

Abstract
In the present study, phospholipase A2 (PLA2)-catalyzed hydrolysis of platelet membrane phospholipids was investigated by measuring PLA2 activity, phospholipid hydrolysis, arachidonic acid release and choline lysophospholipid production in thrombin-stimulated human platelets. Thrombin-stimulated platelets demonstrated selective hydrolysis of arachidonylated plasmenylcholine and plasmenylethanolamine, with little change in diacyl phospholipids. Accelerated plasmalogen hydrolysis was accompanied by increased arachidonic acid and thromboxane B2 release and increased lysoplasmenylcholine production. Thrombin stimulation caused an increase in PLA2 activity measured in the cytosolic fraction with plasmenylcholine only; no increase in activity was measured with phosphatidylcholine. No change in membrane-associated PLA2 activity was observed with either substrate tested. Pretreatment with the Ca2+-independent PLA2-selective inhibitor, bromoenol lactone, inhibited completely any thrombin-stimulated phospholipid hydrolysis. Thus, thrombin stimulation of human platelets activates a cytosolic PLA2 that selectively hydrolyzes arachidonylated plasmalogen phospholipids.
Introduction
Thrombin stimulation of human platelets results in the release of arachidonic acid from membrane phospholipids that is further metabolized by cyclooxygenase and lipoxygenase enzymes resulting in the generation of biologically active eicosanoids, including thromboxane A2 (TxA2). The cleavage of arachidonic acid from the sn-2 position of membrane phospholipids is mediated by the action of phospholipase A2 (PLA2). Multiple classes of PLA2 may be involved in arachidonic acid and TXA2 production in human platelets. Platelets contain a 14-kDa secretory PLA2 (sPLA2) that requires millimolar Ca2+ concentrations for activity and has a broad specificity towards phospholipids with different fatty acyl chains and polar headgroups [1]. However, inhibition of this sPLA2 activity was not found to block arachidonic acid production in thrombin-stimulated platelets [1]. Additionally, a cytosolic PLA2 (cPLA2) that requires micromolar concentrations of intracellular Ca2+ for translocation to membrane phospholipids has been demonstrated in human platelets [2]. In response to thrombin stimulation, platelet cPLA2 is phosphorylated and activated by p38 MAP kinase [3]. To date, there is little evidence for the involvement of a Ca2+-independent PLA2 (iPLA2) in human platelets. However, Lehr and Griessbach [4] have recently reported a decrease in arachidonic acid production in 12-O-tetradecanoylphorbol-13-acetate stimulated platelets pretreated with the iPLA2 inhibitor bromoenol lactone (BEL). In addition, both TxA2 and arachidonic acid release was observed in collagen stimulated platelets isolated from cPLA2-/-/sPLA2-IIA-/- mice, suggesting the involvement of another PLA2 isoform possibly iPLA2 [5].
The most extensively studied iPLA2 isoform to date is an 85 kDa protein that was first identified and characterized in P388D1 macrophages and has been designated iPLA2β [6]. More recently, a novel membrane-associated iPLA2 (iPLA2γ) has been identified. Homology between iPLA2β and iPLA2γ is confined to the ATP binding motif, the serine lipase site (GXSTG) and a region of nine amino acids for which, as yet, there is no known functional significance [7]. In several studies, both iPLA2β and iPLA2γ have demonstrated selectivity for plasmalogen phospholipid substrates [7-9]. A significant amount of arachidonic acid is found in plasmenylethanolamine in human platelets and generation of lysoplasmenylethanolamine has been observed in human platelets stimulated with collagen or U46619 (a TxA2 mimetic) [10,11]. Since no plasmalogen selectivity has been demonstrated for cPLA2, this study was undertaken to determine whether the iPLA2 isoforms contribute to plasmalogen phospholipid metabolism following thrombin stimulation in human platelets.
Methods
Preparation of Platelets
Blood from healthy volunteers was drawn into 0.109M trisodium citrate anticoagulant. Platelet-rich plasma was prepared by centrifuging the whole blood at room temperature for 15 mins at 100 × g. The plasma was gently removed from the precipitated cells and centrifuged at 2000 × g for 20 mins. Platelets were washed and suspended in 140 mM NaCl, 27 mM KCl, 1 mM MgCl2, 5.5 mM glucose, 10 mM HEPES, 1.2 mM CaCl2 immediately prior to stimulation. Platelets were incubated with inhibitors at 37°C where indicated and then incubated with thrombin. For measurement of cellular phospholipids, lysophospholipid and arachidonic acid production, reactions were terminated by the addition of ice-cold methanol and chloroform. For measurement of PLA2 activity and immunoblots, reactions were terminated by the addition of 5 mM EGTA, 1 mM dithiothreitol and 10% glycerol, followed by sonication.
Extraction, separation and analysis of phospholipids (① in Figure 1)
FIGURE 1.

Membrane plasmalogen phospholipids (①) are hydrolyzed at the sn-2 position by iPLA2 (⑤), resulting in the stoichiometric production of a lysoplasmalogen (②) and a free fatty acid (in this case, arachidonic acid, AA, ③). In platelets, AA can be further metabolized to thromboxane A2 (④).
Cellular phospholipids were extracted from washed platelets by the method of Bligh and Dyer at 0-4°C. The chloroform layer was dried under N2 and the lipid residue resuspended in chloroform: methanol 1:1 v/v. . Phospholipids were separated into different classes on an Ultrasphere-Si (5 μm silica), 4.6 × 250 mm high pressure liquid chromatography (HPLC) column (Beckmann Instruments, Fullerton, CA) using gradient elution with a mobile phase comprised of hexane/isopropanol/water [12].
Individual choline and ethanolamine glycerophospholipid molecular species were isolated by reverse phase HPLC on an Ultrasphere ODS (5 μm, C-18) column, 4.6 × 250 mm (Beckmann Instruments, Fullerton, CA) using a gradient elution system with a mobile phase comprised of acetonitrile/methanol/ water with 20 mM choline chloride [13]. Quantification of individual phospholipid molecular species was achieved by determination of lipid phosphorus in reverse phase HPLC column effluents by the method of Itaya and Ui [14].
Measurement of choline lysophospholipids (② in Figure 1)
Choline lysophospholipid measurement was made using a modification of a radiometric assay method described previously [15]. The procedure involves the extraction of lipids from the platelets by the method of Bligh & Dyer [16], followed by the separation of the lysophospholipids from other phospholipids by HPLC. The purified lysophosphatidylcholine (LPC) and lysoplasmenylcholine (LPlasC) fractions as well as known amounts of LPC and LPlasC standards were then acetylated with [3H]-acetic anhydride using 0.33 M dimethylaminopyridine as a catalyst. The acetylated lysophospholipid was then separated by thin layer chromatography and radioactivity quantified by liquid scintillation spectrometry. Standard curves were constructed and LPC and LPlasC levels were derived for all samples and normalized according to protein content as described previously [15].
Measurement of arachidonic acid release (③ in Figure 1)
Platelet-rich plasma was incubated at 37°C with 5 μCi [3H] arachidonic acid for 18 hours. This incubation resulted in greater than 80% incorporation of radioactivity into the platelets. Of the total incorporated radioactivity, 31±2% was incorporated into ethanolamine phospholipids and 52±3% was incorporated into choline phospholipids. Following incubation, unincorporated [3H] arachidonic acid was removed by washing the platelets twice with Tyrode's solution containing 3.6% bovine serum albumin. At the end of the stimulation period, lipids were extracted using the method of Bligh and Dyer [16], and separated by thin layer chromatography on channeled Silica Gel G plates. The area corresponding to [3H] arachidonic acid was scraped and the radioactivity determined by liquid scintillation counting. The amount of [3H] arachidonic acid was expressed as a percentage of the total radioactivity incorporated into the platelet phospholipids.
Measurement of thromboxane B2 production (④ in Figure 1)
Thromboxane B2 production was measured by enzyme-linked immunosorbent assay using an immunoassay kit (R&D Systems Inc., Minneapolis, MN).
Measurement of PLA2 activity (⑤ in Figure 1)
Following stimulation, platelets were sonicated on ice three times for 10 seconds and the sonicate centrifuged at 14,000 × g for 10 minutes. The supernatant was then centrifuged at 100,000 × g for 60 minutes to separate the membrane fraction (pellet) from the cytosolic fraction (supernatant). Phospholipase A2 activity in subcellular fractions was assessed by incubating enzyme with 100 μM (16:0,[3H]18:1) plasmenylcholine or phosphatidylcholine in assay buffer containing 10 mM Tris, 10% glycerol, pH = 7.0 with either 4 mM EGTA or 10 mM Ca2+ at 37°C for 5 minutes in a total volume of 200 μl. Synthesis of radiolabeled phospholipid substrates has been described previously [8]. Reactions were terminated by the addition of butanol and released radiolabeled fatty acid was isolated by thin layer chromatography on channeled Silica Gel G plates using petroleum ether/diethyl ether/acetic acid (70/30/1, v/v) and subsequent quantification by liquid scintillation spectrometry. Protein content of each sample was determined by the Lowry method utilizing freeze dried bovine serum albumin (Bio-Rad Laboratories) as the protein standard as described previously [17].
Immunoblot Analysis of PLA2
Following initial sonication of the platelet suspension, 1 mM sodium orthovanadate, 2 mM phenylmethylsulfonyl fluoride, 20 μg/ml leupeptin, 10 μg/ml aprotinin, and 5 μg/ml pepstatin A were added. Cells were sonicated on ice and centrifuged at 14,000 × g @ 4°C for 10 min to remove cellular debris and nuclei. Cytosolic and membrane fractions were separated by centrifuging the supernatant at 100,000 × g for 60 min. The pellet was resuspended in lysis buffer containing (in mmol/liter) HEPES 20 (pH 7.6), sucrose 250, dithiothreitol 2, EDTA 2, EGTA 2, β-glycerophosphate 10, sodium orthovanadate 1, phenylmethylsulfonyl fluoride 2, leupeptin 20 μg/ml, aprotinin 10 μg/ml and pepstatin A 5 μg/ml and washed twice to minimize contamination of the membrane fraction with cytosolic protein. The final pellet was resuspended in lysis buffer containing 1% Triton X-100. Protein (total protein, cytosol or membrane) was mixed with an equal volume of SDS sample buffer and heated at 95°C for 5 mins prior to loading onto a 10% polyacrylamide gel (Bio-Rad, Richmond, CA). Protein was separated by SDS/PAGE transferred to nitrocellulose membranes (Bio-Rad) at 100 V for 1 hour. Non-specific sites were blocked with Tris Buffered Saline containing 0.05% (v/v) Tween-20 (TBST) and 5% (w/v) nonfat milk and the blocked membrane incubated with antibodies to Ca2+-independent PLA2 beta (Cayman Chemical) or gamma (Aves, custom antibodies), both 1:1000 dilution. After washing, membranes were incubated with horseradish peroxidase-conjugated secondary antibodies (1:10000 dilution) and regions of antibody binding detected using enhanced chemiluminescence (Amersham, Arlington Heights, IL) and exposure to film (Hyperfilm, Amersham)
Results
The major phospholipid classes in human platelets were found to be choline and ethanolamine glycerophospholipids and sphingomyelin (Table 1). Plasmalogen phospholipids account for the majority of ethanolamine glycerophospholipids (55.2 ± 1.5%, Table 1), but are less abundant in choline glycerophospholipids (10.3 ± 1.0%, Table 1). Plasmalogen phospholipids were not detected in phosphatidylserine or phosphatidylinositol. Alkylacyl glycerophospholipids accounted for approximately 1% of ethanolamine and choline glycerophospholipids (Table 1). Separation and identification of individual molecular species demonstrated that 41.2 ± 3.2% of identifiable choline and 66.4 ± 2.6% of ethanolamine glycerophospholipids were arachidonylated at the sn-2 position (Table 2). Comparison of arachidonylated species to total species in each subclass demonstrated that 58% of plasmalogens and 38% of diacyl species in choline glycerophospholipids contained arachidonic acid at the sn-2 position, demonstrating a selective enrichment of arachidonate in plasmalogen phospholipids (Table 2). This enrichment is not as dramatic in ethanolamine glycerophospholipids, where 60% of diacyl and 70% of plasmalogens are arachidonylated (Table 2).
TABLE 1.
Phospholipid classes in human platelets
| Phospholipid Class | nmol phospholipid/ mg proteina | Total Phospholipidb | Plasmalogenc Content | Alkylacyld Content |
|---|---|---|---|---|
| CGP | 36.5 ± 1.9 | 33.8 ± 1.5% | 10.3 ± 1.0% | 0.3 ± 0.1% |
| EGP | 26.1 ± 1.5 | 24.2 ± 1.4% | 55.2 ± 1.5% | 1.1 ± 0.1% |
| PI | 6.6 ± 0.5 | 6.1 ± 0.6% | ND | |
| PS | 13.2 ± 1.2 | 12.2 ± 1.2% | ND | |
| Sphing | 25.6 ± 2.2 | 23.7 + 1.8% | ND | |
Values represent the mean ± SEM of separate measurements obtained using human platelets from six different donations. Abbreviations used: CGP, choline glycerophospholipids; EGP, ethanolamine glycerophospholipids; PI, phosphatidyl inositol; PS, phosphatidyl serine; ND, not detected.
Phospholipid classes were isolated by gradient elution HPLC and quantified by microphosphate assay.
Values are expressed as a percentage of the total phospholipid phosphorus content (108 ± 3 nmol/mg protein).
Plasmalogen content was determined by GLC analysis of the dimethylacetal derivatives expressed as a percentage of the total phospholipid class
Alkylacyl glycerophospholipid content was determined by quantitation of lipid phosphorus in the lysophospholipid fraction remaining after sequential, exhaustive acid- and base-catalyzed hydrolysis and expressed as a percentage of the total phosphorus content of the corresponding phospholipid class.
TABLE 2.
Composition of choline and ethanolamine glycerophospholipid molecular species in unstimulated and thrombin stimulated (0.05 IU/ml, 5 mins) human platelets. Values are expressed in nmol/mg protein and represent mean ± SEM for platelets obtained from 8 different volunteers.
| Composition | Control | Thrombin |
|---|---|---|
| Choline glycerophospholipid molecular species | ||
| (16:0, 20:4) PlsCho | 3.8 ± 0.5 | 1.2 ± 0.3 |
| (16:0, 18:2) PlsCho | 1.2 ± 0.2 | 0.2 ± 0.1 |
| (16:0, 22:6) PtdCho | 1.8 ± 0.2 | 1.6 ± 0.2 |
| (16:0, 20:4) PtdCho | 6.4 ± 0.9 | 5.9 ± 0.4 |
| (18:0, 20:4) PtdCho | 6.5 ± 0.7 | 6.7 ± 0.3 |
| (18:1, 20:4) PtdCho | 3.3 ± 0.3 | 3.2 ± 0.1 |
| (18:2, 20:4) PtdCho | 0.5 ± 0.3 | 0.6 ± 0.2 |
| (16:0, 18:3) PtdCho | 0.8 ± 0.1 | 0.9 ± 0.2 |
| (16:0, 18:2) PtdCho | 9.9 ± 0.7 | 9.7 ± 0.6 |
| (18:0, 18:2) PtdCho | 5.7 ± 0.5 | 5.6 ± 0.4 |
| (18:2, 18:2) PtdCho | 1.6 ± 0.2 | 1.6 ± 0.1 |
| (16:0, 18:1) PtdCho | 7.1 ± 0.3 | 6.8 ± 0.3 |
| Ethanolamine glycerophospholipid molecular species | ||
| (16:0, 20:4) PlsEtn | 3.4 ± 0.4 | 2.0 ± 0.2 |
| (18:0, 20:4) PlsEtn | 5.2 ± 0.4 | 3.4 ± 0.2 |
| (18:1, 20:4) PlsEtn | 1.1 ± 0.2 | 0.5 ± 0.2 |
| (18:0, 18:2) PlsEtn | 1.1 ± 0.1 | 1.4 ± 0.3 |
| (18:1, 18:2) PlsEtn | 2.2 ± 0.2 | 2.2 ± 0.3 |
| (18:0, 18:1) PlsEtn | 0.9 ± 0.2 | 0.5 ± 0.2 |
| (16:0, 20:4) PtdEtn | 1.0 ± 0.1 | 0.5 ± 0.1 |
| (18:0, 20:4) PtdEtn | 5.7 ± 0.5 | 6.0 ± 0.5 |
| (18:1, 20:4) PtdEtn | 0.6 ± 0.2 | 0.9 ± 0.2 |
| (18:2, 20:4) PtdEtn | 0.2 ± 0.1 | 0.2 ± 0.1 |
| (16:0, 18:3) PtdEtn | 0.1 ± 0.1 | 0.1 ± 0.1 |
| (18:1, 18:3) PtdEtn | 0.1 ± 0.1 | 0.1 ± 0.1 |
| (16:0, 18:2) PtdEtn | 1.4 ± 0.2 | 1.1 ± 0.2 |
| (18:0, 18:2) PtdEtn | 1.4 ± 0.2 | 1.0 ± 0.2 |
| (18:2, 18:2) PtdEtn | 0.1 ± 0.1 | 0.1 ± 0.1 |
| (16:0, 18:1) PtdEtn | 1.1 ± 0.2 | 1.3 ± 0.2 |
| (18:0, 18:1) PtdEtn | 0.3 ± 0.1 | 0.3 ± 0.1 |
Individual phospholipid molecular species were isolated by reverse phase HPLC and identified as described under Materials and Methods. The composition of individual molecular species is described by the shorthand notation (a:b, c:d) where a and c represent chain length and b and d represent the number of carbon-carbon double bonds for the aliphatic groups at the sn-1 and sn-2 positions. PlsCho = plasmenylcholine; Ptd = phosphatidylcholine; PlsEtn = plasmenyethanolamine; PtdEtn = phosphatidylethanolamine.
A significant decrease in plasmenylcholine (PlsCho) was observed when human platelets were stimulated with thrombin with no corresponding decrease in phosphatidylcholine (PtdCho) (Table 2). Thrombin stimulation also resulted in a significant decrease in arachidonylated plasmenylethanolamine (PlsEtn) species with a smaller but significant decrease in (16:0, 20:4) phosphatidylethanolamine (PtdEtn) (Table 2). Thus, the production of free arachidonic acid in thrombin-stimulated human platelets is primarily released from the sn-2 position of plasmalogen phospholipids.
Cytosolic and membrane-associated PLA2 activity in unstimulated human platelets was measured using (16:0, [3H]18:1) plasmenylcholine and phosphatidylcholine substrates in the absence and presence of Ca2+. Although PLA2 activity was measurable in the absence of Ca+, it was increased by the addition of 1 mM Ca2+ to the assay buffer in both cytosolic and membrane fraction (Table 3). To determine the role of plasmalogen-selective iPLA2 isoforms in thrombin-stimulated human platelets, we measured PLA2 activity in the absence of Ca2+ in subsequent studies. Thrombin stimulation of human platelets resulted in a significant increase in cytosolic PLA2 activity when measured using (16:0, [3H]18:1) plasmenylcholine substrate; the ED50 of the concentration-response curve was 0.02 ± 0.01 IU/ml thrombin. The increase in cytosolic PLA2 activity occurred within 1 min thrombin stimulation and remained elevated over 15 mins (Figure 1, filled circles). No increase in cytosolic PLA2 activity was measured using (16:0, [3H]18:1) phosphatidylcholine (Figure 1, open squares) demonstrating iPLA2 selectivity for plasmalogen substrates. No change in iPLA2 activity was measured in the membrane fraction of thrombin-stimulated platelets with either substrate (data not shown). Although PLA2 activity could be detected in the absence of Ca2+ in our assay system, platelets suspended in a Ca2+-free buffer and stimulated with thrombin demonstrated no increase in PLA2 activity or arachidonic acid release (data not shown) indicating that thrombin-stimulated PLA2 activity in the intact platelet requires the presence of extracellular Ca2+.
TABLE 3.
Phospholipase A2 activity (nmol-mg protein-1.min-1) in membrane and cytosolic subcellular fractions from human platelets measured using plasmenylcholine or phosphatidylcholine substrates in the absence (4 mM EGTA) or presence (1 mM Ca2+) of calcium. Values represent mean ± SEM for separate measurements from 4 different cell isolations.
| Cell Fraction | Substratea | EGTA | Ca2+ |
|---|---|---|---|
| Membrane | Plasmenylcholine | ||
| 16:0, [3H]18:1 | 1.13±0.16 | 1.77±0.15 | |
| 16:0, [3H]20:4 | 3.21±0.19 | 5.23±0.26 | |
| Phosphatidylcholine | |||
| 16:0, [3H]18:1 | 1.09±0.19 | 2.03±0.33 | |
| 16:0, [3H]20:4 | 3.11±0.26 | 4.03±0.26 | |
| Cytosol | Plasmenylcholine | ||
| 16:0, [3H]18:1 | 0.33±0.09 | 0.39±0.05 | |
| 16:0, [3H]20:4 | 0.74±0.12 | 0.88±0.13 | |
| Phosphatidylcholine | |||
| 16:0, [3H]18:1 | 0.31±0.03 | 0.43±0.05 | |
| 16:0, [3H]20:4 | 0.77±0.08 | 0.92±0.15 |
substrate composition is represented as a:b, c:d where a:b and c:d represent the chain length: number of double bonds for the aliphatic groups at the sn-1 and sn-2 positions, respectively, of the corresponding phospholipid substrate molecule.
To determine whether the accelerated hydrolysis of membrane plasmalogen phospholipids and increased iPLA2 activity in thrombin-stimulated platelets was accompanied by an accumulation of membrane phospholipid metabolites, measurements of free arachidonic acid, TxB2 (the stable metabolite of TxA2) and choline lysophospholipid (lysoplasmenylcholine, LPlsCho and lysophosphatidylcholine, LPC) production were performed. Human platelets stimulated with thrombin (0.05 IU/ml) demonstrated a significant increase in free arachidonic acid (Figure 2A, filled circles) and thromboxane B2 (Figure 2B, filled circles) release that remained elevated over controls for at least 10 min. A time-dependent increase in LPlsCho (Figure 3A, filled circles) was observed with no increase in LPC (Figure 3A, open squares). After 2 min thrombin stimulation, arachidonylated plasmenylcholine content in human platelets decreased by 2.6 nmol/mg protein (3.8±0.5 to 1.2 ± 0.3 nmol/mg protein, Table 2) whereas LPlsCho content increased by 1.5 nmol/mg protein (Figure 3) at the same time interval, suggesting that a significant proportion of LPlsCho catabolized following production. There is no evidence of reacylation of lysoplasmenylcholine in thrombin-stimulated platelets, since total plasmenylcholine content decreases after thrombin stimulation (Table 2). Although we did not measure lysoplasmenylethanolamine production in these experiments, we did measure a decrease in arachidonylated plasmenylethanolamine (9.7 ± 0.3 nmol/mg protein in unstimulated cells to 5.9 ± 0.6 nmol/mg protein, Table 2) after thrombin stimulation. This decrease was not accompanied by a corresponding increase in any other ethanolamine phospholipid molecular species, suggesting that reacylation does not occur in ethanolamine glycerophospholipids.
FIGURE 2.

Thrombin stimulation (0.05 IU/ml) increases cytosolic PLA2 activity in human platelets when measured using (16:0, [3H]18:1) plasmenylcholine substrate (filled circles, solid lines) but not when measured using phosphatidylcholine substrate (open squares, solid lines). Pretreatment of platelets with bromoenol lactone (1 μM, 10 mins) decreased cytosolic PLA2 activity under both basal and thrombin stimulated platelets measured using both plasmenylcholine (filled circles, broken lines) and phosphatidylcholine substrates (open squares, broken lines). Values represent means ± SEM of independent results derived from 8 separate platelet isolates.**p<0.01 when compared with untreated platelets.
FIGURE 3.

Thrombin stimulation (0.05 IU/ml, filled circles) of human platelets results in a time dependent increase in free arachidonic acid (Panel A) and thromboxane B2 (TxB2, Panel B) release. The increase in arachidonic acid and xB2 in thrombin-stimulated platelets is inhibited by pretreatment with bromoenol lactone (open triangles, 1 μM, 10 mins). Values represent means ± SEM of independent results derived from 6 separate platelet isolates.**p<0.01 when compared with controls, ++p<0.01 when compared with both untreated control and thrombin-stimulated values.
To determine whether the accelerated membrane phospholipid hydrolysis was mediated via iPLA2 activity, platelets were incubated with1 μM BEL (a selective inhibitor of iPLA2) [18] for 30 min prior to thrombin stimulation. Pretreatment with BEL significantly decreased basal PLA2 activity measured in the cytosol using both plasmenylcholine and phosphatidylcholine substrates (Figure 1, dotted lines). The thrombin-induced increase in PLA2 activity measured using plasmenylcholine was also inhibited completely by BEL pretreatment (Figure 1, dotted lines). Inhibition of cytosolic PLA2 activity with BEL inhibits completely thrombin-stimulated LPlsCho production (Figure 3B, open bars), arachidonic acid release (Figure 2A, open triangles) and TxB2 production (Figure 2B, open triangles). Additionally, pretreatment with BEL blocked thrombin-induced plasmenylcholine and plasmenylethanolamine hydrolysis (data not shown).
To further support the role of iPLA2 as an important mediator of membrane phospholipid hydrolysis we performed immunoblot analysis to determine the subcellular localization of iPLA2β and iPLA2γ in human platelets. We detected immunoreactive bands using an antibody to iPLA2β at 50kD and iPLA2γ at 55 and 60 kD (Figure 4). The majority of immunoreactive protein for both isoforms was detected in the cytosol (Figure 4). A recent manuscript has described the selective inhibition of iPLA2β and iPLA2γ using the S- and R-enantiomers of BEL respectively [19]. We incubated the cytosolic fraction from human platelets with increasing concentrations of (R)-BEL, (S)-BEL or racemic BEL for 10 minutes prior to assay of iPLA2 activity (Figure 5). Significant inhibition of iPLA2 activity was observed with concentrations of (R)-BEL greater than 1 μM, but no significant inhibition of activity was observed with (S)-BEL at concentrations as high as 10 μM (Figure 5). These data suggest that iPLA2γ is the isoform that contributes to the majority of calcium-independent PLA2 activity in human platelets.
FIGURE 4.

Thrombin stimulation (0.05 IU/ml) of human platelets results in a time-dependent increase in lysoplasmenylcholine (LPlsCho, filled circles in Panel A and filled bars in Panel B) with no corresponding increase in lysophosphatidylcholine (open squares in Panel A and filled bars in Panel B). The increase in LPlsCho in thrombin-stimulated platelets is inhibited completely by pretreatment with bromoenol lactone (1 μM, 10 mins, open bars in Panel B). Values represent means ± SEM of independent results derived from 6 separate platelet isolates.**p<0.01 when compared with controls.
FIGURE 5. Immunoblot with PLA2 isoforms.
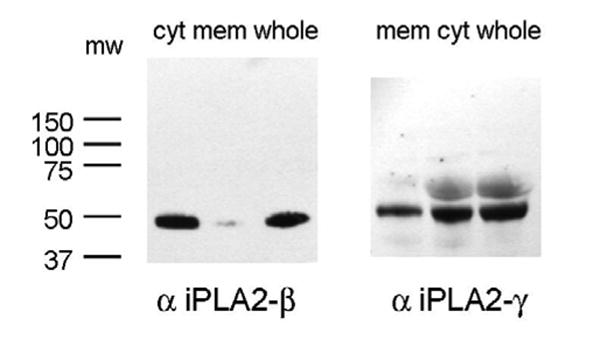
Immunoblot analysis of human iPLA2-beta (left) and -gamma (right) in cytosolic and membrane subcellular fractions and whole platelets. Proteins were separated by SDS-PAGE and transferred to nitrocellulose. Membranes were probed with anti-iPLA2-beta or-gamma antibodies (1:1000 dilution) and incubated with horseradish peroxidase-linked secondary antibodies (1:10,000 dilution). Immunoblots were detected with enhanced chemiluminescence and exposure to film for 5 minutes.
Thus, thrombin stimulation of human platelets results in a significant increase in iPLA2γ-catalyzed plasmalogen phospholipid hydrolysis, resulting in increased release of free arachidonic acid and TxA2 accompanied by an increase in LPlsCho production.
Discussion
In this study, we have demonstrated measurable PLA2 activity in the absence of Ca2+ in the cytosol of human platelets that is largely due to the presence of iPLA2γ. This isoform has recently been identified and characterized [7] and has been found to be uniquely sensitive to (R)-BEL [19]. Human iPLA2-γ has multiple alternative start sites for translation resulting in functional PLA2γ proteins with MW of 63-, 74-, 77- and 88-kDa [7]. Initially, the subcellular localization of iPLA2γ was assumed to be peroxisomal due to the presence of a C-terminal-SKL sequence [7] and recent studies have confirmed that the peroxisomal iPLA2γ is the 63-kDa protein [20,21]. In contrast, other recent studies have demonstrated higher molecular weight forms of iPLA2γ are present in rat heart mitochondria [22] and we have demonstrated that the 88-kDa iPLA2γ isoform is present in rabbit heart and kidney ER and mitochondria [23].
Thrombin stimulation of human platelets has been demonstrated to result in activation of cPLA2 leading to an increase in arachidonic acid release. Two synergistic pathways for cPLA2 activation have been implicated; increased cytosolic Ca2+ results in translocation of cPLA2 to the membrane, and p38 MAP kinase phosphorylates the serine residues on the enzyme [2,3,24]. Activation of both pathways is required for arachidonic acid liberation from membrane phospholipids in response to thrombin stimulation. Platelets are also a source of the group II sPLA2 [25], however, inhibition of this PLA2 isoform did not block arachidonic acid release in thrombin-stimulated platelets [1]. To date, Ca2+-independent PLA2 (iPLA2) has been thought to be a redundant enzyme in human platelets [5]. Using an antibody to iPLA2β, we detected an immunoreactive band at a lower molecular weight than would be expected for this isoform, suggesting that this protein is truncated in platelets. In addition, incubation of platelet cytosolic protein with (S)-BEL did not significantly decrease iPLA2 activity. Taken together, these data suggest that platelet iPLA2β is an inactive protein. The characterization of recently discovered iPLA2 isoforms, such as iPLA2γ, now suggest that this class of PLA2s may play a greater role in platelet function than has been previously appreciated.
The majority of previous studies where direct measurements of platelet cPLA2 activity are made in response to thrombin stimulation use (16:0, [14C]20:4) phosphatidylcholine as substrate. In these studies, we were not able to detect thrombin-stimulated increases in PLA2 activity in platelet cytosol using (16:0, [3H]18:1) phosphatidylcholine as substrate. We measured significant increases in cytosolic PLA2 activity in response to thrombin stimulation when using (16:0, [3H]18:1) plasmenylcholine; this is the first study to demonstrate a preference for plasmalogen phospholipids in human platelets. A preference for plasmalogen substrates has been demonstrated previously in sheep platelets where a cytosolic PLA2 showed substantial preference for ether-linked phospholipid substrates [26]. Membrane phospholipids are categorized into classes, based on the polar headgroup, subclasses, based on the nature of the covalent linkage to the sn-1 position, and molecular species, based on individual aliphatic constituents attached to the sn-1 and sn-2 positions (Figure 7). Plasmalogen phospholipids contain a vinyl ether linkage at the sn-1 position of the glycerol backbone (Figure 7). The significance of a plasmalogen selective iPLA2 in platelets is not completely understood at this time. However, preferential plasmalogen hydrolysis appears to be directly involved in thromboxane A2 generation in human platelets.
FIGURE 7.

Diagrammatic representation of the structure of membrane phospholipids. Membrane phospholipids are categorized into classes, based on the polar headgroup (1); subclasses, based on the nature of the covalent linkage to the sn-1 position (2); and molecular species, based on individual aliphatic constituents attached to the sn-1 and sn-2 positions (3).
Stimulation of human platelet with thrombin results in significant lysoplasmenyethanolamine (LPlsEtn) production (11), providing evidence that a plasmalogen-selective PLA2 may be activated. To date, the only PLA2 isoforms that have demonstrated plasmalogen selectivity are Ca2+-independent [27]. Although we did not measure LPlsEtn in this study, we did measure a significant increase in LPlsCho production in thrombin stimulated platelets (Figure 3). This was accompanied by a significant decrease in arachidonylated plasmenylcholine (Table 2). Thrombin stimulation resulted in a significant decrease in arachidonylated PlsEtn (Table 2) suggesting that LPlsEtn accumulation would occur. Selective hydrolysis of plasmalogen phospholipids has been demonstrated in human platelets stimulated with collagen and U46619 (a TxA2 mimetic) [10,11]. Following thrombin stimulation, we observed a significant decrease in arachidonylated PlsCho with no change in arachidonylated PtdCho. Similarly, 95% of the decrease in arachidonylated ethanolamine glycerophospholipids occurred from PlsEtn. From these data, we can conclude that the majority of arachidonic acid released from thrombin stimulated human platelets is released from plasmalogens.
Although PLA2 activity has been observed previously in platelets and there is evidence that PLA2-catalyzed plasmalogen hydrolysis occurs in response to thrombin [10,11], it was surprising to observe that platelet PLA2 activity and arachidonic acid release was inhibited completely by BEL pretreatment since BEL does not significantly inhibit cPLA2 [18]. Previous studies that have studied cPLA2 activation in thrombin stimulated platelets have commonly used concentrations of thrombin at 1 to 2 IU/ml [3,24,28]. We constructed a concentration-response curve to thrombin and observed significant increases in PLA2 activity at concentrations of thrombin greater than 0.005 IU/ml (data not shown). The ED50 was found to be 0.02 IU/ml and maximal PLA2 activity was observed at 0.5 IU/ml (data not shown). Interestingly, iPLA2 activity decreased when concentrations of thrombin greater than 0.5 IU/ml were used. Human platelets express both protease-activated receptor (PAR)-1 and PAR-4 on their surface [29]. PAR-1 mediates activation of human platelets at low thrombin concentrations, whereas PAR-4 can only mediate platelet activation at higher thrombin concentrations [29]. The presence of the two receptors may allow thrombin to activate distinct signaling pathways, suggested by the fact that there are differences in G protein coupling between the two receptors [30,31]. We suggest that PAR-1 may be coupled to iPLA2 whereas PAR-4 may be coupled to cPLA2 in human platelets and that the use of a lower concentration of thrombin in this study may cleave PAR-1 selectively. This may explain why we failed to observe cPLA2-catalyzed membrane phospholipid hydrolysis as evidenced by the inhibition of phospholipid metabolite production using BEL.
Although our data suggest that iPLA2γ is a major contributor to plasmalogen phospholipid hydrolysis in thrombin-stimulated platelets, we also determined that the presence of extracelluar Ca2+ was requited for thrombin-stimulated iPLA2 activation. This suggests that a Ca2+-dependent step may exist in the signaling process between PAR and iPLA2 activation. In previous studies, we have demonstrated that iPLA2 in ventricular myocytes and endothelial cells is dependent upon protein kinase C (PKC) activity (32, 33). If a Ca2+-dependent PKC phosphorylates iPLA2γ in platelets, this may explain our observation of the lack of iPLA2 activation by thrombin-stimulated platelets suspended in a Ca2+-free buffer.
In conclusion, low concentrations of thrombin activate a cytosolic iPLA2 that hydrolyzes plasmalogen phospholipids in human platelets resulting in production of arachidonic acid, TxA2 and LPlsCho. This is the first study to describe a signaling role for iPLA2γ in platelets and represents a novel therapeutic avenue for manipulation of platelet function.
FIGURE 6.

Inhibition of cytosolic PLA2 activity with (R)-BEL (selectively inhibits iPLA2γ), (S)-BEL (selectively inhibits iPLA2β) and racemic BEL for 10 minutes prior to assay of PLA2 activity using (16:0, [3H]18:1) plasmenylcholine substrate in the presence of 4 mM EGTA. *p<0.05, **p<0.01 when compared to untreated PLA2 activity.
Acknowledgments
This research was supported in part by the American Heart Association (Heartland Affiliate to MHC & JM) and by National Institutes of Health Grant HL-68588 (JM).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bartoli F, Lin HK, Ghomashchi F, Gelb MH. Tight binding inhibitors of 85-kDa phospholipase A2 but not 14 kDa phospholipase A2 inhibit release of free arachidonate in thrombin-stimulated human platelets. J Biol Chem. 1994;269:15625–30. [PubMed] [Google Scholar]
- 2.Kramer RM, Roberts EF, Manetta JV, Hyslop PA, Jakubowski JA. Thrombin-induced phosphorylation and activation of Ca2+-sensitive cytosolic phospholipase A2 in human platelets. J Biol Chem. 1993;268:26796–804. [PubMed] [Google Scholar]
- 3.Borsch-Haubold AG, Bartoli F, Asselin J, Dudler T, Kramer RM, Apitz-Castro R, Watson SP, Gelb MH. Identification of the phosphorylation sites of cytosolic phospholipase A2 in agonist-stimulated human platelets and HeLa cells. J Biol Chem. 1998;273:4449–58. doi: 10.1074/jbc.273.8.4449. [DOI] [PubMed] [Google Scholar]
- 4.Lehr M, Griessbach K. Involvement of different protein kinases and phospholipases A2 in phorbol ester (TPA)-induced arachidonic acid liberation in bovine platelets. Mediators of Inflammation. 2000;9:31–4. doi: 10.1080/09629350050024357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wong DA, Kita Y, Uozumi N, Shimizu T. Discrete role for cytosolic phospholipase A2α in platelets: Studies using single and double mutant mice of cytosolic and group IIA secretory phospholipase A2. J Exp Med. 2002;196:349–57. doi: 10.1084/jem.20011443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ackermann EJ, Kempner ES, Dennis EA. Ca2+-independent cytosolic phospholipase A2 from macrophage-like P388D1 cells. Isolation and characterization. J Biol Chem. 1994;269:9227–33. [PubMed] [Google Scholar]
- 7.Mancuso DJ, Jenkins CM, Gross RW. The Genomic Organization, Complete mRNA Sequence, Cloning, and Expression of a Novel Human Intracellular Membrane-associated Calcium-independent Phospholipase A2. J Biol Chem. 2000;275:9937–45. doi: 10.1074/jbc.275.14.9937. [DOI] [PubMed] [Google Scholar]
- 8.McHowat J, Creer MH. Calcium-independent phospholipase A2 in isolated rabbit ventricular myocytes. Lipids. 1998;33:1203–12. doi: 10.1007/s11745-998-0324-5. [DOI] [PubMed] [Google Scholar]
- 9.Rickard A, Portell C, Kell PJ, Vinson SM, McHowat J. Protease activated receptor stimulation activates a calcium-independent phospholipase A2 in bladder microvascular endothelial cells. Am J Physiol. 2005;288:F714–21. doi: 10.1152/ajprenal.00288.2004. [DOI] [PubMed] [Google Scholar]
- 10.Turini ME, Holub BJ. Eicosanoid/thromboxane A2-independent and –dependent generation of lysoplasmenylethanolamine via phospholipase A2 in collagen-stimulated human platelets. Biochem J. 1993;289:641–6. doi: 10.1042/bj2890641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Turini ME, Holub BJ. The cleavage of plasmenylethanolamine by phospholipase A2 appears to be mediated by the low affinity binding site of the TxA2/PGH2 receptor in U46619-stimulated human platelets. Biochim Biophys Acta. 1994;1213:21–6. doi: 10.1016/0005-2760(94)90217-8. [DOI] [PubMed] [Google Scholar]
- 12.DaTorre SD, Creer MH. Differential turnover of polyunsaturated fatty acids in plasmalogen and diacyl glycerophospholipids of isolated cardiac myocytes. J Lipid Res. 1991;32:1159–72. [PubMed] [Google Scholar]
- 13.McHowat J, Jones JH, Creer MH. Gradient elution reversed-phase chromatographic isolation of individual glycerophospholipid molecular species. J Chromatog B. 1997;702:21–32. doi: 10.1016/s0378-4347(97)00386-1. [DOI] [PubMed] [Google Scholar]
- 14.Itaya K, Ui M. A new micromethod for the colorimetric determination of inorganic phosphate. Clin Chim Acta. 1966;14:361–6. doi: 10.1016/0009-8981(66)90114-8. [DOI] [PubMed] [Google Scholar]
- 15.McHowat J, Liu S, Creer MH. Selective hydrolysis of plasmalogen phospholipids by Ca2+-independent PLA2 in hypoxic ventricular myocytes. Am J Physiol. 1998;274:C1727–37. doi: 10.1152/ajpcell.1998.274.6.C1727. [DOI] [PubMed] [Google Scholar]
- 16.Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Can J Physiol. 1959;37:911–7. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- 17.Markwell MAK, Haas SM, Tolbert NE, Bieber LL. Protein determination in membrane and lipoprotein samples: manual and automated procedures. Methods Enzymol. 1981;72:296–303. doi: 10.1016/s0076-6879(81)72018-4. [DOI] [PubMed] [Google Scholar]
- 18.Zupan LA, Weiss RH, Hazen SL, Parnas BL, Aston KW, Lennon PJ, Getman DP, Gross RW. Structural determinants of haloenol lactone-mediated suicide inhibition of canine myocardial calcium-independent phospholipase A2. J Med Chem. 1993;36:95–100. doi: 10.1021/jm00053a012. [DOI] [PubMed] [Google Scholar]
- 19.Jenkins CM, Han X, Manscuso DJ, Gross RW. Identification of calcium-independent phospholipase A2 (iPLA2)β, and not iPLA2γ, as the mediator of arginine vasopressin-induced arachidonic acid release in A-10 smooth muscle cells. J Biol Chem. 2002;277:32807–32814. doi: 10.1074/jbc.M202568200. [DOI] [PubMed] [Google Scholar]
- 20.Yang J, Han X, Gross RW. Identification of hepatic peroxisomal phospholipase A2 and characterization of arachidonic acid-containing choline glycerophospholipids in hepatic peroxisomes. FEBS Lett. 2003;546:247–50. doi: 10.1016/s0014-5793(03)00581-7. [DOI] [PubMed] [Google Scholar]
- 21.Murakami M, Masuda S, Ueda-Semmyo K, Yoda E, Kuwata H, Takanezawa Y, Aoki J, Arai H, Sumimoto H, Ishikawa Y, Ishii T, Nakatani Y, Kudo I. Group VIB Ca2+-independent phospholipase A2γ promotes cellular membrane hydrolysis and prostaglandin production in a manner distinct from other intracellular phospholipases A2. J Biol Chem. 2005;280:14028–41. doi: 10.1074/jbc.M413766200. [DOI] [PubMed] [Google Scholar]
- 22.Mancuso DJ, Jenkins CM, Sims HF, Cohen JM, Yang J, Gross RW. Complex transcriptional and translational regulation of iPLAgamma resulting in multiple gene products containing dual competing sites for mitochondrial or peroxisomal localization. Eur J Biochem. 2004;271:4709–24. doi: 10.1111/j.1432-1033.2004.04435.x. [DOI] [PubMed] [Google Scholar]
- 23.Kinsey GR, Cummings BS, Beckett CS, Saavedra G, Zhang W, McHowat J, Schnellman RG. Identification and distribution of endoplasmic reticulum iPLA2. Biochem Biophys Res Comm. 2005;327:287–93. doi: 10.1016/j.bbrc.2004.12.016. [DOI] [PubMed] [Google Scholar]
- 24.McNicol A, Shibou TS. Translocation and phosphorylation of cytosolic phospholipase A2 in activated platelets. Thromb Res. 1998;92:19–26. doi: 10.1016/s0049-3848(98)00097-8. [DOI] [PubMed] [Google Scholar]
- 25.Marshall LA, Roshak A. Coexistence of two biochemically distinct phospholipase A2 activities in human platelet, monocyte, and neutrophils. Biochem Cell Biol. 1993;71:331–339. doi: 10.1139/o93-050. [DOI] [PubMed] [Google Scholar]
- 26.Loeb LA, Gross RW. Identification and purification of sheep platelet phospholipase A2 isoforms. Activation by physiologic concentrations of calcium ion. J Biol Chem. 1986;261:10467–70. [PubMed] [Google Scholar]
- 27.Meyer MC, Rastogi P, Beckett CS, McHowat J. Phospholipase A2 inhibitors as potential anti-inflammatory agents. Curr Pharm Des. 2005;11:1301–12. doi: 10.2174/1381612053507521. [DOI] [PubMed] [Google Scholar]
- 28.Tsegaye Y, Daasvatn KO, Holmsen H. Acyl specificity of phospholipases A2 and C in thrombin-stimulated human platelets. Platelets. 2002;13:31–5. doi: 10.1080/09537100120111531. [DOI] [PubMed] [Google Scholar]
- 29.Kahn ML, Nakanishi-Matsui M, Shapiro MJ, Ishihara H, Coughlin SR. Protease-activated receptors 1 and 4 mediate activation of human platelets by thrombin. J Clin Invest. 1999;103:879–87. doi: 10.1172/JCI6042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Coughlin SR. Protease-activated receptors in vascular biology. Thromb Haemost. 2001;86:298–307. [PubMed] [Google Scholar]
- 31.Macfarlane SR, Seatter MJ, Kanke T, Hunter GD, Plevin R. Proteinase-activated receptors. Pharmacol Rev. 2001;53:245–82. [PubMed] [Google Scholar]
- 32.Steer SA, Wirsig KC, Creer MH, Ford DA, McHowat J. Regulation of membrane-associated iPLA2 activity by a novel PKC in ventricular myocytes. Am J Physiol. 2002;283:C1621–6. doi: 10.1152/ajpcell.00109.2002. [DOI] [PubMed] [Google Scholar]
- 33.Meyer MC, Kell PJ, Creer MH, McHowat J. Calcium-independent Phospholipase A2 is regulated by a novel protein kinase C in human coronary artery endothelial cells. Am J Physiol. 2005;288:C475–82. doi: 10.1152/ajpcell.00306.2004. [DOI] [PubMed] [Google Scholar]