

Abstract
The severity of Helicobacter pylori-related disease is correlated with a pathogenicity island (the Cag region of about 26 genes) whose presence is associated with the up-regulation of an IL-8 cytokine inflammatory response in gastric epithelial cells. Statistical analysis of the Cag gene sequences calculated from the complete genome of strain 26695 revealed several unusual features. The Cag7 sequence (1,927 aa) has two repeat regions. Repeat region I runs 317 aa in a form of 𝒜𝒜𝒜 proximal to the protein N terminal; repeat region II extends 907 aa in the middle of the protein sequence consisting of 74 contiguous segments composed from selections among six consensus sequences and includes 58 regularly distributed cysteine residues with consecutive cysteines mostly 12, 18, or 24 aa apart. This “regular” cysteine arrangement may provide a scaffolding of linker elements stabilized by disulfide bridges. When Cag7 homologues from different strains are compared, differences were found almost exclusively in the repeat regions, resulting from deletion and/or insertion of repeating units. These observations suggest that the anomalous repetitive structure of the sequence plays an important role in the conformation of Cag7 gene product and potentially in the function of the pathogenicity island. Other facets of the Cag7 sequence show significant charge clusters, high multiplet count, and extremes of amino acid usage.
Helicobactor pylori (HP) is a Gram-negative spiral-shaped bacterium that colonizes the human stomach. About 50% of humans are infected by HP but only 10% exhibit clinical disease, including chronic gastritis, gastric carcinoma, and peptic ulcer (1). The more severe forms of disease are associated with infection by specific strains called type I. Two type I HP strains have been sequenced in their entirety [strains 26695 (2) and J99 (3)]. Virulent HPs differ from less virulent strains (type II) by the presence of a ∼40-kb block of genes called the Cag pathogenicity island (abbreviated Cag PAI or CagA region; ref. 4). No specific function is established for any gene from the Cag island. However, Cag-positive, but not Cag-negative, strains cause cultured gastric epithelial cells to secrete the proinflammatory cytokine IL-8 (4,5), and this ability is abolished by specific mutation of many of the 26 ORFs found in the Cag island (4–6). Several of these genes are modestly similar to genes of other pathogens that encode subunits of specialized type IV secretory systems that directly deliver bacterial virulence factors to the surface and possibly into host cells. Control of bacterial virulence often is mediated by changes at the DNA sequence level that affect gene regulation or expression (7). Three Cag PAI now have been sequenced from the complete genomes of strains 26695 and J99 and the sequenced cosmid 36 from strain NCTC11638. All three contain an unusual ORF (annotated Cag7 or HP527 in strain 26695), which is significantly variable among HP pathogenic strains, but no mechanisms for this variation have been proposed and no features of the Cag7 sequence have been noted to account for the origin of this variation.
We present here a rigorous statistical analysis of the Cag7 protein (1,927 aa) from strain 26695. Of particular interest, we underscore several sequence features of this protein, including distinctive repeat patterns, a remarkable cysteine residue distribution, a statistically significantly high multiplet count (defined below), a pronounced charge residue cluster (8, 9), extremes of lysine and glutamate amino acid usage, and identification of hydrophobic potential transmembrane segments. Expansion or contraction of the repeats could account for the size variations seen in the ORF of Cag7.
RESULTS
Unusual Sequence Features of Cag7.
The saps (Statistical Analysis of Protein Sequences) program (8) was applied to all the putative proteins encoded from the CagA region of strain 26695. This analysis reveals several unusual sequence features especially for the Cag7 protein, which was found to contain two impressive regions composed of contiguous repeated amino acid sequences.
Repeat I.
Repeat I (Fig. 1), covering amino acid positions 9-325 inclusive, in the pattern 𝒜𝒜*𝒜**, has 𝒜 (130 aa) aligned with 𝒜* (130 aa), showing only three mismatches and 𝒜** (57 aa), a truncated copy of 𝒜*, which matches perfectly over their common 57 aa and, more impressively, in perfect DNA agreement. Remarkably the 𝒜 and 𝒜* differ at only three DNA positions, which all occur in codon site 1. There are no synonymous (silent site) substitutions. The almost perfect DNA identities comparing 𝒜 to 𝒜* or 𝒜** strongly suggest a recent origin to these repeats.
Figure 1.

Alignment of amino acid sequences of 𝒜, 𝒜*, and 𝒜** in repeat I. Matching residues are indicated by dots. 𝒜 and 𝒜* differ at three residues; 𝒜** is 73 aa shorter than 𝒜 or 𝒜*. 𝒜** matches exactly with 𝒜* over their common 57 residues. The numbers to the right of the sequences give their coordinates within the Cag7 protein. DNA conservation with respect to 𝒜 and 𝒜* differ only at codon site 1 of the altered aa. 𝒜* and 𝒜** are identical at the DNA level in their common sequence.
Repeat II.
Repeat II (Figs. 2 and 3) consists of 74 contiguous segments composed from selections among six different consensus sequences, which we call α, β, λ, μ, δ, ɛ, stretching over amino acid positions 477-1383. The underline signifies perfect conservation of the amino acids at that position among the ensemble of sequences of α, of β, etc.
Figure 2.

 |
Figure 3.

Aligned DNA sequences corresponding to the amino acid sequences of group μ are displayed. 1 indicates a position strictly conserved among these sequences, and ★ indicates a position with average conservation index (CI, defined in Fig. 2 legend) exceeding 0.6. The scores for nucleotide comparisons are as follows: identity has value 1, a transition replacement (A ↔ G or C ↔ T) has value 0.3, a substitution A ↔ T or G ↔ C has value −0.3, and a substitution A ↔ C or G ↔ T has value −0.5. The average CI for these sequences over all columns is 0.8, and 87% of the columns show a CI value above 0.6 emphasizing a high level of conservation.
α = C E K L L T P E A (K/R) K L L E (14 aa length). Some α have one or two appended aa, generally E, EE, or QE:
 |
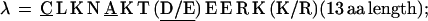 |
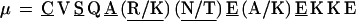 |
 |
In repeat II, a λ sequence is always followed by a β sequence and μ by α:
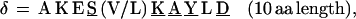 |
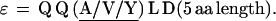 |
The explicit order of the subsequences of repeat II is displayed next:
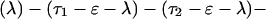 |
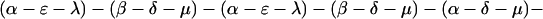 |
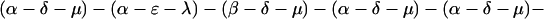 |
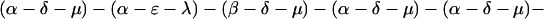 |
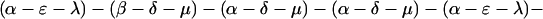 |
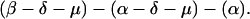 |
The sequences τ1 and τ2 in the above pattern each begin with a cysteine but significantly differ from the consensus sequences α and β, respectively. Each specific α unit aligns substantially with the consensus α, each β unit aligns substantially with the consensus β, etc. The main repeat units occur as triplet groups of sequences of the form
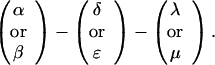 |
The δ sequences invariably are followed by a μ sequence, ɛ sequences are followed invariably by a λ sequence, β sequences are followed by δ sequences, whereas α sequences are followed by either ɛ or δ sequences. It is worth emphasis that DNA conservation in these repeats among the α, β, δ, etc. is very high (see Fig. 3 for μ).
Regular cysteine residue spacings.
Cag7 contains 58 cysteine residues scaffolding repeat II. To underscore the regular distribution of the cysteine residues, we display their spacings. (The notation C-12-C signifies that the positions of the two successive cysteines are 12 residues apart, C-18-C indicates that the two cysteines are 18 residues apart, etc.):
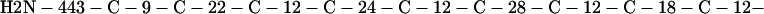 |
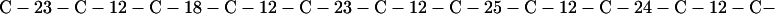 |
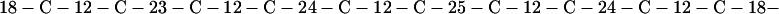 |
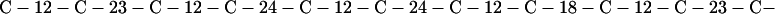 |
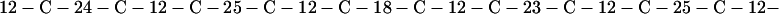 |
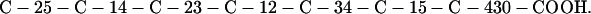 |
We see that there is no cysteine of Cag7 among the initial 443 residues nor in the terminal 430 residues. Otherwise, the cysteines are principally 12 positions apart or sometimes about 18 or 24 positions apart. This cysteine arrangement may implicate a distinctive three-dimensional structural conformation in this part of the protein, probably stabilized by a plethora of disulphide bridges. This cysteine arrangement differs from other classical cysteine arrangements, including kringle patterns, epidermal growth factor domains, fibronectin structures, and zinc fingers.
Comparisons of the Cag7 Sequence Among Different HP Strains.
There are three HP strains from which the CagA region is wholly sequenced. These are available from the complete genome strain 26695, complete genome strain J99, and cosmid 36 strain NCTC 11638 (6). The alignments of the Cag7 protein from the three sources are represented in Fig. 4.
Figure 4.

The Cag7 protein sequence is aligned with the translated protein in cosmid 36 combining ORF14, ORF13, and the intervening part requiring a single base (+1) frame shift after amino acid 682 (counting from the N terminus of ORF14). When introducing the frame shift the DNA sequence encodes a protein that, apart from two gaps, aligns more than 90% with Cag7. The first gap corresponds to amino acids 9–138 of Cag7, consisting of unit 𝒜, the second gap corresponds to amino acids 1114–1182, consisting of two consecutive repeat triplet groups, namely (α-ɛ-λ)–(β-δ-μ) (see text). The Cag7 ortholog jhp0476 in strain J99 is displayed below Cag7. The jhp0476 sequence is missing a segment equivalent to 𝒜 of Cag7, and the unit corresponding to 𝒜** is 16 aa longer. The same two consecutive triplet groups missing from cosmid 36 also are missing from jhp0476, whereas the repeat II in jhp0476 extends longer by 78 aa augmented by the two successive triplet groups (δ-μ-α)–(δ-μ-α). The Cag7 and jhp0476 can be divided into three parts corresponding to ORF14, ORF13, and the intervening piece in cosmid 36. The ★ locate two significantly long uncharged (potential transmembrane) segments, the first traversing amino acid positions 343–370 downstream proximal to repeat I and the second segment of positions 1836-1870 is near the C terminus. + corresponds to a concentrated charge region. The arrows indicate the extent and orientation of the ORFs.
Cag7 matches excellently the two genes ORF14 and ORF13 of cosmid 36 when encoded together with their intervening sequence. The correspondence with ORF14 possesses a deletion of 130 successive residues near the N terminal of Cag7, whereas ORF13 aligns almost perfectly with the C-terminal quarter of Cag7. Notably, the initial 𝒜 of repeat I in Cag7 is the 130-residue segment missing from ORF14. The sequence intervening ORF13 and ORF14 is replete with nonsense codons. However, in introducing a frame shift (skip a guanine at nucleotide position 24186) relative to the cosmid 36 sequence, the amino acid sequence resulting from this translation aligns almost perfectly with the middle part of Cag7, but for an absent block of 69 aa. The missing part is equivalent to the two repeat triplet groups of repeat II in Cag7, those of sequences (α-ɛ-λ)–(β-δ-μ) corresponding to amino acid positions 1114-1182 of Cag7. These alterations suggest that the number of repeat units may be part of the mechanism regulating the expression, conformation, and/or function of the protein. We also guess that the +1 frame shift serves to regulate the expression of Cag7-like genes among different strains, which conceivably also controls the virulence of the bacterium. When the frame shift is present, ORF14 and ORF13 merge into one protein, which is more than 90% identical with Cag7, but with six consecutive repeat subunits missing. When the frame shift is removed, ORF14 and ORF13 lose most of repeat II.
The Cag7 ortholog jhp0476 in strain J99 compared to Cag7 (HP0527) in parallel with cosmid 36 misses 𝒜 of repeat I whereas the unit 𝒜** is 16 aa longer than its counterpart in Cag7. Repeat II of jhp0476 as with cosmid 36 is missing the same triplet groups (α-ɛ-λ)–(β-δ-μ). On the other hand, repeat II in jhp0476 extends 78 aa longer augmented by two triplet groups. The DNAs of these two proteins align with 87% identity.
Possible Role of the Repeat Regions of Cag7 in Pathogenicity.
The repeat lengths of repeats I and II among different strains of HP are markedly variable with different numbers of 𝒜 in repeat I and generally different numbers of repeat units composing repeat II. In fact, comparative analysis of a panel of strains of Cag7 homologues using PCR proceeding from common primers flanking the repeat regions attest experimentally to significant variation in the length of repeats I and II from strain to strain but not in the same strain passed in vitro or in vivo (in the mouse) over time (T.M. and S.F., unpublished data). Consistent with these lines, a survey of HP Cag7 analogs in a collection of several primate isolates revealed significant polymorphism in the repeat I and repeat II lengths. The dramatic DNA identity within the repeat structures putatively generated through recombination or replication strand slippage allows opportunities for changes in the repeat length. Different lengths may produce alternative protein conformations or serve to switch the protein’s expression on and off, thus affording the HP bacterium a means to confound host immune system surveillance.
Significantly High Multiplet Count in the Cag7 Sequence.
A measure of the homopeptide density of a protein sequence is provided by the multiplet count, i.e., the number of distinct homooligopeptide runs of two or more residues. Specifically, multiplet counts refer to the number of homopeptides in protein sequences counting all homodipeptides XX(=X2), homotripeptides YYY(=Y3), homotetrapeptides Z4, etc., where X, Y, Z denotes any amino acid. A statistical assessment of the counts and locations of these multiplets compares the observed multiplet set to the multiplet distribution in a random (shuffled) reconstruction of the protein sequence. A significance test of high multiplet counts would take account of the amino acid composition of the protein sequence under study and is described in Karlin et al. (12). The scarce occurrence of proteins in possession of an abundance of amino acid multiplets stands out in Escherichia coli and in most prokaryotes. The percentage of human proteins with significantly high multiplet counts is about 1.5% with similar percentages observed in mouse and yeast. A greater number of proteins with significantly many multiplets is detected in Drosophila (about 10%), usually associated with developmental regulatory genes (13). Strikingly, Cag7 in HP (strain 26695) and HP (strain J99) is the only protein sequence of HP that carries a significantly high multiplet count. In the case at hand, the bulk of the multiplets concentrate in the two repeat regions (see Fig. 5), where a preponderance of lysine and glutamate doublets KK and EE (or EEE) appear (see Discussion for possible implications).
Figure 5.

The 177-aa multiplets (see text for details) and their distribution in Cag7 are shown. Most of these multiplets occur in the two repeat regions of positions 9-325 and 477-1383, respectively.
It is interesting that significantly high multiplet counts are also present in the genes containing the PGRS repeats of Mycobacterium tuberculosis contemplated also as pathogenicity islands (14, 15). The human neurological disease genes associated with long trinucleotide CAG (glutamine) iterations and other long amino acid runs also are correlated with high multiplet counts (13).
Potential Transmembrane Segments in Cag7.
The Cag7 distinguishes two statistically significant long predominantly hydrophobic uncharged runs, traversing coordinates 343-370 proximal downstream to repeat I and 1836-1870 near the carboxyl end.
DISCUSSION
In the CagA region of the HP genome strain 26695, the Cag7 (HP527) gene (1927 aa) is replete with unusual sequence features. This gene has been noted by other researchers because of its marginal sequence similarity to the virB10 family (percent similarity about 30%) of type IV secretory genes and its necessity for HP’s induction of IL-8 secretion in gastric epithelial cells and the strain-to-strain variation in size. We have found that this variation occurs within two repeat regions in the Cag7 protein. The amino end of Cag7 is distinguished by the long tandem repeat 𝒜𝒜*𝒜** (total length 317 aa). The middle part of Cag7, repeat II, covering amino acid positions 477-1383 consists of 74 subsequences selected from six consensus sequences α (generally 14 aa), β (14 aa), λ (13 aa), μ (13 aa), δ (10 aa), and ɛ (5 aa) (see previous text or Fig. 2 for the explicit sequences). The DNA identity among different representations of the consensus sequences is very high. Other strains of HP maintain polymorphic versions of repeats I and II associated strictly with variation in repeat subunits. The published sequences of Cag7 (HP527) with homologues from two other strains suggest that the strain-to-strain variation could be explained by recombination within the gene mediated by repeat subunits or can, in part, result from replication strand slippage. The three strains also may reflect on Cag7 variation among separate population sources. Thus, strain 26695 comes from a United Kingdom individual, strain J99 comes from a United States individual, and cosmid 36 (strain NCTC11638) was sequenced from an Australian individual. The extent over time of Cag7 variation from a single strain has not been adequately ascertained.
Apart from the striking repeat patterns, Cag7 is extraordinary in other sequence attributes, including high multiplet count, significant charge clusters (not shown), several extreme amino acid usages, and potential of transmembrane segments.
Issues and potential experiments to be considered are:
(i) What part of Cag7 is necessary for virulence? The frame shift in the intervening region between ORF14 and ORF13 of cosmid 36 converts the ORFs into an almost complete homologue of Cag7. The polymorphism resulting from variations of the repeat numbers and lengths may enhance or curtail interactions with the host and serve as a means of shielding the bacterium from an immune system attack. The changes in the repeat numbers may affect how the Cag7 protein surface looks to the immune system and thereby may avoid recognition by antibodies made during previous infections. The almost perfect DNA identities within repeats I and II strongly argues for rather recent changes in repeat numbers. These repeat patterns may indicate a facility of HP for allowing rapid changes prompted by some host immune attacks.
(ii) The regular distribution of cysteine residues in repeat II provides a possible scaffolding involving disulfide bridges cross-linking secondary structures and/or domains of the protein structure. It would be informative to synthesize a triplet unit of the repeat II, say α-ɛ-λ and/or β-δ-μ, and evaluate its secondary structure in an aqueous medium.
(iii) The role of repeats in protein sequences is generally unclear. They may be benign, arising through replication strand slippage or recombination. They may provide flexibility and variation to protein conformation and function in response to environmental stress or host surveillance. They may contribute a regulatory role in gene transcription, translation, and expression. They may facilitate binding capacities in protein–protein and protein–DNA interactions. The pattern of repeat II, coupled to the regular cysteine distribution and an abundance of KK and EE diresidues, may contribute to several of these activities.
(iv) The high multiplet count of Cag7 is dominated by lysine and glutamate doublets, that are especially rife in repeat region II. These conceivably provide opportunities for multiple salt bridges, facilitating conformational stability and/or contributing to protein–protein interactions and/or quaternary structure formations (16).
(v) The main question about Cag7 is what is its function? Its similarity to secretory genes (virB10 family) of other species and its necessity for IL-8 secretion would support the idea that it is a component of a secretory apparatus that delivers a product or products that induces the IL-8 response. The virB10 gene has been proposed to play a regulatory function of the type IV secretory system of Agrobacterium tumefaciens (17). It is noteworthy that the portion containing the repeat regions is absent from members of the virB10 family in all other sequenced species. These observations suggest that the repeat regions and their contractions and expansions play key regulatory roles in the function of the putative HP secretory apparatus.
Acknowledgments
We are happy to acknowledge valuable discussions with Drs. B. E. Blaisdell, L. Brocchieri, A. M. Campbell, J. Mrázek, and N. Salama. This work was supported by National Institutes of Health Grants 5R01GM10452-34 (G.L. and S.K.), 5R01HG00335-11 (G.L. and S.K.), and AI38459 (T.M. and S.F.), National Science Foundation Grant DM59704552 (G.L. and S.K.), and the Cancer Research Fund of the Damon Runyon-Walter Winchell Foundation, DRG-1456 (T.M.).
ABBREVIATION
- HP
Helicobacter pylori
References
- 1.Blaser M J, Parsonnet J. J Clin Invest. 1994;94:4–8. doi: 10.1172/JCI117336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tomb J F, White O, Kerlavage A R, Clayton R A, Sutton G G, Fleischmann R D, Ketchum K A, Klenk H P, Gill S, Dougherty B A, et al. Nature (London) 1997;388:539–547. doi: 10.1038/41483. [DOI] [PubMed] [Google Scholar]
- 3.Alm R A, Ling L S, Moir D T, King B L, Brown E D, Doig P C, Smith D R, Noonan B, Guild B C, deJonge B L, et al. Nature (London) 1999;397:176–180. doi: 10.1038/16495. [DOI] [PubMed] [Google Scholar]
- 4.Censini S, Lange C, Xiang Z, Crabtree J E, Ghiara P, Borodovsky M, Rappuoli R, Covacci A. Proc Natl Acad Sci USA. 1996;93:14648–14653. doi: 10.1073/pnas.93.25.14648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Crabtree J E, Farmery S M, Lindley I J, Figura N, Peichl P, Tompkins D S. J Clin Pathol. 1994;47:945–950. doi: 10.1136/jcp.47.10.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Akopyants N S, Clifton S W, Kersulyte D, Crabtree J E, Youree B E, Reece C A, Bukanov N O, Drazek E S, Roe B A, Berg D E. Mol Microbiol. 1998;28:37–53. doi: 10.1046/j.1365-2958.1998.00770.x. [DOI] [PubMed] [Google Scholar]
- 7.Finlay B B, Falkow S. Microbiol Mol Biol Rev. 1997;61:136–169. doi: 10.1128/mmbr.61.2.136-169.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brendel V, Bucher P, Nourbakhsh I, Blaisdell B E, Karlin S. Proc Natl Acad Sci USA. 1992;89:2002–2006. doi: 10.1073/pnas.89.6.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Karlin S. Curr Opin Struct Biol. 1995;5:360–371. doi: 10.1016/0959-440x(95)80098-0. [DOI] [PubMed] [Google Scholar]
- 10.Brocchieri L, Karlin S. J Mol Biol. 1998;276:249–264. doi: 10.1006/jmbi.1997.1527. [DOI] [PubMed] [Google Scholar]
- 11.Henikoff S, Henikoff J G. Proc Natl Acad Sci USA. 1992;89:10915–10919. doi: 10.1073/pnas.89.22.10915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Karlin S, Brendel V, Bucher P. Mol Biol Evol. 1992;9:152–167. doi: 10.1093/oxfordjournals.molbev.a040704. [DOI] [PubMed] [Google Scholar]
- 13.Karlin S, Burge C. Proc Natl Acad Sci USA. 1996;93:1560–1565. doi: 10.1073/pnas.93.4.1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Karlin S. Curr Opin Microbiol. 1998;1:598–610. doi: 10.1016/s1369-5274(98)80095-7. [DOI] [PubMed] [Google Scholar]
- 15.Cole S T, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, Gordon S V, Eiglmeier K, Gas S, Barry C E, 3rd, et al. Nature (London) 1998;393:537–544. doi: 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- 16.Zhu Z Y, Karlin S. Proc Natl Acad Sci USA. 1996;93:8350–8355. doi: 10.1073/pnas.93.16.8350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Banta L M, Bohne J, Lovejoy S D, Dostal K. J Bacteriol. 1998;180:6597–6606. doi: 10.1128/jb.180.24.6597-6606.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]