

Abstract
Proteolytic cleavage and subsequent activation of protein kinase C (PKC) δ is required for apoptosis induced by a variety of genotoxic agent, including UV radiation. In addition, overexpression of the constitutively active PKCδ catalytic fragment (PKCδ-cat) is sufficient to trigger Bax activation, cytochrome c release, and apoptosis. While PKCδ is a key apoptotic effector, the downstream target(s) responsible for the mitochondrial apoptotic cascade are not known. We found that expression of the active PKCδ-cat in HaCaT cells triggers a reduction in the anti-apoptotic protein Mcl-1, similar to UV radiation. The down-regulation of Mcl-1 induced by PKCδ-cat was not at the mRNA level but was due to decreased protein half-life. Overexpression of Mcl-1 protected HaCaT cells from both UV and PKCδ-cat-induced apoptosis and blocked the release of cytochrome c from the mitochondria, indicating that Mcl-1 down-regulation was required for apoptosis signaling. Indeed, down-regulation of Mcl-1 with siRNA slightly increased the basal apoptotic rate of HaCaT cells and dramatically sensitized them to UV or PKCδ-cat-induced apoptosis. HaCaT cells with down-regulated Mcl-1 had higher activated Bax protein, as measured by Bax cross-linking, indicating that Mcl-1 down-regulation is sufficient for Bax activation. Finally, recombinant PKCδ could phosphorylate Mcl-1 in vitro, identifying Mcl-1 as a direct target for PKCδ. Overall our results identify Mcl-1 as an important target for PKCδ-cat that can mediate its pro-apoptotic effects on mitochondria to amplify the apoptotic signaling induced by a wide range of apoptotic stimuli.
The induction of apoptosis in keratinocytes by UV radiation is an important protective mechanism from sunlight-induced skin tumors (1). UV induces keratinocyte apoptosis primarily via the intrinsic or mitochondrial death effector pathway (2, 3). The UV mitochondrial apoptotic signaling pathway requires the early loss of Mcl-1, which allows the release of cytochrome c and other pro-apoptotic factors from the mitochondria. Release of these pro-apoptotic mediators triggers the activation of caspase-9 and ultimately the activation of effector caspases such as caspase-3 (2, 4). While caspase-3 cleaves a wide range of protein substrates, the widely expressed death substrate PKCδ2 is critical as inhibition of PKCδ cleavage and/or activation protects from progressive caspase activation and apoptosis induced by a variety of stimuli, indicating positive feedback regulation between PKCδ cleavage and caspase activation (5–10). Furthermore, ectopic expression of the constitutively active PKCδ-cat is sufficient to induce apoptosis in several cell types (8, 11–13). PKCδ-cat has been localized to the mitochondria and the nucleus (6, 8) and induces apoptosis involving Bax activation, cytochrome c release, and caspase activation (8, 13). Although several PKCδ targets have been identified, including p73β (14), DNA-PK (12), Rad9 (15), phospholipid scramblases 1 and 3 (16–18), p38δ-ERK1/2 (19), and nuclear lamin B (20), it is unclear how these substrates can directly initiate the mitochondrial apoptotic cascade.
During UV-induced apoptosis, the loss of Mcl-1 is required for the mitochondrial translocation of Bax, release of cytochrome c, and caspase activation (4). The anti-apoptotic activity of Mcl-1 is due to its ability to bind and sequester pro-apoptotic Bcl-2 family proteins such as Bak and Bax (21–23). Upon Mcl-1 loss, Bak and/or Bax are free to oligomerize and mediate pore formation in the mitochondria, allowing the release of cytochrome c (24). In the cell types examined to date, Mcl-1 has a short half-life, and its levels are rapidly modulated during apoptosis at the transcriptional and translational level (25). In normal human keratinocytes, the UV-induced loss of Mcl-1 is at the mRNA level, and no increase in Mcl-1 protein turnover was noted in HeLa cells exposed to UVC (4, 26).
To identify PKCδ targets that can initiate the mitochondrial apoptotic pathway, we have focused on the Bcl-2 family since many of these proteins are regulated by phosphorylation. We previously found that PKCδ-cat triggers the up-regulation and activation of Bax (13). In this study, we find that expression of the active PKCδ-cat causes increased turnover and reduced levels of Mcl-1, and we have identified Mcl-1 as a direct target phosphorylated by PKCδ-cat. These findings can explain the ability of PKCδ-cat to trigger mitochondrial apoptosis and identify Mcl-1 as a key link in the positive feedback loop amplifying the apoptotic cascade of a wide variety of genotoxic agents.
Experimental Procedures
Cell Culture and Retrovirus Infections
The immortalized human keratinocyte cell line HaCaT (27), kindly provided by Dr. Norbert Fusenig (German Cancer Research Center, Heidelberg, Germany), was cultured in Media 154 (Cascade Biologics, Inc., Portland, OR) with 0.07 mm calcium added. Cells were irradiated with 30 mJ/cm2 UV from a bank of four UVB bulbs (FS36T12/UVB-VHO) with the dish lids removed. Cells were infected with retroviruses by spinning at 300 × g for 1 h at 32 °C as described previously (2, 8). For siRNA experiments, cells were infected with pSUPER.retro.puro viruses and selected with 1 μg/ml puromycin for at least 3 days until plated for the experiment.
Retrovirus Production
The catalytic fragment of PKCδ was routinely expressed from an LZRS-based retroviral vector as an estrogen receptor ligand binding domain fusion protein (PKCδ-ER) that is activated by treating the cells with 100 nm 4-hydroxytamoxifen (Tam) (13). The PKCδ-ER fusion protein has some basal PKCδ catalytic fragment activity, and Tam treatment is used to activate it further. In some experiments, the catalytic fragment of PKCδ (PKCδ-cat) in LZRS was used instead of the PKCδ-ER fusion protein (8). The PKCδ-ER virus permitted higher expression because the virus can be produced in the absence of Tam and packaging cell apoptosis is minimized (13). FLAG-tagged Mcl-1 was kindly provided by Dr. W. Douglas Cress (H. Lee Moffitt Cancer Center, Tampa, FL) (28) and was cloned into the Hind III/XhoI sites of the retroviral vector LZRS-Linker (8, 29). Construction of the Bcl-2 retrovirus was described previously (13). As a negative control, the empty retroviral vector LZRS-Linker (Linker) was used. All retroviruses were prepared in Phoenix-Ampho packaging cells (ATCC with permission from Garry P. Nolan, Stanford University Medical Center) by calcium phosphate transfection and selection with 1 μg/ml puromycin (29).
Retroviruses encoding hairpin siRNA were constructed using the pSUPER.retro.puro vector system (OligoEngine, Seattle, WA). A double-stranded hairpin oligonucleotide designed to target the human Mcl-1 cDNA nucleotides 13–33 (AAGAAACGCGGTAATCGGA) (30) was cloned into the BglII/HindIII sites of pSUPER.retro.puro. A control siRNA virus was constructed, which encoded a 19-bp target sequence (GCGCGCTTTGTAGGATTCG) with no significant homology to any gene in the human genome. Correct insertion of the oligonucleotides was confirmed by DNA sequencing. pSUPER viruses were produced in Phoenix-Ampho packaging cells as described for LZRS viruses.
Apoptosis Assays
Apoptosis was assayed by flow cytometry using Annexin V-FITC kit (Beckman Coulter, Inc.) as recommended by the manufacturer. The cells were run on a Coulter Epics XL-MCL flow cytometer to determine the percentage Annexin V-positive cells.
Cell Fractionation and Immunoblotting
Whole cell lysates were prepared by lysing floating and attached cells in 20 mm Tris-HCl, pH 7.5, 1% CHAPS, 5 mm EDTA, and 1× Complete protease inhibitor mixture (Roche Applied Science). Cytoplasmic extracts for cytochrome c release were prepared by trypsinizing the cells, washing once with phosphate-buffered saline, and suspending them in isotonic sucrose buffer: 250 mm sucrose, 10 mm HEPES, pH 7.4, 10 mm KCl, 1.5 mm MgCl2, 1 mm EGTA, 1 mm dithiothreitol, 1× Complete protease inhibitor mixture. Digitonin was added to 0.05%, and the cells mixed gently for 2 min at room temperature. The permeabolized cells were pelleted by spinning at 15,000 × g for 10 min at 4 °C and the cytoplasmic extracts analyzed by SDS-PAGE. Proteins were transferred to nitrocellulose, stained with antibodies, and visualized with ECL (Amersham Biosciences) or on an Odyssey Infrared Imaging System (LI-COR Biosciences, Inc., Lincoln, NE). Antibodies used for immunoblotting were Mcl-1 (sc-819, Santa Cruz Biotechnology, Santa Cruz, CA) at 1:500, β-actin (691001, MP Biomedicals, Aurora, OH) at 1:4000, cytochrome c (K2016–1, ApoAlert cell fractionation kit, BD Biosciences) at 1:500, and FLAG (M2, Sigma) at 1:2000. Mcl-1 was detected using monoclonal antibody 4602, clone RC13, (Chemicon International, Inc., Temecula, CA) in Fig. 1B. For quantitation of band intensities, blots were imaged on the Odyssey Infrared Imaging System.
FIGURE 1. PKCδ-cat and UV trigger loss of Mcl-1 in HaCaT cells.

A, HaCaT cells were infected with either control Linker virus (L) or PKCδ-ER virus and treated with 100 nm Tam for 3 days to activate PKCδ-ER or exposed to UV (30 mJ/cm2, 18 h) as indicated. Western blot analysis for Mcl-1 protein was performed, and the percentage of Mcl-1 relative to Linker virus control is shown. Western blot for β-actin confirmed equal protein loading. B, HaCaT cells were infected with either control Linker virus (L) or PKCδ-ER virus (PKCδ) and treated with 100 nm Tam as indicated for 3 days or exposed to UV (30 mJ/cm2, 18 h). Western blot and quantitation using two different antibodies are shown. The right panel used sc-819 (Santa Cruz Biotechnology), and the right panel used monoclonal antibody 4602 (clone RC13, Chemicon). C, quantitative analysis of Mcl-1 protein level measured from six independent Western blots experiments as described for A. The Mcl-1 levels in Linker-infected cells were normalized to 100%. Error bars represent standard deviation.
Kinase Reactions
Recombinant human PKCδ (PanVera Corp., Madison, WI), activated with 10 nm 12-O-tetradecanoylphorbol-13-acetate, or the active PKCδ-cat were added to FLAG-Mcl-1 or FLAG-Bcl-2 immunoprecipitated from transfected Phoenix-Ampho cells using anti-FLAG-agarose and incubated in the presence of [γ-32P]ATP and for 10 min at 30 °C. To generate the active PKCδ-cat, recombinant PKCδ was incubated with recombinant active caspase-3 (Alexis Biochemicals, San Diego, CA) as described previously (8). After the kinase reaction, the agarose beads were washed with immunoprecipitation buffer and boiled in SDS sample buffer. Phosphorylated proteins were run on SDS-PAGE and transferred to nitrocellulose for detection by autoradiography and Western blotting.
Protein Half-life Determination
The half-life of Mcl-1 was determined by treating cells with 10 μg/ml cyclohexamide for 0–6 h to inhibit protein synthesis and then preparing cell lysates to determine Mcl-1 levels by Western blotting. Mcl-1 protein levels were quantified by densitometry scanning and analysis using Scion Image. Mcl-1 band intensities for each treatment condition were normalized so that the percent of Mcl-1 in the no cyclohexamide group equaled 100%, and semi-log plots were generated in Microsoft Excel. Linear regression was performed and the half-life calculated from the fitted line equation.
Ribonuclease Protection Assays
RNA was isolated from HaCaT cells using TRIzol reagent (Invitrogen). The RNA was used in the Ribo-Quant multi-probe RNase protection assays system (Pharmingen) with the hAPO-2c probe set as described in the manufacturer's instructions.
Results
Expression of PKCδ-cat Reduces Steady-state Mcl-1 Protein Levels
Mcl-1 is an anti-apoptotic Bcl-2 family protein that can bind to and sequester pro-apoptotic proteins such as Bax and Bak (21–23). Since apoptosis induced by both PKCδ-cat and UV involves the activation of Bax (13), we examined Mcl-1 protein levels in HaCaT cells induced to undergo apoptosis by either PKCδ-cat expression or UV irradiation. To induce the activity of PKCδ-cat, we infected HaCaT cells with a retrovirus encoding a PKCδ-cat/estrogen receptor ligand binding domain fusion protein (PKCδ-ER). This PKCδ-ER fusion protein has some basal PKCδ enzymatic activity that can be activated further by treating the cells with Tam (13). As a negative control, cells were infected with an empty virus (Linker) and also treated with Tam. The Western blot in Fig. 1A shows that total Mcl-1 levels were reduced to 57% of control in cells infected with the PKCδ-ER virus, and Mcl-1 was reduced further (41% of control) when the PKCδ-ER was activated with Tam. Consistent with previous reports, UV caused a dramatic reduction in Mcl-1 levels, which was almost undetectable 18 h after UV irradiation. Fig. 2B shows a similar down-regulation of Mcl-1 by PKCδ-ER and UV using a different Mcl-1 antibody that primarily recognizes the short form of Mcl-1, Mcl-1S. On average (n = 6 experiments), Mcl-1 was reduced to 41% of control in PKCδ-ER expressing cells treated with Tam, with UV reducing Mcl-1 levels to 17% of control (Fig. 1C).
FIGURE 2. PKCδ-cat and UV reduce Mcl-1 protein half-life in HaCaT cells.

A, HaCaT cells were infected with either Linker, PKCδ-ER, or the catalytically inactive PKCδ(K378A)-ER virus and treated with Tam to activate PKCδ-ER or exposed to UV as indicated. Three days after virus infection or immediately after UV exposure, protein synthesis was blocked with 10 μg/ml of cyclohexamide (CHX), and cells were harvested at different time points (0, 0.5, 1, 2, 4, and 6 h). Western blots for Mcl-1 were performed and the intensity of Mcl-1 protein bands determined and normalized to β-actin. The half-life of Mcl-1 protein was estimated as described under “Materials and Methods.” B, Mcl-1 protein half-life was determined as described for A, and data averaged from three experiments are shown. Error bars represent standard deviation. The asterisks indicate significant differences between the treated groups and the Linker + Tam group (p < 0.05). C, the half-life of Mcl-1 protein in HeLa cells was determined as described for A. Cells were treated with CHX alone, exposed to UV, or exposed to UV and treated with CHX and the half-life of Mcl-1 calculated from Mcl-1 Western blots. Note that in HeLa cells Mcl-1 has a short half-life (∼40 min) that is not affected by UV.
Reduced Mcl-1 Levels Triggered by PKCδ-cat Are Due to Increased Protein Turnover, Not Decreased mRNA Levels
Mcl-1 protein and mRNA are reported to have a short half-life, and Mcl-1 levels can be regulated at the level of transcription, translation, and protein turnover (4, 22, 25). To determine how PKCδ-cat reduces Mcl-1 steady state protein levels, we measured Mcl-1 protein half-life by treating HaCaT cells with the protein synthesis inhibitor cyclohexamide for 0–6 h and quantified Mcl-1 levels by Western blot. As shown in Fig. 2, A and B, Mcl-1 protein had a half-life of 4 h 20 min in control cells (Linker virus plus Tam), and activation of PKCδ-ER with Tam reduced the half-life to 2 h 30 min. UV also reduced the half-life of Mcl-1 protein to 2 h, but the kinase inactive PKCδ(K378A)-ER mutant did not significantly affect Mcl-1 turnover. As a control, we also measured the half-life of Mcl-1 in HeLa cells which are reported to have rapidly turned over Mcl-1 (half-life of ∼40 min) that is not influenced by UV radiation (4). Fig. 2C confirms that the half-life of Mcl-1 in HeLa cells is ∼40 min and is not decreased further by UV, indicating that cell type differences exist for the regulation and turnover of Mcl-1.
We also examined Mcl-1 mRNA levels to determine whether PKCδ-cat or UV caused a decrease which could contribute to the reduced Mcl-1 protein levels. The ribonuclease protection assay in Fig. 3 shows that PKCδ-ER plus Tam did not cause an appreciable decrease in Mcl-1 mRNA levels. The mRNA levels of other Bcl-2 family members (Bcl-w, Bcl-xL, Bid, Bik, Bak, and Bax) were also not affected. UV caused a generalized decrease in mRNA levels for all Bcl-2 family members examined. Together, these results indicate that PKCδ-cat triggers loss of Mcl-1 by destabilizing the protein.
FIGURE 3. PKCδ-cat does not reduce Mcl-1 mRNA levels.

HaCaT cells were infected with Linker, PKCδ-ER, or PKCδ(K378A)-ER viruses and treated with Tam to activate PKCδ-ER or exposed to UV (30 mJ/cm2) as indicated. RNA was isolated 3 days after viral infection or 18 h after UV exposure and an RNase protection assay performed. Hybridization to L32 and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as control to ensure equal levels of input RNA. Note that Mcl-1 mRNA levels were not decreased by PKCδ-cat (PKCδ-ER+Tam).
Ectopic Expression of Mcl-1 Inhibits Apoptosis Induced by PKCδ-cat
To determine whether the reduction in Mcl-1 triggered by PKCδ-cat is required for apoptosis induction, we overexpressed Mcl-1 in HaCaT cells by retroviral transduction and triggered apoptosis by expressing PKCδ-cat or exposing cells to UV radiation. Fig. 4A confirms the retroviral overexpression of the full-length Mcl-1 (Mcl-1L) in HaCaT cells. In addition a fainter ∼30-kDa band was detected that corresponds in size to the alternative spliced Mcl-1S isoform (31), although the identity of this lower molecular weight Mcl-1 requires additional characterization. Fig. 4B shows that PKCδ-cat expression or UV irradiation induced morphological cell death and the overexpression of Mcl-1 partially protected from the morphological cell death. Fig. 4C shows that Mcl-1 expression protected from PKCδ-cat and UV induced apoptosis (p < 0.05 and 0.01, respectively) as measured by Annexin V binding. A kinase-inactive mutant of PKCδ-cat, [PKCδ(K378A)-ER], did not trigger apoptosis, confirming that kinase activity is required for PKCδ-cat to induce apoptosis.
FIGURE 4. Mcl-1 protects from PKCδcat and UV apoptosis.

A, HaCaT cells were infected with either control Linker (L) or Mcl-1 (M) viruses and Mcl-1 protein levels determined by Western blotting. Note the overexpression of Mcl-1L and Mcl-1S proteins in Mcl-1-infected cells. B, HaCaT cells were infected with the control Linker or Mcl-1 retroviruses in the morning and in the afternoon re-infected with PKCδ-ER as indicated. The next day cells were treated with Tam for 3 days and phase contrast pictures taken. The cells irradiated with UV (30 mJ/cm2) were photographed after 18 h. Note that the morphological cell death in the PKCδ-ER+Tam and UV-irradiated cells was partially inhibited by Mcl-1. C, HaCaT cells were infected with Linker or Mcl-1 virus and then re-infected with either PKCδ-ER or PKCδ(K378A)-ER viruses and treated with Tam to activate PKCδ-ER or irradiated with UV as indicated. Apoptosis was assayed measured by Annexin V/propidium iodide staining. Note the significant reduction of PKCδ-ER and UV apoptosis by Mcl-1. The catalytically inactive PKCδ(K378A)-ER virus did not induce apoptosis. Data shown are averaged from three independent experiments with error bars representing the standard deviation.
The release of cytochrome c from the mitochondria is a key event in the activation of caspase-9 and is under negative regulation by anti-apoptotic Bcl-2 family proteins such as Mcl-1 (4). To determine whether Mcl-1 expression can inhibit the release of cytochrome c caused by PKCδ-cat (13), we assayed cytoplasmic cytochrome c levels by Western blot. As shown in Fig. 5, PKCδ-ER plus Tam triggered the release of cytochrome c into the cytoplasm, and this could be almost completely blocked by Mcl-1 expression. Mcl-1 expression also blocked UV-induced release of cytochrome c from the mitochondria. Note that the caspase inhibitor Z-VAD did not inhibit cytochrome c release triggered by PKCδ-cat, indicating that cytochrome c release is an early, caspase-independent event. Together these results suggest that the loss of Mcl-1 triggered by PKCδ-cat may facilitate the release of cytochrome c and subsequent apoptotic signaling.
FIGURE 5. Mcl-1 blocks PKCδ-cat-induced cytochrome c release.

HaCaT cells were infected with Linker, PKCδ-ER, and/or Mcl-1 viruses, treated with Tam to activate PKCδ-ER, or exposed to UV as indicated. The general caspase inhibitor Z-VAD was added at 10 μg/ml as indicated. Cytosolic fractions were analyzed by Western blotting for release of cytochrome c from the mitochondria. Note that PKCδ-cat and UV triggered cytochrome c release that was inhibited by Mcl-1. Z-VAD did not prevent cytochrome c release. A Western blot for β-actin was done as a control of protein loading.
Suppression of Mcl-1 with siRNA Sensitizes HaCaT Cells to Apoptosis
To determine whether the reduction in Mcl-1 caused by PKCδ-cat is sufficient to trigger apoptosis, we infected HaCaT cells with a retrovirus encoding a siRNA targeting Mcl-1 and assessed apoptosis. Fig. 6A shows that the Mcl-1 siRNA reduced Mcl-1 protein levels to 42–67% of control, with an average reduction to 55% of control (n = 3 experiments). Note that this level of inhibition is similar to the reduction in Mcl-1 triggered by PKCδ-ER (41% of control, Fig. 1). In Fig. 6B, quantitation of apoptosis by Annexin V binding shows that Mcl-1 siRNA alone cause a small but statistically significant (p < 0.05) increase in apoptosis over a 2-day time period. However in HaCaT cells expressing the catalytic fragment of PKCδ or exposed to UV radiation, the Mcl-1 siRNA caused a substantial increase in apoptosis that was highly significant (p < 0.001).
FIGURE 6. Suppression of Mcl-1 with siRNA sensitizes HaCaT cells to apoptosis.

A, HaCaT cells were infected with control (C) or Mcl-1 siRNA retroviruses and cultured in the presence of 1 μg/ml puromycin for 2 days. Western blots for Mcl-1 and β-actin were performed. Data from three independent experiments are shown, with the percent of Mcl-1 in each experiment normalized to the control. B, HaCaT cells infected with either control or Mcl-1 siRNA viruses were selected with puromycin and then re-infected with PKCδ-cat virus or exposed to UV as indicated. Three days after the PKCδ-cat infection or 18 h after UV irradiation, apoptosis was assayed by Annexin V/propidium iodide staining. Each condition in the experiment was performed in triplicate, with error bars denoting standard deviation. C, HaCaT cells infected with control or Mcl-1 siRNA viruses were selected with puromycin and re-infected with PKCδ-ER virus as indicated. PKCδ activity was induced by addition of Tam to the media of PKCδ-ER-infected cells. Three days after PKCδ-ER infection or 18 h after UV irradiation, the heavy membrane fraction was isolated and a Western blot for Bax performed. Total Bax levels were quantified by densitometry and are indicated at the bottom of the Western blot. Note the increase in total Bax levels, especially the dimer (2×) and trimer (3×) forms, indicative of Bax activation.
Mcl-1 can bind and sequester the pro-apoptotic protein Bax (21), and the induction of apoptosis by the catalytic fragment of PKCδ or UV involves the up-regulation and activation/oligomerization of Bax (13). We determined whether partial down-regulation of Mcl-1 with siRNA was sufficient to activate Bax. The Bax cross-linking experiment in Fig. 6C shows that the Mcl-1 siRNA induced the appearance of Bax dimers (2×) and increased the total amount of Bax 2.4-fold. Activation of the PKCδ-cat and UV irradiation also increased the Bax dimer and total Bax levels, and the Mcl-1 siRNA further enhanced the oligomerization and up-regulation of Bax. Taken together, these results indicate that even a partial reduction in Mcl-1 levels can dramatically sensitize cells to apoptotic stimuli.
PKCδ Directly Phosphorylates Mcl-1
Since our results demonstrated that the catalytic fragment of PKCδ causes enhanced turnover of Mcl-1, and phosphorylation of Mcl-1 by a variety of stimuli has been reported to regulate its stability and activity (32–34), we determined whether PKCδ can directly phosphorylate Mcl-1. Mcl-1, or Bcl-2 as a control, was immunoprecipitated from Phoenix-Ampho packaging cells, incubated with recombinant PKCδ in a kinase assay, and the phosphorylated proteins analyzed by SDS-PAGE and autoradiography. Fig. 7A shows that PKCδ phosphorylated a protein at ∼45 kDa in the FLAG immunoprecipitate from Mcl-1-transfected cell lysate, and this band corresponds in size to the Mcl-1 in Fig. 7B. No phosphorylation of Bcl-2 (∼30 kDa) or Mcl-1 in the absence of exogenous PKCδ was detected, indicating that the phosphorylation of Mcl-1 was specific and due to PKCδ. Similar results were obtained with PKCδ-cat generated by caspase-3 cleavage (data not shown). A nonspecific band at ∼85 kDa reacting with the Mcl-1 antibody was not phosphorylated by PKCδ. These results indicated that Mcl-1 is a direct substrate for PKCδ-cat.
FIGURE 7. Direct phosphorylation of Mcl-1 protein in vitro by PKCδ-cat.
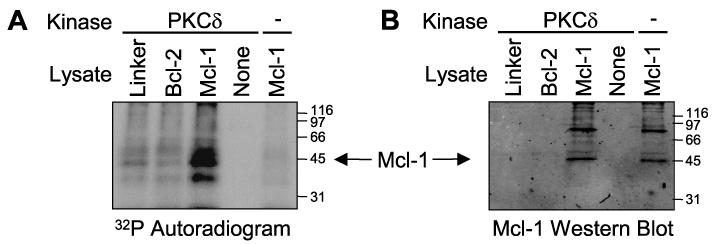
Lysates from Phoenix-Ampho cells transfected with Linker, FLAG-Bcl-2, or FLAG-Mcl-1 plasmids were immunoprecipitated with anti-FLAG-agarose. Immunoprecipitated proteins were used as substrates with or without recombinant PKCδ in the presence of [γ-32P]ATP. A kinase reaction using anti-FLAG agarose in the absence of a cell lysate (None) was also included as a negative control. Proteins were resolved on SDS-PAGE and transferred to nitrocellulose. A, autoradiogram showing phosphorylated proteins. Note the prominent phosphorylated protein at ∼42 kDa in the Mcl-1 lysate incubated with PKCδ. B, Mcl-1 Western blot corresponding to A localizing immunoprecipitated Mcl-1. Note that the phosphorylated protein in FLAG-Mcl-1-expressing cells corresponds in size to Mcl-1. As a negative control, FLAG-tagged Bcl-2 (∼30 kDa) was not phosphorylated.
Discussion
PKCδ is a widely expressed death substrate activated by caspase-3 cleavage in cells triggered to undergo apoptosis by a variety of agents. In this study, we have identified Mcl-1, an anti-apoptotic Bcl-2 family member, as an important target for apoptosis mediated by PKCδ-cat. Ectopic expression of PKCδ-cat caused the down-regulation of Mcl-1 by increasing Mcl-1 protein turnover (Fig. 2), and the loss of Mcl-1 was required for PKCδ-cat-induced cytochrome c release (Fig. 5) and apoptosis (Fig. 4). The loss of Mcl-1 is also involved in UV-induced apoptosis (4), and we previously demonstrated that the cleavage and activation of PKCδ were also partially required for the down-regulation of Mcl-1 by UV radiation (10). This study extends these observations by demonstrating that PKCδ-cat is sufficient to cause Mcl-1 down-regulation and provides mechanistic insight into the down-regulation of Mcl-1 by PKCδ-cat.
The expression of Mcl-1 is under multiple types of regulation at both the transcriptional and translation levels and responds rapidly to environmental clues and apoptotic stimuli (25, 35, 36). Expression of PKCδ-cat caused ∼50% decrease in steady-state Mcl-1 levels, while UV caused almost complete loss of Mcl-1 (Fig. 1), consistent with the much stronger apoptotic response of HaCaT cells to UV radiation than PKCδ-cat. Both PKCδ-cat and UV decreased the half-life of Mcl-1 protein (Fig. 2), and UV, but not PKCδ-cat, reduced Mcl-1 mRNA levels. In contrast, UV has been reported to trigger the loss of Mcl-1 in HeLa cells by inhibiting its synthesis and had no effect of Mcl-1 protein turnover (4). To validate our Mcl-1 half-life measurement method, we confirmed that UV did not increase Mcl-1 turnover in HeLa cells (Fig. 2C), indicating cell type-specific regulation for Mcl-1 down-regulation. Understanding the apoptotic regulation of Mcl-1 in HaCaT cells, a human keratinocyte cell line, is highly relevant to UV skin carcinogenesis as these non-tumorigenic cells harbors UV signature mutations in p53 and resemble the premalignant cells frequently targeted by UV radiation (27, 37). In normal human keratinocytes, UV caused a reduction in Mcl-1 mRNA and protein levels, consistent with our results in HaCaT cells shown in Figs. 1 and 3 (26). Mcl-1 transcription is regulated by E2F1, CREB, and ETS transcription factors (38).
The down-regulation of an anti-apoptotic protein such as Mcl-1 is not necessarily sufficient to induce apoptosis as most cells, including HaCaT cells, express multiple pro-survival Bcl-2 family members (4, 39, 40). Mcl-1 knock-out mice were embryonic lethal, and Mcl-1 has a specific survival role for hematopoietic stem cells (41, 42). Using a retrovirus expressing a hairpin siRNA against Mcl-1, we were able to down-regulate Mcl-1 levels ∼50%, a level comparable with that triggered by PKCδ-cat (Figs. 1 and 6). We found a small but statistically significant increase in basal apoptosis and total Bax levels (Fig. 6). However, the down-regulation of Mcl-1 with siRNA dramatically sensitized HaCaT cells to PKCδ-cat and UV-induced Bax activation and apoptosis (Fig. 6). This synergy may be due to either more complete Mcl-1 loss by the combination of PKCδ-cat and Mcl-1 siRNA or the involvement of additional apoptosis inducing signals initiated by PKCδ-cat. Since ∼50% reduction of Mcl-1 by siRNA did not trigger apoptosis to the same extent as the active PKCδ-ER virus, which also reduced Mcl-1 levels ∼50%, our results suggest that additional signals initiated by PKCδ-cat cooperate with Mcl-1 loss to trigger apoptosis. In addition, more complete reduction of Mcl-1 by siRNA in other cell types was also not sufficient to induce high levels of apoptosis or cell death (4, 22).
We also demonstrated that Mcl-1 is directly phosphorylated by PKCδ-cat in vitro (Fig. 7). Since kinase activity was required for increased turnover of Mcl-1 (Fig. 2) and apoptosis (Fig. 4), phosphorylation is a likely mechanism for targeting Mcl-1 for degradation. Furthermore, both PKCδ-cat and Mcl-1 are localized to the mitochondria, putting the active kinase and its substrate at the same subcellular localization (4, 8). Mcl-1 can be phosphorylated by several protein kinases. 12-O-Tetradecanoylphorbol-13-acetate treatment induced phosphorylation at a conserved extracellular signal-regulated kinase (ERK) site in the PEST region of Mcl-1, and G2/M arresting drugs or a protein phosphatase 1/2A inhibitor also induced an ERK-independent phosphorylation of Mcl-1 (32, 34, 43). Mcl-1 was phosphorylated at Ser-121 and Thr-163 by c-Jun NH2-terminal kinase in response to H2O2, and this phosphorylation was associated with inactivation of Mcl-1 and enhanced apoptosis (33). Mcl-1 was also phosphorylated by glycogen synthase kinase-3 at Ser-159 in response to interleukin-3 withdrawal or AKT inhibition leading to increased ubiquitinylation and degradation (44). Mcl-1 can be degraded by the proteasome and is also subject to cleavage by caspases and granzyme B (4, 43–45). The BH3 domain-containing Mule/ARF-BP1 E3 ubiquitin ligase is necessary and sufficient for Mcl-1 polyubiquitination and proteasome targeting (46). We found that the proteasome inhibitor MG132 significantly increased Mcl-1 protein levels (data not shown), suggesting a role for the ubiquitin proteasome pathway in Mcl-1 degradation in HaCaT cells.
During apoptosis induced by UV and other stimuli, PKCδ cleavage and activation are caspase-dependent and occur only after caspase activation, while Mcl-1 loss is an early event required for caspase activation (4, 5, 8, 11, 47, 48). These kinetics make it impossible for PKCδ-cat to mediate the initial Mcl-1 down-regulation that permits Bax/Bak activation, cytochrome c release, and caspase activation. However, the ability of PKCδ-cat to phosphorylate and increase the turnover of Mcl-1 can explain why PKCδ cleavage and activation are required for a robust apoptotic response, as this would establish a positive feedback loop between PKCδ cleavage and activation, Mcl-1 loss, and further caspase activation.
Mcl-1 is unique among Bcl-2 family members in that it is responsive to a variety of growth factors or phorbol ester treatment (49) and is required for early pre-implantation and development in the mouse and survival of hematopoietic stem cells (41, 42). Mcl-1 also has functions in cell cycle control, indicating it has non-apoptotic functions (50). The multiple regulatory features and cell type-specific regulatory mechanisms of Mcl-1 are consistent with this multifaceted functions and may be required to integrate inputs from diverse signaling pathways, including the PKCδ apoptotic pathway discussed here. Taken together, our results identify Mcl-1 as a key link in the positive feedback loop targeted by PKCδ-cat in cells destined to undergo apoptosis.
Acknowledgments
We thank all members of the Skin Cancer Research Program for their help with this project, especially Drs. Brian J. Nickoloff and Jian-Zhong Qin for helpful discussions. We also thank Dr. W. Douglass Cress (H. Lee Moffitt Cancer Center, Tampa, FL) for furnishing the Mcl-1 cDNA.
Footnotes
This work was supported by a grant from the Potts Foundation and National Institutes of Health Grant CA83784 (to M. F. D.).
The abbreviations used are: PKC, protein kinase C; Tam, 4-hydroxytamoxifen; PKCδ-cat, PKCδ catalytic fragment; ERK, extracellular signal-regulated kinase; siRNA, small interfering RNA; ER, estrogen receptor; CHAPS, 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonic acid; Z, benzyloxycarbonyl.
References
- 1.Grossman D, Kim PJ, Blanc-Brude OP, Brash DE, Tognin S, Marchisio PC, Altieri DC. J Clin Invest. 2001;108:991–999. doi: 10.1172/JCI13345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sitailo LA, Tibudan SS, Denning MF. J Biol Chem. 2002;277:19346–19352. doi: 10.1074/jbc.M200401200. [DOI] [PubMed] [Google Scholar]
- 3.Takasawa R, Tanuma S. Apoptosis. 2003;8:291–299. doi: 10.1023/a:1023629023696. [DOI] [PubMed] [Google Scholar]
- 4.Nijhawan D, Fang M, Traer E, Zhong Q, Gao W, Du F, Wang X. Genes Dev. 2003;17:1475–1486. doi: 10.1101/gad.1093903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Denning MF, Wang Y, Nickoloff BJ, Wrone-Smith T. J Biol Chem. 1998;273:29995–30002. doi: 10.1074/jbc.273.45.29995. [DOI] [PubMed] [Google Scholar]
- 6.DeVries TA, Neville MC, Reyland ME. EMBO J. 2002;21:6050–6060. doi: 10.1093/emboj/cdf606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fujii T, Garcia-Bermejo ML, Bernabo JL, Caamano J, Ohba M, Kuroki T, Li, Yuspa SH, Kazanietz MG. J Biol Chem. 2000;275:7574–7582. doi: 10.1074/jbc.275.11.7574. [DOI] [PubMed] [Google Scholar]
- 8.Denning MF, Wang Y, Tibudan S, Nickoloff BJ, Qin JZ. Cell Death Diff. 2002;9:40–52. doi: 10.1038/sj.cdd.4400929. [DOI] [PubMed] [Google Scholar]
- 9.Matassa AA, Carpenter L, Biden TJ, Humphries MJ, Reyland ME. J Biol Chem. 2001;276:29719–29728. doi: 10.1074/jbc.M100273200. [DOI] [PubMed] [Google Scholar]
- 10.D'Costa AM, Denning MF. Cell Death Differ. 2005;12:224–232. doi: 10.1038/sj.cdd.4401558. [DOI] [PubMed] [Google Scholar]
- 11.Ghayur T, Hugunin M, Talanian RV, Ratnofsky S, Quinlan C, Emoto Y, Pandey P, Datta R, Huang Y, Kharbanda S, Allen H, Kamen R, Wong W, Kufe D. J Exp Med. 1996;184:2399–2404. doi: 10.1084/jem.184.6.2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bharti A, Kraeft SK, Gounder M, Pandey P, Jin S, Yuan ZM, Lees-Miller SP, Weichselbaum R, Weaver D, Chen LB, Kufe D, Kharbanda S. Mol Cell Biol. 1998;18:6719–6728. doi: 10.1128/mcb.18.11.6719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sitailo LA, Tibudan SS, Denning MF. J Invest Dermatol. 2004;123:434–443. doi: 10.1111/j.0022-202X.2004.23403.x. [DOI] [PubMed] [Google Scholar]
- 14.Ren J, Datta R, Shioya H, Li Y, Oki E, Biedermann V, Bharti A, Kufe D. J Biol Chem. 2002;277:33758–33765. doi: 10.1074/jbc.M110667200. [DOI] [PubMed] [Google Scholar]
- 15.Yoshida K, Wang HG, Miki Y, Kufe D. EMBO J. 2003;22:1431–1441. doi: 10.1093/emboj/cdg134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu J, Chen J, Dai Q, Lee RM. Cancer Res. 2003;63:1153–1156. [PubMed] [Google Scholar]
- 17.Frasch SC, Henson PM, Kailey JM, Richter DA, Janes MS, Fadok VA, Bratton DL. J Biol Chem. 2000;275:23065–23073. doi: 10.1074/jbc.M003116200. [DOI] [PubMed] [Google Scholar]
- 18.He Y, Liu J, Durrant D, Yang HS, Sweatman T, Lothstein L, Lee RM. Cancer Res. 2005;65:10016–10023. doi: 10.1158/0008-5472.CAN-05-1688. [DOI] [PubMed] [Google Scholar]
- 19.Efimova T, Broome AM, Eckert RL. Mol Cell Biol. 2004;24:8167–8183. doi: 10.1128/MCB.24.18.8167-8183.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cross T, Griffiths G, Deacon E, Sallis R, Gough M, Watters D, Lord JM. Oncogene. 2000;19:2331–2337. doi: 10.1038/sj.onc.1203555. [DOI] [PubMed] [Google Scholar]
- 21.Zhou P, Qian L, Kozopas KM, Craig RW. Blood. 1997;89:630–643. [PubMed] [Google Scholar]
- 22.Cuconati A, Mukherjee C, Perez D, White E. Genes Dev. 2003;17:2922–2932. doi: 10.1101/gad.1156903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Willis SN, Chen L, Dewson G, Wei A, Naik E, Fletcher JI, Adams JM, Huang DC. Genes Dev. 2005;19:1294–1305. doi: 10.1101/gad.1304105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Letai A, Bassik MC, Walensky LD, Sorcinelli MD, Weiler S, Korsmeyer SJ. Cancer Cell. 2002;2:183–192. doi: 10.1016/s1535-6108(02)00127-7. [DOI] [PubMed] [Google Scholar]
- 25.Iglesias-Serret D, Pique M, Gil J, Pons G, Lopez JM. Arch Biochem Biophys. 2003;417:141–152. doi: 10.1016/s0003-9861(03)00345-x. [DOI] [PubMed] [Google Scholar]
- 26.Qin JZ, Bacon P, Panella J, Sitailo LA, Denning MF, Nickoloff BJ. J Cell Physiol. 2004;200:155–166. doi: 10.1002/jcp.20017. [DOI] [PubMed] [Google Scholar]
- 27.Boukamp P, Petrussevska RT, Breitkreutz D, Hornung J, Markham A, Fusenig NE. J Cell Biol. 1988;106:761–771. doi: 10.1083/jcb.106.3.761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ma Y, Cress WD, Haura EB. Mol Cancer Ther. 2003;2:73–81. [PubMed] [Google Scholar]
- 29.Kinsella TM, Nolan GP. Hum Gene Ther. 1996;7:1405–1413. doi: 10.1089/hum.1996.7.12-1405. [DOI] [PubMed] [Google Scholar]
- 30.Han J, Goldstein LA, Gastman BR, Rabinovitz A, Rabinowich H. J Biol Chem. 2005;280:16383–16392. doi: 10.1074/jbc.M411377200. [DOI] [PubMed] [Google Scholar]
- 31.Bae J, Leo CP, Hsu SY, Hsueh AJ. J Biol Chem. 2000;275:25255–25261. doi: 10.1074/jbc.M909826199. [DOI] [PubMed] [Google Scholar]
- 32.Domina AM, Vrana JA, Gregory MA, Hann SR, Craig RW. Oncogene. 2004;23:5301–5315. doi: 10.1038/sj.onc.1207692. [DOI] [PubMed] [Google Scholar]
- 33.Inoshita S, Takeda K, Hatai T, Terada Y, Sano M, Hata J, Umezawa A, Ichijo H. J Biol Chem. 2002;277:43730–43734. doi: 10.1074/jbc.M207951200. [DOI] [PubMed] [Google Scholar]
- 34.Domina AM, Smith JH, Craig RW. J Biol Chem. 2000;275:21688–21694. doi: 10.1074/jbc.M000915200. [DOI] [PubMed] [Google Scholar]
- 35.Croxton R, Ma Y, Song L, Haura EB, Cress WD. Oncogene. 2002;21:1359–1369. doi: 10.1038/sj.onc.1205157. [DOI] [PubMed] [Google Scholar]
- 36.Rahmani M, Davis EM, Bauer C, Dent P, Grant S. J Biol Chem. 2005;280:35217–35227. doi: 10.1074/jbc.M506551200. [DOI] [PubMed] [Google Scholar]
- 37.Lehman TA, Modali R, Boukamp P, Stanek J, Bennett WP, Welsh JA, Metcalf RA, Stampfer MR, Fusenig N, Rogan EM, Harris CC. Carcinogenesis. 1993;14:833–839. doi: 10.1093/carcin/14.5.833. [DOI] [PubMed] [Google Scholar]
- 38.Croxton R, Ma Y, Cress WD. Oncogene. 2002;21:1563–1570. doi: 10.1038/sj.onc.1205232. [DOI] [PubMed] [Google Scholar]
- 39.Chaturvedi V, Qin JZ, Denning MF, Choubey D, Diaz MO, Nickoloff BJ. J Biol Chem. 1999;274:23358–23367. doi: 10.1074/jbc.274.33.23358. [DOI] [PubMed] [Google Scholar]
- 40.Bowen AR, Hanks AN, Allen SM, Alexander A, Diedrich MJ, Grossman D. J Invest Dermatol. 2003;120:48–55. doi: 10.1046/j.1523-1747.2003.12010.x. [DOI] [PubMed] [Google Scholar]
- 41.Rinkenberger JL, Horning S, Klocke B, Roth K, Korsmeyer SJ. Genes Dev. 2000;14:23–27. [PMC free article] [PubMed] [Google Scholar]
- 42.Opferman JT, Iwasaki H, Ong CC, Suh H, Mizuno S, Akashi K, Korsmeyer SJ. Science. 2005;307:1101–1104. doi: 10.1126/science.1106114. [DOI] [PubMed] [Google Scholar]
- 43.Derouet M, Thomas L, Cross A, Moots RJ, Edwards SW. J Biol Chem. 2004;279:26915–26921. doi: 10.1074/jbc.M313875200. [DOI] [PubMed] [Google Scholar]
- 44.Maurer U, Charvet C, Wagman AS, Dejardin E, Green DR. Mol Cell. 2006;21:749–760. doi: 10.1016/j.molcel.2006.02.009. [DOI] [PubMed] [Google Scholar]
- 45.Han J, Goldstein LA, Gastman BR, Froelich CJ, Yin XM, Rabinowich H. J Biol Chem. 2004;279:22020–22029. doi: 10.1074/jbc.M313234200. [DOI] [PubMed] [Google Scholar]
- 46.Zhong Q, Gao W, Du F, Wang X. Cell. 2005;121:1085–1095. doi: 10.1016/j.cell.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 47.Mizuno K, Noda K, Araki T, Imaoka T, Kobayashi Y, Akita Y, Shimonaka M, Kishi S, Ohno S. Eur J Biochem. 1997;250:7–18. doi: 10.1111/j.1432-1033.1997.00007.x. [DOI] [PubMed] [Google Scholar]
- 48.Emoto Y, Manome Y, Meinhardt G, Kisaki H, Kharbanda S, Robertson M, Ghayur T, Wong WW, Kamen R, Weichselbaum R, Kufe D. EMBO J. 1995;14:6148–6156. doi: 10.1002/j.1460-2075.1995.tb00305.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Michels J, Johnson PW, Packham G. Int J Biochem Cell Biol. 2005;37:267–271. doi: 10.1016/j.biocel.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 50.Fujise K, Zhang D, Liu J, Yeh ET. J Biol Chem. 2000;275:39458–39465. doi: 10.1074/jbc.M006626200. [DOI] [PubMed] [Google Scholar]