B) Polymerization of F40W and F135W mutants and wt EcFtsZ (25 μM) in a solution containing 50 mM Mes (pH 6.5), 50 mM KCl, and 10 mM MgCl2, followed by turbidity changes at 30°C. The addition of GTP is indicated by the arrow. (C) In vivo complementation was estimated from the number of colonies obtained after titration at the restrictive temperature (42°C) compared with the number of colonies obtained at the permissive temperature (30°C). C+ corresponds to the transformation of E. coli VIP-2(DE3) strain with wt EcFtsZ, and C− with the vector alone (see Materials and Methods).
Table 2.
Critical concentration and slope values for polymerization of wt and mutant derivatives of EcFtsZ

The results indicate that the Cr values and the slope for the F40W mutant were in the same range as that of wt FtsZ, whereas a higher critical concentration and a reduced value for the slope were achieved for the F135W mutant. The F40W mutant showed a longer extension of polymerization, which agrees with the lower GTPase activity (Fig. 3A,B). The I294W mutant showed a baseline with higher turbidity than wt FtsZ after polymerization with GTP, and the addition of this nucleotide had no effect (Fig. 4B). As it will be shown later the turbidity of I294W mutant was produced by large sheet-like polymers. Complementation assays of the mutants under nonpermissive conditions in the strain Escherichia coli VIP2(DE3), which lacks chromosomal ftsZ, were carried out in order to determine in vivo functionality. F135W and F40W mutants presented similar complementation to that of wt EcFtsZ within the experimental error (Fig. 3C). Interestingly, Figure 4C shows that the I294W mutant was unable to complement the defect of VIP2(DE3) under nonpermissive conditions.
Figure 4.

Functional characterization of wt EcFtsZ and the F275W, I294W, and F275A/I294W mutants. (A) GTP hydrolysis activity was started by the addition of 1 mM GTP and stopped by adding 10% perchloric acid. Each protein (25 μM) sample was incubated in buffer containing 50 mM Mes (pH 6.5), 10 mM MgCl2, and 50 mM KCl for 5 min at 30°C. Percentages of activity are respect to EcFtsZ wt. The errors were obtained by linear regression analysis. (B) Polymerization of I294W and F275A/I294W mutants and wt EcFtsZ at 12 μM, and F275W (22 μM) in a solution containing 50 mM Mes (pH 6.5), 50 mM KCl, and 10 mM MgCl2, followed by turbidity changes at 30°C. The addition of GTP is indicated by the arrow. (C) In vivo complementation was estimated from the number of colonies obtained after titration at the restrictive temperature (42°C) compared with the number of colonies obtained at the permissive temperature (30°C). C+ corresponds to the transformation of E. coli VIP-2(DE3) strain with wt EcFtsZ, and C− with the vector alone (see Materials and Methods).
Given that the in vitro polymerization and GTPase activity of F135W and I294W mutants were altered, the protein stability of the mutants was analyzed through unfolding/folding curves using GdmCl.
Stability of wt FtsZ and mutant derivatives followed by GdmCl unfolding/folding assays
The GdmCl equilibrium unfolding curves of wt and F135W and I294W mutants of EcFtsZ, as determined by CD, showed that raising the concentration of the chaotropic agent to 0.6 M released the nucleotide completely. The apo-protein denaturation at concentrations >0.6 M GdmCl showed a cooperative behavior. The inserted table (Fig. 5A) shows that the transition midpoint (GdmCl50%) for the mutants and wt present similar values around 1.2 M GdmCl. These values were obtained adjusting the experimental points to a cooperative denaturation curve using Equation 1 as indicated in Materials and Methods. It has been described that this parameter is altered when the protein stability is affected by structural perturbations, such as point mutations (Khorasanizadeh et al. 1996; Hannemann et al. 2002). To follow the stability of each domain and the unfolding/folding behavior of the mutants, the fluorescence properties of tryptophan were characterized. The equilibrium unfolding process of FtsZ was not affected by the tryptophan substitutions at F135 or I294, and the global stability of the secondary structure remained similar to that of wt FtsZ. Figure 5, B and C, shows that the unfolding curves of F135W and I294W mutants as tracked by fluorescence measurements were similar and that the refolding curves showed the same pattern observed in the denaturation curves reaching the same fluorescence intensity and maximum emission wavelength compared with the native state. Interestingly, the F135W mutant showed a small hyperfluorescence state that was more prominent in the F275W mutant (Fig. 5D). Figure 5, B and D, shows that the maximum of the hyperfluorescence effect is produced at the same concentration of GdmCl at which the nucleotide is released. This result indicates that tryptophan senses the structural changes induced by the apo-protein conformation. The shape and the overlap of the denaturation/renaturation curves are indicative of chemically reversible unfolding/folding processes. In addition, the average transition midpoint for the F135W, F275W, and I294W mutants occurs at a similar concentration of GdmCl as for wt FtsZ (1.2 M). However, the stability of F40W was reduced, as shown by a lower value for the transition midpoint (1.0 M GdmCl) (data not shown). It is important to point out that the same unfolding behavior observed from the CD experiments was detected following the fluorescence of a tryptophan residue located in the amino or in the carboxyl domain. Taken together, the results suggest that the stability of both domains in the protein is determined by a common structural feature that could be the stronger interdomain interface interaction.
Figure 5.

Secondary structure and stability analysis of wt and mutant derivatives of EcFtsZ. (A) Equilibrium unfolding curves followed by circular dichroism of (♦) wt EcFtsZ and (○) F135W and (Δ) I294W mutants (7.5 μM) at 25°C in 50 mM Tris-HCl (pH 7.5). Each point corresponds to the unfolded protein fraction calculated from experimental values of ellipticity at 222 nm using GdmCl as denaturant agent. (+) mole of nucleotide released per mole of FtsZ. The inserted tables show the transition midpoints [GdmCl]50% (GdmCl concentration value for 50% of denatured protein) of wt and mutant FtsZ estimated from the unfolding curves. (B) Equilibrium (○) unfolding and (•) refolding curves determined by tryptophan fluorescence of the F135W mutant (12 μM) in 50 mM Tris-HCl (pH 7.5) at 25°C. (C) Equilibrium (△) unfolding and (▴) refolding curves followed by tryptophan fluorescence of the I294W (mutant 12 μM) in 50 mM Tris-HCl (pH 7.5) at 25°C. (D) Equilibrium (□) unfolding and (▪) refolding curves followed by tryptophan fluorescence of the F275W mutant (12 μM) in 50 mM Tris-HCl (pH 7.5) at 25°C. Each folded and refolded protein fraction was calculated from the normalized fluorescence intensity at maximum emission wavelength of 343, 340, and 339 nm for F135W, F275W, and I294W mutants, respectively. Equilibrium refolding curves were under the same experimental conditions used for unfolding, where GdmCl was diminished by fast dilution. The inserted tables in B and C show the transition midpoint ([GdmCl]50%) values determined from the unfolding and refolding curves as tracked by tryptophan fluorescence.
The effect of the point mutation I294W on the functional properties of FtsZ was reverted by the double mutation I294W/F275A
To determine the nature of the inhibitory effect of the point mutation I294W on the functional properties of FtsZ, GTPase activity (13% of wt) and polymerization (permanently polymerized) were analyzed (Fig. 4A,B). The interaction of tryptophan at position 294 with the surrounding amino acids was evaluated in the 3D structure of the model of EcFtsZ. Figure 6A shows that the tryptophan in the I294W mutant was very close to F275 located in the H10-helix, with both aromatic rings separated by a distance of 4.5 Å, probably forming an aromatic stack. This interaction is located between the H10-helix and S9-strand, a flexible region of FtsZ (Fig. 6A). It seems reasonable to postulate that the inhibitory effect produced by this mutation could be in part attributed to this aromatic interaction. If this hypothesis is correct, the simultaneous presence of aromatic residues at positions 275 and 294 should not be found in FtsZ from different species. A multiple alignment of 474 FtsZ sequences from different organisms, which is partly presented in Figure 6B, demonstrates that aromatic residues can be found at either positions 275 or 294, but interestingly, aromatic residues are never present in both positions.
Figure 6.
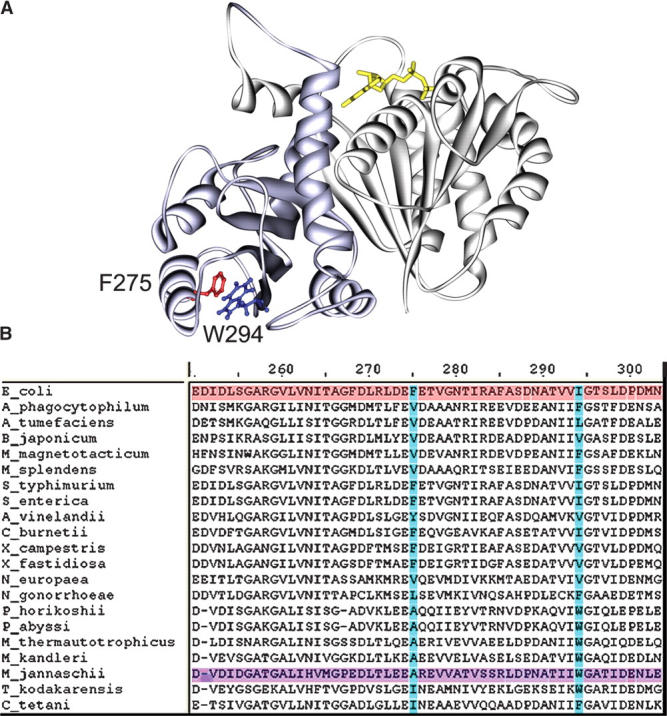
Cα ribbon representation of the I294W mutant in the structural model of EcFtsZ. (A) (light gray) The amino domain; (blue) the carboxyl domain. The residues F275 and W294 are presented in detail showing the stacking formed according to our model. GTP is projected in yellow sticks. The image was made using DS Modeling v1.1 and PovRay 3.6 (as in Fig. 1). (B) A multiple alignment of FtsZ sequence fragments where EcFtsZ (red) and MjFtsZ (violet) are highlighted. Positions 275 and 294 are highlighted in blue.
To examine whether phenylalanine 275 is involved in the effect produced by the I294W mutation, this phenylalanine was mutated to either tryptophan or alanine. F275W mutants fully complement in vivo, although the modified FtsZ polymerizes in vitro with reduced efficiency (Fig. 4C,B). The Cr of the F275W mutant was approximately one order of magnitude higher than that of wt FtsZ, and the slope was similar (Table 2). These data indicate that this mutation diminished the polymerization capacity but had no effect on the shape of the polymers, discarding a specific effect of tryptophan in the environment of I294. To test the hypothetical interaction between F275 and W294 in the mutant, a double mutant was designed in which F275 was changed to alanine (small side-chain residue), which is found at position 275 in several sequences of FtsZ. One example is MjFtsZ, which has tryptophan and alanine located in the equivalent positions of I294 and F275 in EcFtsZ. Figure 4A shows that the GTP hydrolytic activity of the double mutant increased to 21% of wt levels, instead of just 13% achieved for the I294W mutant. The double mutant presented lower turbidity than the I294W mutant (Fig. 4B). Figure 7A shows that the mutant I294W produced large sheet-like polymers. Glutamate (1 M) increased the turbidity of both the I294W mutant and the double mutant (data not shown). Figure 7B shows that in the presence of glutamate, I294W polymerizes, forming large sheets that are not affected by GTP or GDP (Fig. 7C). In the double mutant, glutamate induced a small amount of amorphous aggregates (Fig. 7E), and the addition of GTP induced single and double filaments and sheets (Fig. 7F). Surprisingly, the double mutant fully reestablished cell division by in vivo complementation (Fig. 4C).
Figure 7.

Polymerization products of EcFtsZ mutant I294W and double mutant F275A/I294W. Electron micrograph of the polymerization products of the I294W and F275A/I294W mutants (7.5 μM). (A,D) I294W and I294W/F275A polymerization controls in buffer (50 mM Mes at pH 6.5, 50 mM KCl, 10 mM MgCl2) without GTP or sodium glutamate at 30°C. The polymerization reaction was initiated with GTP at a final concentration of 1 mM. (B,E) I294W and F275A/I294W mutants before the addition of GTP, respectively, in the presence of 1 M sodium glutamate. (C,F) I294W and F275A/I294W mutants 5 min after the addition of GTP, respectively, in the presence of 1 M sodium glutamate. Bars, 100 nm.
These results show that the flexibility of S9 where I294 is located has a dramatic influence on the functionality of the protein. In order to analyze the relationship of the GTPase activity with the flexibility of the protein, a structural comparison between GTP/GDP forms of FtsZ was carried out.
Structural changes of GTP/GDP forms
To compare two crystallized conformations of the same protein, it is important to have the same space group because protein contacts belonging to different space groups may affect the comparison between conformations. For this reason, the two crystal structures of FtsZ chosen for the comparison of GTP and GDP forms had the same space group in the crystals. Another important point is that these proteins were crystallized in the GDP form, and GTP was exchanged into the crystal. Under these restrictions, the structures of MtbFtsZ bound to GTP (1RLU.pdb) and GDP (1RQ7.pdb) (Leung et al. 2004) and MjFtsZ crystallized in GTP (1W58.pdb) and GDP (1FSZ.pdb) conformation (Lowe 1998) were analyzed. The sequence identity among these proteins is 48%, with a RMSD (root mean square deviation) for the structural superposition of 1.45 Å between the 1W58.pdb and 1RLU.pdb. Figure 8 shows the secondary structural changes given by the difference in position of Cα of the polypeptide chain in the GTP and GDP states. Analysis of these data shows that GTP hydrolysis induces movements that are greater than the noise (RMSD shown in Table 3) in the polypeptide chain, mainly in the external regions of FtsZ. The amino acids involved in movements >0.5 Å are shown in Table 4; specifically, they are located in the amino domain near the GTP-binding site in the T2 and T3 loops and in the H6–H7 loop. More remarkable are the motions of other residues of the carboxyl domain associated with the phosphorylation state of the nucleotide. According to the nomenclature given by Oliva et al. (2004), these movements are located in the H8–S7, S8–H10, and S9–S10 loops. These results indicate that the carboxyl domain also shows flexibility, a finding that has not been reported previously.
Figure 8.

Comparison of MjFtsZ and MtbFtsZ structures in GTP and GDP states. A bar plot of the average Cα–Cα distance between structural equivalent amino acids versus secondary structural element in MjFtsZ (black bars) and MtbFtsZ (white bars). The symbol (*) indicates secondary structural elements that have been reported to move between GTP and GDP conformations (Leung et al. 2004; Oliva et al. 2004).
Table 3.
Parameters from the crystal structure of FtsZ in GTP and GDP conformations

Table 4.
Principal changes in Cα position between GTP/GDP conformations

Discussion
In this study, using single tryptophan fluorescence, both amino and carboxyl domains of EcFtsZ showed similar unfolding/folding characteristics, and the interaction between these domains was determined to be permanent. GTPase activity and polymerization of FtsZ were found to be dependent on the structural flexibility of the carboxyl domain of the protein. Thus, the I294W mutation located in the flexible carboxyl domain appears to form an aromatic interaction with phenylalanine 275, affecting both the functionality in vitro and in vivo. On the other hand, the double mutant F275A/I294W partially recovers both the GTPase activity and the polymerization capacity in the presence of glutamate, and furthermore, it fully recovers in vivo functionality.
Structural stability and folding of EcFtsZ
Whereas the structural stability of proteins is generally not affected when mutations are introduced in flexible zones, their functionality can be altered (Hilser et al. 1998; Freire 1999). The results presented in this study indicate that the content of secondary structures of the F135W and I294W mutants was similar, suggesting that no significant secondary structure rearrangements were caused by these substitutions. In addition, the unfolding of wt FtsZ followed by changes in the secondary structure was the same as that of the mutants, as analyzed by the fluorescence of a single tryptophan located in each domain, showing that the structural stability of FtsZ remained unaltered in the tryptophan substitution mutants.
Using the intrinsic properties of tryptophan, the environmental characteristics of the tertiary structure of FtsZ around these residues introduced into the amino (F135W) or carboxyl (I294W) domains of the mutants allowed the stability and tracking of the unfolding/folding process to be determined. The unfolding and refolding of the mutants with tryptophan in the amino or in the carboxyl domain showed similar fluorescence curves, indicating the same global stability for the whole protein. These results are compatible with a cooperative folding mechanism where both domains fold with similar stabilities depending on the global structure of the protein, although they may have different stabilities separately.
The unfolding/folding processes in the F135W and I294W mutants did not show the presence of intermediates. In contrast, other studies on EcFtsZ monitoring the fluorescence of ANS (8-anilinonaphthalene-1-sulphonate) and FITC (fluorescein isothiocyanate) probes report the presence of intermediates (Santra and Panda 2003). However, this apparent contradiction could be explained by the CD results (Fig. 4B) of the mutants, confirming the presence of the apo-protein at 0.6 M GdmCl (Andreu et al. 2002). This value is coincident with the maximum value for the minor hyperfluorescence shown by the F135W mutant and for the more pronounced hyperfluorescence of the F275W mutant that could correspond to the intermediates that appear upon denaturation with urea (Santra and Panda 2003). The possibility of local arrangements taking place during the initial steps of FtsZ unfolding in regions close to the nucleotide binding site that are far from the interdomain zone would give greater accessibility to the action of denaturant agents. These changes can be attributed to local unfolding of regions that are more susceptible to denaturant agents than the interdomain region, generating the intermediates seen by Santra and Panda (2003), and by the CD unfolding curves obtained in this study. This kind of behavior has been observed in other proteins and has been defined as local cooperativity related to conformational changes (Hilser et al. 1998; Freire 1999).
Influence of the interdomain interaction and flexibility on FtsZ function
The thermal factor analysis of the implicit flexibility and the exchange of GDP by GTP showed minor displacements (Tables 3 and 4; Fig. 8) mainly in specific external regions of the protein including those close to the GTP-binding site, and interestingly, in zones of the carboxyl domain far from the GTP-binding site.
A hypothetical model for polymerization and depolymerization of FtsZ correlating the flexible zones of EcFtsZ will be discussed based on a double filament (Oliva et al. 2003). It is known that the hydrolysis of GTP is induced by the interaction of the amino longitudinal face of one monomer with the carboxyl longitudinal face of the other (Sossong et al. 1999). The surface of this interaction, which falls closer to the non-obligate classification, allows a conformational change from GTP to GDP that weakens the structure of the polymer (Table 1). It is assumed that this conformational change spreads through the flexible zones of the molecule during depolymerization. Thus, GTP stabilizes the straight conformation in the polymer, while GDP destabilizes it. Therefore, a mutation such as I294W located in a flexible region of the carboxyl domain should alter the equilibrium between these conformations, affecting polymerization or depolymerization, depending on the conformation favored. Consistent with our hypothesis, the functionality of the I294 mutant was almost totally abolished; this mutant remained permanently polymerized, had very low GTPase activity, and showed residual in vivo complementation. Also the point mutation F135W, located in a flexible zone of the amino domain, reduced the functionality of the protein (diminished polymerization, reduced GTPase to 70%, and showed poor in vivo complementation). In agreement with our results, it was reported that the replacement of amino acid T92 by tryptophan in the flexible loop T3 in MjFtsZ diminished the functionality of the protein (Diaz et al. 2001). On the other hand, the F40W mutant, with a tryptophan located in the rigid zone of the amino domain, was almost totally functional (polymerization in vitro was almost identical to wt FtsZ, the GTPase activity was 80%, and it fully complemented in vivo), where the small differences could be due to the small change in secondary structure induced by a steric effect of tryptophan (Fig. 2B). These results support the hypothesis that a small modification in the flexible zone affects the function of FtsZ. Previous work reported a significant alteration in GTPase activity of MjFtsZ when the W319 residue, located in a structurally equivalent position of the I294 residue in the 3D structure of EcFtsZ, was changed to tyrosine (Oliva et al. 2003). These results and the fact that the I294W mutation did not affect the structural stability of FtsZ, as demonstrated by the CD and fluorescence experiments, suggest that the strong effect on functionality caused by the I294W mutation could be the result of an alteration in the flexibility. This effect could be explained through an aromatic stacking interaction between tryptophan 294 and phenylalanine 275 in the mutant, which could stabilize the GTP conformation. This is confirmed by the full recovery of the in vivo complementation and the increment in the rate of GTP hydrolysis in the F275A/I294W double mutant, in which the stacking would disappear.
A rigid C-terminal region in the “GTP conformation” could stabilize the protofilaments as occurs with the stabilization of the polymers in the I294W mutant (Fig. 7A). The fine regulation of the longitudinal interactions should be carried out by specific amino acid residues at the interface between the monomers and the state of phosphorylation of the nucleotide. This model requires flexible zones in the amino and carboxyl domains. The importance of the interdomain interaction for a functional conformation was demonstrated in TmFtsZ, in which the GTPase activity was recovered in the presence of both domains that were purified separately (Oliva et al. 2004). These results indicate a strong interaction between them that could induce a conformational change that, in turn, would be responsible for the correct flexibility of functional FtsZ.
Materials and Methods
Reagents
Guanidinium chloride (GdmCl), MgCl2, glycerol, and EDTA were purchased from Winkler Ltda. Tris Base ultrapure was supplied by US Biological and Mes (2-morpholinoethanesulfonic acid) from AppliChem. KCl and ammonium sulfate were purchased from Merck. Monosodium glutamate was acquired from Fluka Chemie AG, and IPTG (isopropyl-1-thio-β-D-galactopyranoside) was supplied by Sigma Chemical Co.
Model building
The FtsZ structure from P. aeruginosa (1OFU.pdb chain A) (Cordell et al. 2003), sharing 58% sequence identity with the sequence of EcFtsZ, was obtained from the Protein Data Bank and used as the template structure to construct a 3D model of EcFtsZ, with the program Modeller v6.2 (Sánchez and Sali 1997). The model was constructed using the default “model” routine, and the reliability was evaluated with PROSAII (Sippl 1993) and Verify3D software.
Characterization of the FtsZ interdomain interface
The interdomain interfaces of PaFtsZ (1OFU.pdb chain A), MtbFtsZ (1RQ7.pdb), MjFtsZ (1FSZ.pdb), and the model of EcFtsZ 3D structures were analyzed (Lowe 1998; Cordell et al. 2003; Leung et al. 2004; Oliva et al. 2004). Ser177 in EcFtsZ and the analog residues for the other structures were used as the domain limit. The number of interdomain H-bonds per 100 Å2 of ΔASA (change in the accessible solvent area) was calculated using HBPLUS in which H-bonds are defined according to standard geometric criteria (McDonald and Thornton 1994). The determination of the ΔASA value and the percentage of hydrophobic residues in the interdomain interface were obtained via the Protein–Protein Interaction Server v1.5 (Jones and Thornton 1996), where neither water molecules nor GTP was included.
Conformational changes related to GTP hydrolysis
The putative amino acids involved in conformational changes were obtained from the structural comparison of MtbFtsZ (1RLU.pdb and 1RQ7.pdb) in GTP and GDP conformations and MjFtsZ (1W58.pdb and 1FSZ.pdb) in both conformations. Tables of Cα distances were obtained from the structural comparison made with Align3d routine, from Modeller v6.2.
Mutant construction and protein purification
pMFV57 was constructed starting from pMFV56 (Rivas et al. 2000) in which the gene β-lactamase was cloned into the EcoRV site interrupting the gene encoding for Kn resistance. EcFtsZ mutants were constructed by site-directed mutagenesis using the Quick Change Mutagenesis Kit from Stratagene. All plasmid DNAs were purified with E.Z.N.A Plasmid Miniprep Kit I from Omega Bio-tek. Each ftsZ gene with the corresponding point mutation was completely sequenced. Recombinant EcFtsZ and FtsZ mutants were overexpressed in the E. coli BL21(DE3) strain by induction with IPTG. The cells were grown at 37°C with agitation in 2 L of LB broth containing 100 μg/mL Ap until an OD600 of 0.7 was reached; then 0.4 mM IPTG was added, and the cells were grown for an additional 4 h. The proteins were purified as described in Beuria et al. (2003) with some modifications. Briefly, the cells were centrifuged at 7140g in a SORVALL RC-3B centrifuge with an H6000A rotor for 45 min, and the pellet was suspended in buffer A (50 mM Tris-HCl at pH 8.0, 50 mM KCl, 1 mM EDTA, and 10% glycerol) and lysed by sonication with five pulses of 45 W and two pulses of 75 W. The lysed cells were centrifuged at 92,000g for 90 min, and the supernatant was saturated with 25% ammonium sulfate. The solution was centrifuged at 9391g at 4°C, and the pellet was dissolved in buffer A then dialyzed against buffer A to remove the salt excess. Mono Q anion-exchange chromatography was performed, and the protein eluted with a salt gradient (Buffer B: 50 mM Tris-HCl at pH 8.0, 50 mM KCl, 1 mM EDTA, 1 M KCl, and 10% glycerol). A monosodium glutamate polymerization and depolymerization cycle was performed using a solution of 50 mM PIPES (pH 6.5), 1 M glutamate, 10 mM CaCl2, 10 mM MgCl2, and 2 mM GTP for 30 min at 37°C, and the polymers obtained were centrifuged at 9391g for 30 min at 27°C (Beuria et al. 2003). The protein was suspended in 5 mL of buffer A, washed twice with the same volume of buffer, and concentrated in a Centripep system and stored at −80°C. The protein concentration and the nucleotide content were determined by absorption at 254 and 280 nm in 6 M GdmCl with phosphate buffer (pH 6.5) (Rivas et al. 2000). The Bradford method was calibrated for FtsZ. The content of GDP or GTP was determined by HPLC (high-performance liquid chromatography) in a SUPELCOSIL LC-18-D13 column.
Polymerization turbidity
The assay was performed in a solution at 30°C containing 50 mM Mes (pH 6.5), 50 mM KCl, and 10 mM MgCl2 using the protein concentration indicated in the figures. The samples were incubated for 3 min at 30°C, and the reaction was started by adding 1 mM GTP. The turbidity of the polymerization reactions was monitored at 90° by illuminating the sample at 350 nm in a Perkin-Elmer LS-50 spectrofluorimeter with a 2% transmission filter in the emission beam.
GTPase activity
The protein was incubated for 3 min at 30°C in a solution containing 50 mM Mes (pH 6.5), 50 mM KCl, and 10 mM MgCl2; 1 mM GTP was added to initiate the reaction. The reactions were stopped at different times by adding 10% perchloric acid and neutralized with 1 M KHCO3; the bubbles of CO2 were removed by centrifugation in a Speed-Vac for 5 min. The denatured protein and the insoluble KClO4 salt were removed by centrifugation at 17,530g for 10 min, and the supernatants were analyzed by HPLC in a SUPELCOSIL LC-18-D13 column. The separation of GDP from GTP was undertaken using a mobile phase containing 4 mM TBAP (tetrabutyl ammonium dihydrogen phosphate), 0.2 M K2HPO4, and 0.1 M glacial acetic acid. The nucleotide content was calculated from the area of the peak associated with GDP and GTP, and GDP was normalized with respect to the total nucleotide content (GDP/[GDP + GTP]).
In vivo assay
To evaluate the functionality in vivo of the F40W, F135W, F275W, I294W, and I294W/F275A mutants of EcFtsZ, an assay measuring the viability of cells carrying these mutations with respect to the wild type was performed using as a host the strain E. coli VIP2(DE3)/pLAR9. VIP2(DE3)/pLAR9 is VIP2/pLAR9 (Pla et al. 1991) lysogenized by the λDE3 Lysogenization Kit (Novagen). In this strain, the ftsZ chromosomal copy is interrupted by an insertion, and it is complemented with a copy encoded in the thermosensitive plasmid pLAR9. Overnight cultures of VIP2(DE3)/pLAR9 grown at the permissive temperature (30°C) were transformed with plasmid pMFV57 containing ftsZ wt or the point-mutated ftsZ genes. The transformed cells were grown at 30°C in LB broth containing 100 μg/mL Ap, 50 μg/mL Kn, and 50 μg/mL Cm until an OD600 of 0.6 was attained. Serial dilutions were plated at both the permissive temperature (30°C) in LB-Ap/Kn/Cm and nonpermissive temperature (42°C) in LB-Ap/Kn, and the number of colonies was counted.
Circular dichroism and fluorescence spectroscopies
CD (circular dichroism) spectra were recorded in a Jasco 600 spectropolarimeter using cells of 1 mm pathlength. Each sample and buffer were measured with four scans, using a 20-nm/min scan speed, 1-nm bandwidth, and 2-sec time constant. Emission fluorescence spectra were obtained with a Perkin Elmer LS-50 spectrofluorimeter. Three scans were recorded for each measurement, using a 295-nm excitation wavelength and 140-nm/min scan speed. CD and fluorescence emission spectra were analyzed with GRAMS software and plotted with SigmaPlot of Systat Software Inc.
Protein unfolding and refolding
Unfolding and refolding experiments were measured by CD (EcFtsZ 7.4 μM) and fluorescence (EcFtsZ 12 μM) in solutions containing 50 mM Tris-HCl (pH 7.5) with different GdmCl concentrations. The samples were left on ice for 1 h, to reach the equilibrium avoiding thermal denaturation, then incubated for 15 min at 25°C and carefully filtrated through a 0.2-μm filter before the measurement. A control for each sample (solution without protein) was treated in the same way. The native spectra were obtained from the sample without GdmCl. For the refolding experiments, the protein was previously denatured in 6 M GdmCl and equilibrated for 30 min at 4°C. An aliquot of this denatured sample was rapidly diluted (100 times) in the same buffer used for denaturation with the corresponding GdmCl concentrations and kept on ice for 1 h; after 15 min at 25°C, CD and fluorescence were measured. The emission spectra were measured observing the change in fluorescence intensity and in the red shift when the GdmCl concentration was increased. To calculate the value of [GdmCl]50%, which corresponds to the concentration of GdmCl at 50% of unfolding or refolding, a fit of the experimental data to the following equation was undertaken using SigmaPlot software:
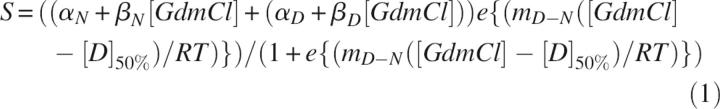 |
where S is the observed spectroscopic signal; αN is the signal of the native state without GdmCl, and βN = dαN/d[GdmCl] (the slope); αD and βD are the corresponding quantities for the denatured state; m D−N is a constant of proportionality; and [D]50% is the GdmCl concentration in which the protein is 50% unfolded (Staniforth et al. 1993).
Electron microscopy
FtsZ samples were adsorbed to carbon-coated grids, negatively stained with 1% uranyl acetate, and observed in a Zeiss EM-109 model electron microscope.
Correlated mutations analysis
The 1fsz.hssp alignment obtained from the HSSP database, available in May 2005, was analyzed by PLOTCORR software (Pazos et al. 1997).
Acknowledgments
We thank José Manuel Andreu, David Jameson, and Michael Handford for critically reading the manuscript. This work was supported by FONDECYT Grants 1050677 and 7060162.
Footnotes
Reprint requests to: Octavio Monasterio, Laboratorio de Biología Estructural y Molecular, Departamento de Biología, Facultad de Ciencias, Universidad de Chile, Las Palmeras 3425, Ñuñoa, Casilla 653, Santiago, Chile; e-mail: monaster@uchile.cl; fax: 56-2-276-3870.
Abbreviations: ΔASA, change in the accessible solvent area; GdmCl, guanidinium chloride; EcFtsZ, Escherichia coli FtsZ; MjFtsZ, Methanococcus jannaschii FtsZ; TmFtsZ, Thermotoga maritima FtsZ; PaFtsZ, Pseudomona aeruginosa FtsZ; MtbFtsZ, Mycobacterium tuberculosis FtsZ; Mes, 2-Morpholinoethanesulfonic acid; IPTG, isopropyl-1-thio-β-D-galactopyranoside; CD, circular dichroism; TBAP, tetrabutyl ammonium dihydrogen phosphate; RMS, root mean square deviation; ANS, 8-anilinonaphthalene-1- sulphonate; FITC, fluorescein isothiocyanate; HPLC, high-performance liquid chromatography; Ap, ampicillin; Kn, kanamycin; Cm, chloramphenicol; Cr, critical concentration.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.072807607.
References
- Andreu J.M., Oliva, M.A., and Monasterio, O. 2002. Reversible unfolding of FtsZ cell division proteins from archaea and bacteria. Comparison with eukaryotic tubulin folding and assembly. J. Biol. Chem. 277: 43262–43270. [DOI] [PubMed] [Google Scholar]
- Beuria T.K., Krishnakumar, S.S., Sahar, S., Singh, N., Gupta, K., Meshram, M., and Panda, D. 2003. Glutamate-induced assembly of bacterial cell division protein FtsZ. J. Biol. Chem. 278: 3735–3741. [DOI] [PubMed] [Google Scholar]
- Buey R.M., Diaz, J.F., and Andreu, J.M. 2006. The nucleotide switch of tubulin and microtubule assembly: A polymerization-driven structural change. Biochemistry 45: 5933–5938. [DOI] [PubMed] [Google Scholar]
- Chen Y. and Erickson, H.P. 2005. Rapid in vitro assembly dynamics and subunit turnover of FtsZ demonstrated by fluorescence resonance energy transfer. J. Biol. Chem. 280: 22549–22554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y., Bjornson, K., Redick, S.D., and Erickson, H.P. 2005. A rapid fluorescence assay for FtsZ assembly indicates cooperative assembly with a dimer nucleus. Biophys. J. 88: 505–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordell S.C., Robinson, E.J., and Lowe, J. 2003. Crystal structure of the SOS cell division inhibitor SulA and in complex with FtsZ. Proc. Natl. Acad. Sci. 100: 7889–7894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creighton T.E. 1993. Proteins: Structures and molecular properties, 2nd ed, pp. 1–47. Freeman, New York.
- Diaz J.F., Kralicek, A., Mingorance, J., Palacios, J.M., Vicente, M., and Andreu, J.M. 2001. Activation of cell division protein FtsZ. Control of switch loop T3 conformation by the nucleotide γ-phosphate. J. Biol. Chem. 276: 17307–17315. [DOI] [PubMed] [Google Scholar]
- Errington J., Daniel, R.A., and Scheffers, D.J. 2003. Cytokinesis in bacteria. Microbiol. Mol. Biol. Rev. 67: 52–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freire E. 1999. The propagation of binding interactions to remote sites in proteins: Analysis of the binding of the monoclonal antibody D1.3 to lysozyme. Proc. Natl. Acad. Sci. 96: 10118–10122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaskin F., Cantor, C.R., and Shelanski, M.L. 1974. Turbidimetric studies of the in vitro assembly and disassembly of porcine neurotubules. J. Mol. Biol. 89: 737–755. [DOI] [PubMed] [Google Scholar]
- Hannemann F., Bera, A.K., Fischer, B., Lisurek, M., Teuchner, K., and Bernhardt, R. 2002. Unfolding and conformational studies on bovine adrenodoxin probed by engineered intrinsic tryptophan fluorescence. Biochemistry 41: 11008–11016. [DOI] [PubMed] [Google Scholar]
- Henrick K. and Thornton, J.M. 1998. PQS: A protein quaternary structure file server. Trends Biochem. Sci. 23: 358–361. [DOI] [PubMed] [Google Scholar]
- Hilser V.J., Dowdy, D., Oas, T.G., and Freire, E. 1998. The structural distribution of cooperative interactions in proteins: Analysis of the native state ensemble. Proc. Natl. Acad. Sci. 95: 9903–9908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huecas S. and Andreu, J.M. 2004. Polymerization of nucleotide-free, GDP- and GTP-bound cell division protein FtsZ: GDP makes the difference. FEBS Lett. 569: 43–48. [DOI] [PubMed] [Google Scholar]
- Jones S. and Thornton, J.M. 1996. Principles of protein–protein interactions. Proc. Natl. Acad. Sci. 93: 13–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khorasanizadeh S., Peters, I.D., and Roder, H. 1996. Evidence for a three-state model of protein folding from kinetic analysis of ubiquitin variants with altered core residues. Nat. Struct. Biol. 3: 193–205. [DOI] [PubMed] [Google Scholar]
- Leung A.K., White, E.L., Ross, L.J., Reynolds, R.C., DeVito, J.A., and Borhani, D.W. 2004. Structure of Mycobacterium tuberculosis FtsZ reveals unexpected, G protein-like conformational switches. J. Mol. Biol. 342: 953–970. [DOI] [PubMed] [Google Scholar]
- Lowe J. 1998. Crystal structure determination of FtsZ from Methanococcus jannaschii . J. Struct. Biol. 124: 235–243. [DOI] [PubMed] [Google Scholar]
- Lu C., Reedy, M., and Erickson, H.P. 2000. Straight and curved conformations of FtsZ are regulated by GTP hydrolysis. J. Bacteriol. 182: 164–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C., Stricker, J., and Erickson, H.P. 2001. Site-specific mutations of FtsZ—Effects on GTPase and in vitro assembly. BMC Microbiol. 1: 7. doi: 10.1186/1471-2180-1-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutkenhaus J. 1990. Regulation of cell division in E. coli . Trends Genet. 6: 22–25. [DOI] [PubMed] [Google Scholar]
- Marrington R., Small, E., Rodger, A., Dafforn, T.R., and Addinall, S.G. 2004. FtsZ fiber bundling is triggered by a conformational change in bound GTP. J. Biol. Chem. 279: 48821–48829. [DOI] [PubMed] [Google Scholar]
- McDonald I.K. and Thornton, J.M. 1994. Satisfying hydrogen bonding potential in proteins. J. Mol. Biol. 238: 777–793. [DOI] [PubMed] [Google Scholar]
- Mingorance J., Rueda, S., Gomez-Puertas, P., Valencia, A., and Vicente, M. 2001. Escherichia coli FtsZ polymers contain mostly GTP and have a high nucleotide turnover. Mol. Microbiol. 41: 83–91. [DOI] [PubMed] [Google Scholar]
- Mukherjee A. and Lutkenhaus, J. 1998. Dynamic assembly of FtsZ regulated by GTP hydrolysis. EMBO J. 17: 462–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee A. and Lutkenhaus, J. 1999. Analysis of FtsZ assembly by light scattering and determination of the role of divalent metal cations. J. Bacteriol. 181: 823–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nooren I.M. and Thornton, J.M. 2003. Structural characterisation and functional significance of transient protein–protein interactions. J. Mol. Biol. 325: 991–1018. [DOI] [PubMed] [Google Scholar]
- Oliva M.A., Huecas, S., Palacios, J.M., Martin-Benito, J., Valpuesta, J.M., and Andreu, J.M. 2003. Assembly of archaeal cell division protein FtsZ and a GTPase-inactive mutant into double-stranded filaments. J. Biol. Chem. 278: 33562–33570. [DOI] [PubMed] [Google Scholar]
- Oliva M.A., Cordell, S.C., and Lowe, J. 2004. Structural insights into FtsZ protofilament formation. Nat. Struct. Mol. Biol. 11: 1243–1250. [DOI] [PubMed] [Google Scholar]
- Pazos F., Helmer-Citterich, M., Ausiello, G., and Valencia, A. 1997. Correlated mutations contain information about protein–protein interaction. J. Mol. Biol. 271: 511–523. [DOI] [PubMed] [Google Scholar]
- Pla J., Sanchez, M., Palacios, P., Vicente, M., and Aldea, M. 1991. Preferential cytoplasmic location of FtsZ, a protein essential for Escherichia coli septation. Mol. Microbiol. 5: 1681–1686. [DOI] [PubMed] [Google Scholar]
- Rivas G., Lopez, A., Mingorance, J., Ferrandiz, M.J., Zorrilla, S., Minton, A.P., Vicente, M., and Andreu, J.M. 2000. Magnesium-induced linear self-association of the FtsZ bacterial cell division protein monomer. The primary steps for FtsZ assembly. J. Biol. Chem. 275: 11740–11749. [DOI] [PubMed] [Google Scholar]
- Romberg L. and Mitchison, T.J. 2004. Rate-limiting guanosine 5′-triphosphate hydrolysis during nucleotide turnover by FtsZ, a prokaryotic tubulin homologue involved in bacterial cell division. Biochemistry 43: 282–288. [DOI] [PubMed] [Google Scholar]
- Sánchez R. and Sali, A. 1997. Evaluation of comparative protein structure modeling by MODELLER-3. Proteins (Suppl.) 1: 50–58. [DOI] [PubMed] [Google Scholar]
- Santra M.K. and Panda, D. 2003. Detection of an intermediate during unfolding of bacterial cell division protein FtsZ: Loss of functional properties precedes the global unfolding of FtsZ. J. Biol. Chem. 278: 21336–21343. [DOI] [PubMed] [Google Scholar]
- Sippl M.J. 1993. Recognition of errors in three-dimensional structures of proteins. Proteins 17: 355–362. [DOI] [PubMed] [Google Scholar]
- Sossong T.M., Brigham-Burke, M.R., Hensley, P., and Pearce Jr, K.H. 1999. Self-activation of guanosine triphosphatase activity by oligomerization of the bacterial cell division protein FtsZ. Biochemistry 38: 14843–14850. [DOI] [PubMed] [Google Scholar]
- Staniforth R.A., Burston, S.G., Smith, C.J., Jackson, G.S., Badcoe, I.G., Atkinson, T., Holbrook, J.J., and Clarke, A.R. 1993. The energetics and cooperativity of protein folding: A simple experimental analysis based upon the solvation of internal residues. Biochemistry 32: 3842–3851. [DOI] [PubMed] [Google Scholar]
- Taniuchi H. and Anfinsen, C.B. 1969. An experimental approach to the study of the folding of staphylococcal nuclease. J. Biol. Chem. 244: 3864–3875. [PubMed] [Google Scholar]
- Vicente M., Rico, A.I., Martinez-Arteaga, R., and Mingorance, J. 2006. Septum enlightenment: Assembly of bacterial division proteins. J. Bacteriol. 188: 19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]