

Abstract
Identification of genes and genomic regions under directional natural selection has become one of the major goals in evolutionary genetics, but relatively little work to this end has been done by applying hitchhiking mapping to wild populations. Hitchhiking mapping starts from a genome scan using a randomly spaced set of molecular markers followed by a fine-scale analysis in the flanking regions of the candidate regions under selection. We used the hitchhiking mapping approach to narrow down a selective sweep in the genomic region flanking a candidate locus (Stn90) in chromosome VIII in the three-spined stickleback (Gasterosteus aculeatus). Twenty-four microsatellite markers were screened in an ∼800-kb region around the candidate locus in three marine and four freshwater populations. The patterns of genetic diversity and differentiation in the candidate region were compared to those of a putatively neutral set of markers. The Bayesian FST-test indicated an elevated genetic differentiation, deviating significantly from neutral expectations, at a continuous region of ∼20 kb upstream from the candidate locus. Furthermore, a method developed for an array of microsatellite markers rejected neutrality in a region of ∼90 kb flanking the candidate locus supporting the selective sweep hypothesis. Likewise, the genomewide pattern of genetic diversity differed from the candidate region in a bottleneck analysis suggesting that selection, rather than demography, explains the reduced genetic diversity at the candidate interval. The neutrality tests suggest that the selective sweep had occurred mainly in the Lake Pulmanki population, but the results from bottleneck analyses indicate that selection might have operated in other populations as well. These results suggest that the narrow interval around locus Stn90 has likely been under directional selection, but the region contains several predicted genes, each of which can be the actual targets of selection. Understanding of the functional significance of this genomic region in an ecological context will require a more detailed sequence analysis.
UNDERSTANDING the genetic basis of evolutionary change is of fundamental interest in evolutionary biology (Orr 2005a,b; Ungerer et al. 2007; Stinchcombe and Hoekstra 2007). However, the molecular basis of the specific mutations underlying evolutionary shifts in mean trait values has seldom been uncovered in any detail (but see Colosimo et al. 2005; Storz et al. 2007). In recent years, QTL-mapping studies have shed light on the genetic architecture of some ecologically important morphological traits (e.g., Colosimo et al. 2004, 2005; Shapiro et al. 2004). From the methodological point of view, a standard QTL-mapping approach requires that the association between the phenotype and genotype can be established (Mackay 2001). However, many traits underlying adaptive divergence are not always easily detectable at the phenotypic level and not well-suited to QTL mapping (Schlötterer 2003). In such cases, one possible approach to tackle the genetic basis of adaptation is to use neutral genetic markers to identify targets of natural selection (Schlötterer 2002, 2003). Based on principles of population genetics, natural selection is expected to create a skew in allele frequencies of the genes under selection and also on the flanking neutral sites—commonly known as genetic hitchhiking (Maynard Smith and Haigh 1974; Schlötterer 2003). Theory further predicts that natural selection leaves predictable “footprints” in the degree of genetic differentiation and diversity of linked neutral markers, which are distinguishable from neutral processes (Nielsen 2005). Genomic regions under directional selection are expected to show decreased within population diversity and increased among population differentiation, whereas the effects of balancing selection are expected to be roughly opposite (e.g., Nielsen 2005; Charlesworth 2006).
The identification of targets of natural selection can be compromised by several factors. First, separating the footprints of selection from those resulting from neutral processes—such as random genetic drift and population bottlenecks—can be challenging (Schlötterer 2003; Storz 2005; Teshima et al. 2006). These caveats can be avoided by screening a large number of markers spaced across an organism's whole genome to characterize the background levels of marker variability and differentiation (Schlötterer 2003). Second, as the analysis of genome scan data involves multiple statistical tests, identification of false-positive footprints of selection may become an issue (Wiehe et al. 2007). One way to alleviate this particular problem is to genotype more loci in the genomic regions near a candidate locus. It is unlikely that a signature of selection emerging from a particular genomic region is false if it is detected in more than one marker locus (Wiehe et al. 2007). However, while the analysis of markers flanking genomic regions of the candidate locus does not completely rule out demographic explanations, it is expected to result in a considerable reduction in the number of false positives (Thornton and Jensen 2007; Wiehe et al. 2007). Conducting this type of fine-scale analyses at the genomic level is becoming more feasible due to the increased availability of the whole-genome sequences for various organisms in recent years (Benson et al. 2007; Storz and Hoekstra 2007).
In a standard hitchhiking mapping approach, one performs a first-pass genome scan with a randomly spaced set of markers scattered throughout the organism's genome (Harr et al. 2002; Schlötterer 2003). This kind of analysis can identify loci showing footprints of natural selection and provide a starting point for a finer-scale analysis around these candidate loci. The adjacent marker loci can provide further proof of selection in a particular genomic region, but can also help to narrow down the genomic interval at which selection is operating (Wiehe et al. 2007). Previous studies suggest that this approach might be useful in identifying genes involved in domestication, artificial selection, or in resistance to a drug treatment (Kohn et al. 2000; Nair et al. 2003; DuMont and Aquadro 2005; Olsen et al. 2006; Pool et al. 2006; Sutter et al. 2007). However, fine-scale mappings around candidate loci identified in first-pass genome scans have rarely been conducted in wild populations (but see Harr et al. 2002; Ihle et al. 2006).
Recently, H. S. Mäkinen, J. M. Cano and J. Merilä (unpublished results) performed a microsatellite genome scan to detect genomic regions under natural selection in marine and freshwater populations of the three-spined stickleback (Gasterosteus aculeatus). Several candidate loci showing signals of both directional and balancing selection were identified. Altogether, five loci were interpreted as potentially being linked to genomic regions under directional selection. Of these, three loci (Stn365, 380, and 381) were linked to the Eda gene, coding for the number of lateral plates, and showed the strongest signal of natural selection. One microsatellite locus, Stn90, located on chromosome VIII and not linked to the Eda locus, showed a strong signal of directional selection and was interpreted to be linked to gene(s) important for adaptive divergence. Thus, the genomic region containing Stn90 was chosen for a more detailed analysis.
Here we have investigated this finding further, focusing on three specific objectives. First, we aimed to confirm the signature of selection in the genomic region flanking the candidate locus Stn90 by genotyping 24 microsatellite markers in an ∼800-kb interval around Stn90. Second, using this densely spaced set of markers, we intended to narrow down the chromosomal region showing the selective imprint with fine-scale mapping. Our third aim was to identify the actual target gene of natural selection by using the putative homologies and genescan gene predictions annotated in the whole three-spined stickleback genome sequence. This approach is novel in the sense that most of the explorative genome-scan studies have not elucidated the signal from the candidate outliers probably due to lack of suitable genomic resources. Furthermore, fine-scale hitchhiking mapping studies have been rarely performed in wild fish populations.
MATERIALS AND METHODS
Study populations:
Twenty-four individuals were genotyped from three marine and four freshwater three-spined stickleback populations (Figure 1). The marine populations were sampled from the Baltic Sea (Merirastila), the North Sea (Orrevatnet), and in the pelagic region from the Barents Sea. Three of the freshwater populations were located in Fennoscandia: Lake Vättern in southern Sweden, Lake Kevo and Lake Pulmanki in the Finnish Lapland. One of the freshwater populations (River Neretva) was located in the Adriatic Sea region. This population has a different evolutionary history than the Fennoscandian populations: it diverged from its marine ancestors probably during the late Pleistocene whereas the Fennoscandian populations are of postglacial origin (∼10,000 years ago; Mäkinen et al. 2006). Inclusion of the River Neretva population should increase the background level of genetic differentiation and thus give more support for significant outliers in neutrality tests based on allele frequencies.
Figure 1.—

The sampling locations of the seven study populations. The marine locations are indicated in boldface type.
Microsatellite development and genotyping:
Twenty-four microsatellite loci, located in an 852.57-kb region flanking the Stn90 locus were developed, hereafter referred to as a candidate marker data set (appendix a). The average spacing of the markers was one marker per every 35.52 kb. In a 39.94-kb region flanking the Stn90 locus, a more densely spaced set of markers was used with an average density of one marker for every 4.71 kb (appendix a). The microsatellite markers were developed using the whole-genome sequence available at the Ensembl Genome Browser (Hubbard et al. 2007; http://www.ensembl.org/Gasterosteus_aculeatus/index.html). The primer sequences, repeat motifs, and their genomic positions are listed in appendix a. The microsatellite genotypes in H. S. Mäkinen, J. M. Cano and J. Merilä (unpublished results) were used as reference data. Initially, this data set comprised 103 microsatellite loci and two indel markers. Of these, 20 loci potentially affected by selection were excluded from the present analyses and the remaining 85 loci were considered as putatively neutral reference loci (hereafter referred to as the neutral marker data set). Both data sets are provided as supplemental files in Microsatellite Analyzer input format at http://www.genetics.org/supplemental/.
DNA was extracted from pelvic fins using a silica fine-based method embedded in 96-well filter plates with slight modifications to the original protocol of Elphinstone et al. (2003). To facilitate high throughput genotyping, a commercial multiplex PCR kit (QIAGEN, Valencia, CA) was used, which allowed the use of similar PCR conditions for all loci. PCR was carried out in a 10-μl volume consisting of 2 pmol of each primer, 1× QIAGEN multiplex PCR master mix, 0.5× Q-solution and ∼20 ng of template DNA. The PCR cycling started with an activation step of 15 min at 95°, followed by 30 cycles of 30 sec at 94°, 90 sec at 53°, and 60 sec at 72°, and a final extension at 72° for 10 min. The forward primers were labeled with FAM, HEX, and TET fluorescent dyes for the visualization of the PCR products and a GTTT tail was added to the 5′-end of the reverse primers to enhance the 3′-adenylation (Brownstein et al. 1996). Before the electrophoresis in the Megabase 1000 capillary sequencer the PCR products were diluted 1:50 with MQ-water and mixed with ET-ROX size standard according to the manufacturer's instructions (Amersham Biosciences, Piscataway, NJ). The alleles were scored with the program Fragment Profiler 1.2 and were manually edited by T. Shikano.
Genetic data analysis:
The expected heterozygosities and the allelic richness were estimated for the neutral and candidate marker data sets separately. Deviations from Hardy–Weinberg (HW) expectations were assessed by 1000 permutations over loci and populations. Population differentiation was estimated using the θ estimator of FST (Weir and Cockerham 1984) and the 95% confidence intervals were determined by 1000 permutations. The analysis of linkage disequilibrium was conducted between all loci in each population. All the above calculations were performed with FSTAT 2.9.3.2 (Goudet 2001).
Marker loci subject to directional selection are expected to show higher-than-average differentiation (FST) in allele frequencies and reduced genetic diversity as compared to neutral expectations (Lewontin and Krakauer 1973; Beaumont and Nichols 1996; Kauer et al. 2003; Beaumont and Balding 2004). Selection tends to affect only locus-specific patterns of genetic diversity and differentiation, whereas neutral processes—such as random genetic drift—have genomewide effects. In practice, marker loci in the tails of the distribution of given test statistics are considered to be potentially affected by selection. Several model-based and model-free methods have been developed to detect targets of natural selection on the basis of the above assumptions and are detailed in the following.
The first type of test assumes that marker loci subject to divergent directional selection have a higher-than-average level of population differentiation (FST). In this context, the hierarchical structure of FST has been modeled in the Bayesian framework as log (Fij/1− Fij) = αi + βj + γij, where αi is a locus effect, βj is a population effect, and γij is a locus-by-population effect (Beaumont and Balding 2004). The interpretations of the potential outliers are based on the locus effect (αi): under neutrality, the locus effects are expected to be zero, while positive values are indicative of directional selection and negative values indicative of balancing selection. Statistically significant locus effects were estimated from the posterior distribution: the locus was considered to be under directional selection if the 2.5% quantile was positive and under balancing selection if its 97.5 quantile was negative (Beaumont and Balding 2004). All computations were performed with the program BAYESFST and the locus effects were estimated from 2000 draws from the posterior distribution generated by MCMC simulation. The locus effects were summarized with the R package using the functions provided with the distribution package of BAYESFST (http://www.reading.ac.uk/Statistics/genetics/software.html). The analyses were repeated twice to check whether independent runs converged to similar parameter estimates. The major advantage of the Bayesian approach is that the model is flexible in the assumptions of the population structure in comparison to the frequentist method (Beaumont and Nichols 1996; Beaumont and Balding 2004). The Bayesian method allows FST to vary among populations whereas the frequentist method, as implemented in the program FDIST, assumes fixed FST among study populations (Beaumont and Nichols 1996; Beaumont and Balding 2004). Although simulation studies have shown that both methods give almost similar results, the Bayesian method was preferred given its flexibility in the assumptions about the population structure (Beaumont and Balding 2004).
The second type of test is based on the assumption of reduced genetic diversity at the vicinity of a selective sweep (Maynard Smith and Haigh 1974; Schlötterer 2002). The Ln RH test assumes that the empirical distribution of the logarithmic ratio of locus-specific genetic diversities (θ = 4Neμ) between two populations is roughly normally distributed (Kauer et al. 2003). Ninety-five percent of the standardized Ln RH estimates are expected to lie between −1.96 and 1.96 and loci outside this distribution are considered to deviate from neutrality (Kauer et al. 2003). Recently, Wiehe et al. (2007) suggested a modification of the Ln RH test to accommodate an array of microsatellite loci in a genomic interval. The proposed approach starts with standardizing the Ln RH values and identifying the locus with the most extreme Ln RH value in an array of microsatelllite loci. Then all loci upstream and downstream from the extreme Ln RH value are selected until the first locus with a positive value (or negative value depending on in which population selection has occurred) is found, resulting in K loci to be included in the analysis. After dropping the locus with the extreme Ln RH value from the analysis, the parameter 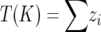 , which sums the standardized Ln RH values for the number of K loci, is calculated. Then a P-value is estimated assuming that T(K) follows a normal distribution with a mean 0 and standard deviation √K (Wiehe et al. 2007). One concern in this method is that it assumes independency (linkage equilibrium) of the analyzed loci. On the other hand, simulation studies have shown that Ln RH values at loci even with a 1-kb distance are only weakly correlated (Wiehe et al. 2007) although it is worth noting that this result was obtained using parameters from Drosophila melanogaster populations and might not be applicable to G. aculeatus. However, an analysis of linkage disequilibrium indicated significant linkage disequilibrium at the candidate marker data set but not after correction for multiple tests. The results of the Ln RH test might be biased if many loci have been subject to selection increasing the standard deviation and only the extreme values would be deviating significantly from neutral expectations (Storz 2005; Wiehe et al. 2007). To overcome such bias, the Ln RH estimates were standardized as suggested in Wiehe et al. (2007) using the neutral marker data set from H. S. Mäkinen, J. M. Cano and J. Merilä (unpublished results). The calculations were performed with the program MSA Analyzer (Dieringer and Schlötterer 2003) using the “constrained gene diversity” option and Excel spreadsheet to carry out the standardization. The pairwise comparisons were conducted between the adjacent marine and freshwater populations reflecting the likely colonization history (Mäkinen et al. 2006).
, which sums the standardized Ln RH values for the number of K loci, is calculated. Then a P-value is estimated assuming that T(K) follows a normal distribution with a mean 0 and standard deviation √K (Wiehe et al. 2007). One concern in this method is that it assumes independency (linkage equilibrium) of the analyzed loci. On the other hand, simulation studies have shown that Ln RH values at loci even with a 1-kb distance are only weakly correlated (Wiehe et al. 2007) although it is worth noting that this result was obtained using parameters from Drosophila melanogaster populations and might not be applicable to G. aculeatus. However, an analysis of linkage disequilibrium indicated significant linkage disequilibrium at the candidate marker data set but not after correction for multiple tests. The results of the Ln RH test might be biased if many loci have been subject to selection increasing the standard deviation and only the extreme values would be deviating significantly from neutral expectations (Storz 2005; Wiehe et al. 2007). To overcome such bias, the Ln RH estimates were standardized as suggested in Wiehe et al. (2007) using the neutral marker data set from H. S. Mäkinen, J. M. Cano and J. Merilä (unpublished results). The calculations were performed with the program MSA Analyzer (Dieringer and Schlötterer 2003) using the “constrained gene diversity” option and Excel spreadsheet to carry out the standardization. The pairwise comparisons were conducted between the adjacent marine and freshwater populations reflecting the likely colonization history (Mäkinen et al. 2006).
Simulation studies have shown that population bottlenecks can mimic the effect of a selective sweep (Teshima et al. 2006; Wiehe et al. 2007). This opens an opportunity to further understand the patterns of the genetic diversity around the candidate locus Stn90. A simple working hypothesis is to assume that if selection had shaped the distribution of the genetic diversity flanking Stn90, then the effective population size in the candidate region might show decline in comparison to the genomewide patterns. To tell apart such effects, bottleneck analyses were conducted separately for the neutral and the candidate marker data sets. The detection of bottlenecks is based on the expectation that the number of alleles and heterozygosity are reduced in a bottlenecked population. However, a reduction in population size is expected to reduce the number of alleles faster than the heterozygosity (Cornuet and Luikart 1996). In practice, in the bottleneck analysis the equilibrium heterozygosity (HEQ) simulated from the number of alleles assuming a constant population size is compared to the actual heterozygosity (HE) in the population. If the equilibrium heterozygosity (HEQ) exceeds the actual heterozygosity then this type of heterozygosity excess would be indicative of a recent population bottleneck (Cornuet and Luikart 1996). The bottleneck estimations were carried out in the program BOTTLENECK 1.2.02 using 1000 coalescent simulations and assuming a two-phase mutation model (TPM) as suggested for microsatellite data (Cornuet and Luikart 1996). The bottleneck analyses were also conducted assuming a step-wise mutation model (SMM), which is the most conservative model for testing heterozygosity excess (Cornuet and Luikart 1996). The statistical significance of the deviations at equilibrium and observed heterozygosities were assessed with Wilcoxon signed-rank tests.
The putative protein homologies based on the genescan gene predictions at the candidate interval were identified with protein–protein Blast searches at the NCBI nonredundant protein database. The molecular function and biological processes of the homologies were classified according to the gene ontology (GO) categories (Harris et al. 2006).
RESULTS
The basic population genetic estimates for the data are summarized in Table 1. There were no marked differences in the mean heterozygosities, allele numbers, or FIS estimates for the putatively neutral marker data set and the candidate marker data set. However, in the Lake Pulmanki population the heterozygosity in the candidate marker data set was clearly lower (HE = 0.33) than in the neutral marker data set (HE = 0.67). The average population differentiation was significantly higher (FST = 0.24, 95% C.I. 0.19–0.3) for the loci in the candidate marker data set than for the putatively neutral set of loci (FST = 0.16, 95% C.I. 0.15–0.18).
TABLE 1.
Basic population genetic parameters estimated as average across all populations separately for the putatively neutral data set (85 loci) and for the loci flanking the candidate locus
| HE Neutral | HE Candidate | ANeutral | ACandidate | FIS Neutral | FIS Candidate | |
|---|---|---|---|---|---|---|
| Merirastila | 0.76 | 0.65 | 10.6 | 10.9 | 0.025 | 0.035 |
| Orrevatnet | 0.65 | 0.55 | 5.4 | 3.8 | 0.011 | 0.026 |
| Barents | 0.73 | 0.61 | 8.9 | 8.6 | 0.008 | −0.011 |
| Lake Vättern | 0.73 | 0.56 | 9.5 | 8.9 | 0.011 | −0.003 |
| Lake Pulmanki | 0.67 | 0.33 | 6.8 | 4.7 | −0.003 | 0.034 |
| Lake Kevo | 0.56 | 0.48 | 5.5 | 5.2 | 0.008 | −0.037 |
| River Neretva | 0.52 | 0.53 | 6.4 | 8.2 | 0.033 | 0.023 |
| Mean | 0.77 | 0.67 | 18.0 | 18.1 | 0.016 | 0.009 |
HE, expected heterozygosity; A, number of alleles; FIS, inbreeding coefficient.
The result of the original data, which was used as a starting point for this study, is shown in Figure 2a (H. S. Mäkinen, J. M. Cano and J. Merilä, unpublished results). The Bayesian FST-test identified seven loci in the 852.57-kb region flanking the candidate locus Stn90 showing footprints of directional selection in the analysis including all seven populations at the 5% significance level (Figures 2b and 3, appendix b). Most of the significant loci were concentrated on the genomic interval ∼19.4 kb upstream from the candidate locus (Figure 3). Furthermore, two loci, one 43.3 kb upstream (E) and the other two 48.5 kb (Q) downstream from the candidate locus showed a signal of directional selection (Figure 3). Interestingly, downstream from the candidate there was a region with low FST and one locus (O) was affected by balancing selection (Figures 2b and 3). An analysis without Lake Pulmanki indicated that only two loci (Stn90 and J) were directional selection outliers in the candidate interval, but several loci (G, M, O, and P) appeared in the lower tail of the FST distribution (Figure 2c). Excluding the distantly located River Neretva population from the analysis resulted in a roughly similar distribution of FST estimates, but the locus Q downstream from the candidate did not deviate from neutrality anymore, whereas an additional locus (C) appeared as an outlier (Figure 2d). The low FST region was apparent also in this comparison but none of the loci were significant balancing selection outliers. Excluding both River Neretva and Lake Pulmanki populations from the Bayesian FST-test indicated that the loci in the vicinity of the candidate appeared in the upper tail of the distribution, but were no longer statistically significant (Figure 2e). An additional analysis including all 105 loci as reference data showed almost the same number and identities of outlier loci but locus L was no longer a statistically significant outlier (FST = 0.19, P = 0.036).
Figure 2.—





(a–e) Results of the Bayesian FST-tests. The solid line indicates the critical cutoff for the P-value at the 5% level.
Figure 3.—

Schematic overview of the genomic region (∼800 kb) flanking the candidate locus Stn90. The shaded bars show the Bayesian FST estimates (locus effects) for the loci spanning in this interval. Asterisks denote significant P-values at the 5% level on the top of the shaded bars. The Ln RH estimates are indicated as solid and open squares. The solid squares indicate Ln RH values, which were included in the multilocus Ln RH test. The dashed lines indicate the expected neutral distribution (from −1.96 to 1.96) of the Ln RH values. The solid boxes indicate Ensembl gene predictions and their putative protein homologies.
In the Ln RH analysis, a significant reduction in genetic diversity around the candidate locus was identified only in the Barents Sea and Lake Pulmanki comparison (Figure 3, appendix b). In the other marine–freshwater comparisons, there were only 1–2 loci deviating from neutrality in the candidate region (data not shown). Lower genetic diversity in the Lake Pulmanki than in the Barents Sea population indicates that a selective sweep had occurred in the Lake Pulmanki population. However, one locus (E) showed an opposite pattern: the genetic diversity was reduced below neutral expectations in the Barents Sea population (Figure 3, appendix b). Altogether, 50% (12/24) of the loci in the genomic interval flanking the candidate locus displayed a lower-than-average level of genetic diversity (Figure 3). Ten of the significant loci were located in the nearby genomic regions of the candidate locus and the remaining loci again upstream or downstream from the candidate locus. The highest Ln RH estimate (3.83) was observed in locus G located 27.9 kb upstream of the candidate locus. Using the criteria recently introduced by Wiehe et al. (2007), 13 loci were included in the multilocus Ln RH test (Figure 3). Locus H in this array was monomorphic and thus excluded from the analysis. The multilocus test statistics for these loci were T (12) = 18.58 (after dropping the locus with the highest Ln RH value) and resulted in a highly significant P-value (P < 0.001), i.e., the cumulative probability assuming normal distribution with mean = 0 and standard deviation (SD) = 3.46. Thus, the reduction of the genetic diversity in this genomic region is strongly deviating from neutrality. For comparative purposes, Ln RH tests were carried out between L. Pulmanki and other marine reference populations (Merirastila and Orrevatnet). The test statistics for loci H–Q in the Merirastila and Lake Pulmanki comparison were T (9) = 12.35, SD = 3.0 (P < 0.001) and in the Orrevatnet and Lake Pulmanki comparison for loci E–Q, T (14) = 14.65, SD = 3.74 (P < 0.001). Likewise, using the standard deviation and mean derived from the 105-loci data set for the standardization for Ln RH statistics had only marginal effect on the Ln RH estimates.
The results of the bottleneck tests were roughly concordant with a priori expectations (see materials and methods). In the analysis of the putatively neutral data set only the coastal population from the North Sea (Orrevatnet) deviated from the heterozygosity expected at a constant population size (P = 0.008, Table 2). When the analysis was performed for the candidate marker data set, statistically significant bottleneck signatures were found in all populations (Table 2). Assuming the SMM-model indicated similar patterns in the neutral data set, no indications of a bottleneck were found in the Orrevatnet population. An analysis of the candidate marker data set revealed bottlenecks in all of the freshwater populations and in the Orrevatnet population. It is therefore likely that the populations (except Orrevatnet) have not experienced reductions in population size during their history, but other evolutionary forces such as selection might have shaped the patterns of genetic diversity in the candidate region at least in the freshwater populations. Similar patterns were recovered when the bottleneck analyses were conducted with the 105-loci data set.
TABLE 2.
Results of the bottleneck analyses for the putatively neutral and candidate loci data sets
| Neutral
|
Candidate
|
|||||
|---|---|---|---|---|---|---|
| HEQ (SMM) | HEQ (TPM) | HE | HEQ (SMM) | HEQ (TPM) | HE | |
| Merirastila | 0.82 | 0.80 | 0.76 | 0.54 | 0.52*** | 0.56 |
| Orrevatnet | 0.67 | 0.64** | 0.65 | 0.47*** | 0.46*** | 0.61 |
| Barents | 0.79 | 0.73 | 0.77 | 0.49 | 0.47*** | 0.54 |
| Lake Vättern | 0.79 | 0.73 | 0.78 | 0.46** | 0.44*** | 0.54 |
| Lake Pulmanki | 0.72 | 0.70 | 0.67 | 0.39*** | 0.38*** | 0.55 |
| Lake Kevo | 0.65 | 0.63 | 0.52 | 0.42** | 0.41*** | 0.52 |
| River Neretva | 0.68 | 0.65 | 0.55 | 0.48** | 0.47*** | 0.59 |
The average of the equilibrium heterozygosity based on the observed number alleles (HEQ) assuming either a step-wise mutation model (SMM) or a two-phase mutation model (TPM), and the expected heterozygosity (HE).
**P < 0.01; ***P < 0.001.
The putative protein homologies of the genes, position in chromosome VIII, molecular functions, and biological processes in the candidate genomic region are listed in Table 3. In general, the candidate region was relatively gene rich containing several putative homologies to known genes. The level of protein homology varied from 74% (ABCA1, Gallus gallus) to 93% (GAMT, Danio rerio). The putative homologies had various molecular functions in RNA and DNA binding (Dazap1), protein binding (ABCA1 and JAK3), and in transcription factor activity (Arid3A). Also the biological processes of candidate genes were variable including functions in spermatogenesis (Dazap1), regulation of body size (GAMT), regulation of transcription (Arid3A and JAK3), and in metabolism (ABCA1).
TABLE 3.
Some of the putative protein homologies in the candidate interval including their molecular functions and biological processes according to the Gene Ontology categories
| Protein homology gene | Location | Molecular function | Biological process |
|---|---|---|---|
| Dppe9 (Monodelphis domestica, 86%) | 507309–514916 | — | — |
| Dazap1 (Danio rerio, 89%) | 517721–533991 | RNA binding, DNA binding | Spermatogenesis |
| GAMT (D. rerio, 93%) | 538945–542200 | — | Regulation of body size, spermatogenesis |
| Arid3A (Homo sapiens, 84%) | 567695–586061 | Transcription factor activity | Regulation of transcription, DNA dependent |
| ABCA1 (Gallus gallus, 74%) | 608799–628295 | Protein binding | Metabolism, transport |
| JAK3 (Tetraodon fluviatilis, 85%) | 670476–676889 | Protein binding | Regulation of transcription |
DISCUSSION
The main aim of this study was to confirm the footprint of selection emerging from the previously identified candidate locus Stn90. Additional markers in the nearby regions showed an elevated degree of allele frequency differentiation and reduced genetic diversity consistent with the selective-sweep scenario. Thus, consistency between the Bayesian FST and the Ln RH test in multiple loci may be considered as evidence for natural selection in the candidate genomic region. Population bottleneck tests in the flanking region of the candidate locus revealed an excess of heterozygosity relative to the expectations of a constant population size, suggesting reduction of the local effective population size. Thus, selection might have shaped the genetic diversity of the candidate region in other study populations as well, but this result depends on the parameter choice in the bottleneck analysis. The continuous genomic region suggested to be affected by the selective sweep was ∼92-kb long, but deviations from neutrality occurred also upstream and downstream of this region. The results demonstrate that the hitchhiking mapping approach starting from a candidate locus identified in the first-pass genome was useful in narrowing down the genomic region experiencing a selective sweep in wild three-spined stickleback populations. The actual gene(s) causing the footprint of selection remains to be identified since the genomic interval identified as a target of selection contained several predicted genes and putative protein homologies. However, some candidate genes in this interval would be interesting targets for more detailed genomic analysis.
Hitchhiking mapping—methodological considerations:
The majority of the previous studies have focused on selective sweeps in marker loci flanking a priori known genes and little work has been done to identify novel candidate genes underlying adaptive divergence. These examples show selective sweeps resulting from domestication (Clark et al. 2004; Olsen et al. 2006), artificial selection (Pollinger et al. 2005; Sutter et al. 2007), and pesticide treatment (Nair et al. 2003; Nash et al. 2005). In these cases selection is assumed to have been strong and recent, and thus, relatively easily detectable according to theoretical expectations (De Kovel 2006). In malaria parasites (Plasmodium falciparum) the introduction of an antimalarial drug in the 1970s has led to strongly reduced levels of genetic diversity in a 100-kb region surrounding resistance locus dhfr (Nair et al. 2003). In domestic dog breeds, most of the phenotypic evolution has occurred during the past 200 years as a result of strong artificial selection facilitating the identification of signatures of selection (Pollinger et al. 2005). When it comes to wild populations, the selection history can be of older origin making detection of genomic imprints of selection difficult if mutation and recombination have restored the genetic diversity back to the background level (De Kovel 2006). According to the simulation studies by De Kovel (2006), intermediate-strength selection would be detectable in large populations only within 200–400 generations after the divergence.
In most Fennoscandian three-spined stickleback populations selection might originate from the period after the last glaciation, which is ∼10,000 years and roughly 5000 generations assuming a two-year generation interval (Mäkinen et al. 2006). The transition from the marine to freshwater environment is one of the best characterized sources of divergent selection (Bell and Foster 1994; McKinnon and Rundle 2002; Colosimo et al. 2005). Yet, the fact that we found a clear signature of selection apparently related to freshwater–marine divergence suggests that the hitchhiking approach seems to work also in wild three-spined stickleback populations, where selection might originate from the relatively long freshwater isolation. Nevertheless, the signal of selection emerged mainly from Lake Pulmanki and evidence for selection in other freshwater populations was not compelling suggesting that the adaptive significance of the candidate interval might not be universal in the marine–freshwater divergence.
Another important methodological implication of this study is that it confirms selection in a candidate region, which was found to be an outlier but only marginally significant in the first-pass genome scan (H. S. Mäkinen, J. M. Cano and J. Merilä, unpublished results). Using a method developed for an array of microsatellites resulted in a very low probability that the reduction in the genetic diversity at the candidate region would be due to neutral processes. Therefore, this approach holds promise in narrowing down the genomic interval under selection even in wild populations and is in line with results obtained from D. melanogaster populations (Harr et al. 2002). Also in natural populations of house mouse (Mus musculus) the hitchhiking mapping approach has revealed a candidate region for a selective sweep (Ihle et al. 2006). A further sequence analysis in this genomic region revealed a lowered nucleotide diversity in a 20-kb region of a β-defensin locus 6 (Ihle et al. 2006). The genomic interval where the candidate gene of interest might be located by hitchhiking mapping seems to be relatively narrow compared to QTL mapping. For example, Colosimo et al. (2005) looked for the gene responsible in controlling the lateral plate number in three-spined sticklebacks in a QTL cross and were able to locate the gene to a 539-kb candidate interval in the initial analysis. Thus, the hitchhiking mapping approach, at least in our case, provided a narrower (∼92-kb) genomic region to search for the actual gene underlying adaptive divergence. On the other hand, this study demonstrates that it would be difficult to identify a single gene underlying adaptive divergence even with the whole-genome sequence information. Another challenge would be to link this genomic region with phenotypic variation highlighting the distinct weakness of the hitchhiking approach. In a very strict sense, without the information on phenotypic variation, it is still possible that the genomic region with a signal of a selective sweep could be a demographic artifact. Recently, Thornton and Jensen (2007) emphasized an ascertainment bias problem associated with selecting genomic regions for a more detailed analysis. In a simulation model including both population bottleneck and selective sweep the tails of the empirical distribution seemed to be enriched by “false” signals of selection. It is, however, reasonable to assume that our study populations have not encountered “true” bottlenecks during their history, but merely bottlenecks in the candidate interval. Furthermore, empirical genome scans typically identify only a handful of outlier loci (1.4–9.5%, reviewed in Stinchcombe and Hoekstra 2007), which could be selected for further analysis. In our case, Stn90 was a logical starting point as the signal of selection was roughly comparable to the Eda-associated loci, with known adaptive significance in the same study populations (Cano et al. 2006).
The bottleneck analyses indicated a lower genetic diversity at the candidate interval than would be expected assuming constant population size in all study populations. Using the putatively neutral data set for the same analyses suggested that the populations have not experienced actual bottlenecks, the Orrevatnet population being an exception in this respect. However, when using the more conservative mutation model (SMM instead of TPM), the bottleneck analyses indicated that marine populations (Merirastila and Barents) fitted to the expectations of a constant population size in the candidate region. The indications of bottlenecks are in contrast with the Bayesian FST and Ln RH neutrality tests, which suggest that the signal of selection is mainly emerging from the Lake Pulmanki population. Therefore, it might appear that selection has been operating in the candidate interval in every population, or at least in freshwater populations but has been strong enough only in Lake Pulmanki to leave an imprint detectable by the neutrality tests. A roughly similar pattern of divergence has been observed in X-linked and autosomal microsatellites in African and cosmopolitan D. melanogaster populations (Schöfl and Schlötterer 2006). Cosmopolitan populations showed indications of bottlenecks in X-linked microsatellites but not in autosomal microsatellites probably due to selection. Recent theoretical work suggests that if the selective sweep involves multiple origins of the beneficial allele (“soft” sweeps) then some of the ancestral genetic diversity may be retained in the population (Pennings and Hermisson 2006). This is in contrast with the traditional view of the selective sweep (“hard”), which is considered to involve only a single origin of a beneficial allele resulting in a more drastic reduction of the linked neutral variability (Pennings and Hermisson 2006). This scenario might explain why especially the Ln RH test, which is based on the reduction of θ, failed to detect selection in other freshwater populations than Lake Pulmanki. Other bottleneck-mimicking processes, such as migration from a divergent population are not likely explanations for the observed patterns as the freshwater populations are geographically isolated and the marine populations are genetically relatively uniform (Mäkinen et al. 2006). Deviations from Hardy–Weinberg assumptions are also not likely causes for the heterozygosity excesses, because the genotype frequencies followed the HW equilibrium expectations.
The genomic size of the selective sweep:
Depending on the method employed, the estimated size of the genomic region affected by selection varied between 19.4 kb (FST-test) and 92 kb (Ln RH) flanking the candidate locus. The regions overlapped only in an ∼20-kb region upstream of the candidate locus, but both methods identified loci deviating from neutral expectations at down- and upstream genomic regions outside the continuous region. It has been suggested that the region affected by selection is determined mainly by the strength of selection, local recombination rate, population history, and the age of the beneficial allele (Nordborg and Tavare 2002). Empirical studies have reported variable-sized (30–260-kb) chromosomal regions with reduced genetic diversity in flanking sites of a selected locus, which is comparable to the interval detected here. In cases of strong selection, as found in the Waxy gene in rice (Oryzas sativa), the region showing signatures of a selective sweep spanned ∼260 kb (Olsen et al. 2006). In P. falciparum the region underlying selection around resistance loci depended strongly on the strength of selection. In the Laos population, experiencing weak selection for resistance to an antimalarial drug, the genome regions affected by hitchhiking were smaller (34–69 kb) than in the Thailand population influenced by strong selection (98–268 kb; Nash et al. 2005). In our study, the selective sweep seems not to be restricted only to the loci in the vicinity of the candidate locus. Other loci with significant deviations from neutrality were found several kilobases apart from the candidate locus indicating that the genomic interval contains other selected regions as well. Taking into account all of the loci significant in the Ln RH test between the Barents Sea and Lake Pulmanki comparison would expand the region where selection has operated to ∼192 kb. Unfortunately, estimation of the selection coefficient would require information of the mutation and recombination rates, as well as effective population size and this data is lacking at the moment. However, the selection in this genomic region might be fairly strong given that the size of the region is comparable to the Nash et al. (2005) findings.
The genome interval contained several predicted genes and they have putative protein homologies to the known genes, but without more detailed sequence analysis it would be premature to conclude which would be the actual gene(s) underlying selection. However, protein homologies suggest some interesting candidate genes for further studies. For example, two predicted genes have putative homologies to genes (DAZ and GAMT) involved in spermatogenesis. Genes involved in spermatogenesis have been found to be under selection in humans (Zhang et al. 2007) and in Drosophila species (Civetta et al. 2006). Thus, spermatogenesis-related genes might also be under selection in G. aculeatus populations. Maybe the most exciting candidate gene for a more detailed analysis would be the GAMT, which has been found to also be involved in body size regulation in mouse (M. musculus; Vitarius et al. 2006). The analysis of body shape in the same populations analyzed here indicates that this trait is under genetic control and has a high adaptive value (Leinonen et al. 2006). Furthermore, traits related to morphology have been found to be under strong directional selection in the previous studies (Colosimo et al. 2005; H. S. Mäkinen, J. M. Cano and J. Merilä, unpublished results). It is also possible that the genomic interval in question contains a cluster of genes with similar functions affecting the same trait given the relatively high number of genes and the large region affected by selection.
Conclusions:
We have demonstrated that a candidate locus identified in a genomewide scan can be used as a starting point for a finer-scale mapping in wild populations. Using a densely spaced set of microsatellite markers in combination with a recently developed multilocus analysis method revealed that the patterns in genetic diversity at the candidate genomic region deviate from neutral expectations. This suggests that narrowing down genomic regions affected by directional selection is possible in wild three-spined stickleback populations, and this approach is not—at least in our case—compromised by mutation and recombination. The results further suggest that the observed selective sweep has occurred in a freshwater population, and thereby adds support to the earlier contention (H. S. Mäkinen, J. M. Cano and J. Merilä, unpublished results) that the selective sweep may be related to freshwater adaptation. However, our top-down approach based on hitchhiking mapping did not allow identification of a single candidate gene even with the access to information from the whole-genome sequence. The complexity of the pattern found can be due to selection acting on a cluster of functionally related genes. A future challenge would be to link the causal polymorphisms to an ecological context and to verify the signal of natural selection.
Acknowledgments
We thank John Loehr for proofreading and Anti Vasemägi and two anonymous referees for constructive comments on an earlier version of this manuscript. Our research was supported by the Finnish Graduate School in Population Genetics and the Academy of Finland.
APPENDIX A: PRIMER SEQUENCES, POSITIONS IN CHROMOSOME VIII, AND THE REPEAT MOTIFS
| Locus | Position (bp) | Repeat motif | Forward primer 5′–3′ | Reverse primer 5′–3′ |
|---|---|---|---|---|
| Gaest7 | 9348674 | (TG)21 | CTGAAGCAGAAAGTGCTCA | TGGTCTATTACTGATGCTCAAA |
| A | 9443885 | (CA)17 | CCCAACAGGTATCAACATAAAC | AAAGACACCAAACCCCTAAT |
| B | 9488797 | (AC)14 | TGAGCCGGACAAATAGAG | AGTCTTCGGTCAAAAGTGAT |
| C | 9510432 | (TG)15 | GAAAAGTCTGTGCAGGTCTC | CAGTGAGCCAGGTGTGTAA |
| D | 9516682 | (TAT)6 | ACGACTAAATCAATGTCCCA | TTAAACGAAGCTGACACACA |
| E | 9531673 | (AAAG)4 | ATCGACTACTTCCCCATACTG | CAGTTGTAGGTTTCTGTGCAA |
| F | 9543566 | (AC)13 | CTTGAGAACTTGTATGTATGGG | TGACTTTTGAGTGATGATGG |
| G | 9547035 | (TA)14 | GAAGGAAAGGATGGAAAGTC | ATCACGTTACAAGGAAACCTC |
| H | 9551511 | (TCC)6 | CCTAGTTGCACTTTATGTTGTC | CTACAATAAGGGTTTGGCTCT |
| I | 9555637 | (AGG)6 | TTCAGGACGGACAAAATACT | CTGAGTAGAAGAAACCACCAAC |
| J | 9561953 | (CTCAT)4 | AGCGTCAACACAATACACAG | ATGTCTTCATGTAACCACAGTC |
| K | 9567752 | (TGC)7 | AAACACTAAAAGGGGAAAGG | AGCCGCCTCTAACAAGAC |
| L | 9571427 | (TG)17 | GAGATGGTGGTTGAGACAGA | GCCTCGGAATAGATTGATTAAC |
| Stn90a | 9575008 | (AC)12 | TGAGCTAAATTTGACTGCCG | ATTTACACCTGCCAACCACC |
| M | 9579874 | (GT)37 | TCACTAACAGCCCCTTCC | GGGAGTTGGCATTAAACATT |
| N | 9585973 | (CTG)9 | GGACCTGAGTGTGTTGGG | CGGGACTGGTACTGCTTC |
| O | 9597139 | (TG)37 | CAAAATGAGATGGACGAGA | GTACACATGACAATGCACATC |
| P | 9607259 | (TA)10 | TCAAGTAGAACCTGTCAAGGA | ACTGGACTGTAATGCACTGTTA |
| Q | 9623459 | (AG)15 | CAAACTGTATTTCTAGCACTCACC | TTTCATGGAGAGCAGCGT |
| R | 9636370 | (GT)21 | GGAGCTTACTGCCTAACTCA | TACCTTCGTTCTACTCTCACCT |
| S | 9702839 | (TA)16 | ACTTATTTTGTGACGGTAGAGC | ATCACGTTAAAGCCAAAGAG |
| T | 9766526 | (TG)48 | CGTAGTGAGTTGGATTAGCATA | GTGACGGACGAGATACACA |
| U | 9827512 | (GT)10(TT)(GT)4 | CTGCAACCTAAAAGACATCAC | AGAGAATAACCGTGGAGACAC |
| V | 9895278 | (CA)19 | CATGCCGATGTTTTCACT | ACAATACCTGGCCTAAATCTC |
| Stn239a | 10201248 | (CA)20 | CTCTGAAACATGCAGACATTGG | TGTTGATCTATCCCTTTGGG |
APPENDIX B: BASIC POPULATION GENETIC ESTIMATES FOR THE LOCI IN THE CANDIDATE INTERVAL
| Locus | Genomic position (bp) | Bayes FST (P-value) | Ln RH | AR | HE | FIS |
|---|---|---|---|---|---|---|
| GAest7 | 9348557 | 0.09 | −1.09 | 11.7 | 0.90 | 0.1 |
| A | 9443885 | 0.12 | 0.45 | 14.2 | 0.91 | 0.04 |
| B | 9488797 | 0.12 | 0.57 | 14.2 | 0.92 | 0.05 |
| C | 9510432 | 0.18 | 2.13 | 6.7 | 0.80 | 0.04 |
| D | 9516682 | 0.20 | −0.03 | 6.1 | 0.79 | −0.03 |
| E | 9531673 | 0.30 (0.023) | −2.06 | 2.2 | 0.29 | −0.12 |
| F | 9543566 | 0.14 | 1.28 | 10.2 | 0.87 | 0.03 |
| G | 9547035 | 0.07 | 3.83 | 15.8 | 0.92 | −0.04 |
| H | 9551511 | 0.18 | −0.52 | 1.1 | 0.01 | 0.0 |
| I | 9555637 | 0.28 (0.012) | 2.45 | 2.9 | 0.53 | −0.04 |
| J | 9561953 | 0.28 (0.007) | 2.36 | 3.5 | 0.58 | −0.01 |
| K | 9567752 | 0.26 (0.006) | 1.98 | 4.9 | 0.65 | −0.08 |
| L | 9571427 | 0.21 (0.022) | 2.18 | 9.9 | 0.65 | −0.06 |
| Stn90 | 9575008 | 0.29 (0.003) | 2.14 | 4.6 | 0.72 | −0.04 |
| M | 9579874 | 0.07 | 3.12 | 21.7 | 0.95 | −0.02 |
| N | 9585973 | 0.11 | 0.12 | 1.3 | 0.02 | −0.02 |
| O | 9597139 | 0.05 (0.998) | 2.38 | 21.8 | 0.96 | 0.0 |
| P | 9607259 | 0.08 | 3.33 | 18.7 | 0.94 | 0.1 |
| Q | 9623459 | 0.27 (0.012) | −0.70 | 5.7 | 0.62 | −0.08 |
| R | 9636370 | 0.19 | −0.13 | 4.7 | 0.47 | −0.036 |
| S | 9702839 | 0.09 | 3.39 | 16.8 | 0.92 | 0.07 |
| T | 9766526 | 0.09 | 1.61 | 17.1 | 0.85 | 0.02 |
| U | 9827512 | 0.25 | −0.52 | 2.3 | 0.17 | −0.06 |
| V | 9895278 | 0.17 | 1.35 | 7.8 | 0.72 | 0.06 |
| Stn239 | 10201248 | 0.09 | 1.79 | 19.4 | 0.938 | 0.05 |
Bayes FST indicates the locus effect (α) and the significant P-values in the global comparison. Ln RH values are from the Barents Sea–L. Pulmanki comparison and underlined values were chosen for the multilocus Ln RH test. Note that locus H was monomorphic and it was excluded from the multilocus test. AR, allelic richness; HE, expected heterozygosity; and FIS, inbreeding coefficient.
References
- Beaumont, M. A., and D. J. Balding, 2004. Identifying adaptive genetic divergence among populations from genome scans. Mol. Ecol. 13: 969–980. [DOI] [PubMed] [Google Scholar]
- Beaumont, M. A., and R. A. Nichols, 1996. Evaluating loci for use in the genetic analysis of population structure. Proc. R. Soc. Lond. B 263: 1619–1626. [Google Scholar]
- Bell, M. A., and S. A. Foster (Editors), 1994. Introduction to the evolutionary biology of the threespine stickleback, pp. 1–26 in The Evolutionary Biology of the Threespine Stickleback. Oxford University Press, Oxford.
- Benson, D. A., I. Karsch-Mizrachi, D. J. Lipman, J. Ostell and D. L. Wheeler, 2007. GenBank. Nucleic Acids Res. 35: D21–D25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brownstein, M. J., J. D. Carpten and J. R. Smith, 1996. Modulation of non-templated nucleotide addition by taq DNA polymerase: primer modifications that facilitate genotyping. Biotechniques 20: 1004–1008. [DOI] [PubMed] [Google Scholar]
- Cano, J. M., C. Matsuba, H. Mäkinen and J. Merilä, 2006. The utility of QTL-linked markers to detect selective sweeps in natural populations—a case study of the EDA gene and a linked marker in threespine stickleback. Mol. Ecol. 15: 4613–4621. [DOI] [PubMed] [Google Scholar]
- Charlesworth, D., 2006. Balancing selection and its effects on sequences in nearby genome regions. PLoS Genet. 2: e64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Civetta, A., S. Rajakumar, B. Brouwers and J. Bacik, 2006. Rapid evolution and gene-specific patterns of selection for three genes of spermatogenesis in Drosophila. Mol. Biol. Evol. 23: 655–662. [DOI] [PubMed] [Google Scholar]
- Clark, R., E. Linton, J. Messing and J. Doebley, 2004. Pattern of diversity in the genomic region near the maize domestication gene tb1. Proc. Nat. Acad. Sci. USA 101: 700–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colosimo, P. F., C. L. Peichel, K. Nereng, B. K. Blackman, M. D. Shapiro et al., 2004. The genetic architecture of parallel armor plate reduction in threespine sticklebacks. PLoS Biol. 2: E109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colosimo, P. F., K. E. Hosemann, S. Balabhadra, G. Villarreal, Jr., M. Dickson et al., 2005. Widespread parallel evolution in sticklebacks by repeated fixation of ectodysplasin alleles. Science 307: 1928–1933. [DOI] [PubMed] [Google Scholar]
- Cornuet, J. M., and G. Luikart, 1996. Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics 144: 2001–2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Kovel, C. G., 2006. The power of allele frequency comparisons to detect the footprint of selection in natural and experimental situations. Genet. Sel. Evol. 38: 3–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dieringer, D., and C. Schlötterer, 2003. Microsatellite analyser (MSA): a platform independent analysis tool for large microsatellite data sets. Mol. Ecol. Notes 3: 167–169. [Google Scholar]
- DuMont, V., and C. Aquadro, 2005. Multiple signatures of positive selection downstream of notch on the X chromosome in Drosophila melanogaster. Genetics 171: 639–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elphinstone, M. S., G. N. Hinten, M. J. Anderson and C. J. Nock, 2003. An inexpensive and high-throughput procedure to extract and purify total genomic DNA for population studies. Mol. Ecol. Notes 3: 317–320. [Google Scholar]
- Goudet, J., 2001. FSTAT, a program to estimate and test gene diversities and fixation indices version 2.9.3. http://www.unil.ch/izea/softwares/fstat.html.
- Harr, B., M. Kauer and C. Schlötterer, 2002. Hitchhiking mapping: a population-based fine-mapping strategy for adaptive mutations in Drosophila Melanogaster. Proc. Natl. Acad. Sci. USA 99: 12949–12954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris, M. A., J. I. Clark, A. Ireland, J. Lomax, M. Ashburne et al., 2006. The gene ontology (GO) project in 2006. Nucleic Acids Res. 34: D322–D326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbard, T. J., B. L. Aken, K. Beal, B. Ballester, M. Caccamo et al., 2007. Ensembl 2007. Nucleic Acids Res. 35: D610–D617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ihle, S., I. Ravaoarimanana, M. Thomas and D. Tautz, 2006. An analysis of signatures of selective sweeps in natural populations of the house mouse. Mol. Biol. Evol. 23: 790–797. [DOI] [PubMed] [Google Scholar]
- Kauer, M. O., D. Dieringer and C. Schlötterer, 2003. A microsatellite variability screen for positive selection associated with the “out of Africa” habitat expansion of Drosophila melanogaster. Genetics 165: 1137–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohn, M. H., H. J. Pelz and R. K. Wayne, 2000. Natural selection mapping of the warfarin-resistance gene. Proc. Natl. Acad. Sci. USA 97: 7911–7915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leinonen, T., J. M. Cano, H. Mäkinen and J. Merilä, 2006. Contrasting patterns of body shape and neutral genetic divergence in marine and lake populations of threespine sticklebacks. J. Evol. Biol. 19: 1803–1812. [DOI] [PubMed] [Google Scholar]
- Lewontin, R. C., and J. Krakauer, 1973. Distribution of gene frequency as a test of the theory of the selective neutrality of polymorphisms. Genetics 74: 175–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay, T. F., 2001. The genetic architecture of quantitative traits. Annu. Rev. Genet. 35: 303–339. [DOI] [PubMed] [Google Scholar]
- Mäkinen, H. S., J. M. Cano and J. Merilä, 2006. Genetic relationships among marine and freshwater populations of the European three-spined stickleback (Gasterosteus aculeatus) revealed by microsatellites. Mol. Ecol. 15: 1519–1534. [DOI] [PubMed] [Google Scholar]
- Maynard Smith, J. M., and J. Haigh, 1974. The hitch–hiking effect of a favourable gene. Genet. Res. 23: 23–35. [PubMed] [Google Scholar]
- McKinnon, J. S., and H. D. Rundle, 2002. Speciation in nature: the threespine stickleback model systems. Trends Ecol. Evol. 17: 480–488. [Google Scholar]
- Nair, S., J. T. Williams, A. Brockman, L. Paiphun, M. Mayxay et al., 2003. A selective sweep driven by pyrimethamine treatment in southeast Asian malaria parasites. Mol. Biol. Evol. 20: 1526–1536. [DOI] [PubMed] [Google Scholar]
- Nash, D., S. Nair, M. Mayxay, P. N. Newton, J. P. Guthmann et al., 2005. Selection strength and hitchhiking around two anti-malarial resistance genes. Proc. R. Soc. Lond. B Biol. Sci. 272: 1153–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen, R., 2005. Molecular signatures of natural selection. Annu. Rev. Genet. 39: 197–218. [DOI] [PubMed] [Google Scholar]
- Nordborg, M., and S. Tavare, 2002. Linkage disequilibrium: what history has to tell us. Trends Genet. 18: 83–90. [DOI] [PubMed] [Google Scholar]
- Olsen, K. M., A. L. Caicedo, N. Polato, A. McClung, S. McCouch et al., 2006. Selection under domestication: evidence for a sweep in the rice waxy genomic region. Genetics 173: 975–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr, H. A., 2005. a The genetic basis of reproductive isolation: insights from Drosophila. Proc. Natl. Acad. Sci. USA 102(Suppl 1): 6522–6526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr, H. A., 2005. b The genetic theory of adaptation: a brief history. Nat. Rev. Genet. 6: 119–127. [DOI] [PubMed] [Google Scholar]
- Peichel, C. L., K. S. Nereng, K. A. Ohgi, B. L. Cole, P. F. Colosimo et al., 2001. The genetic architecture of divergence between threespine stickleback species. Nature 414: 901–905. [DOI] [PubMed] [Google Scholar]
- Pennings, P. S., and J. Hermisson, 2006. Soft sweeps III: the signature of positive selection from recurrent mutation. PLoS Genet. 2: 1998–2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollinger, J. P., C. D. Bustamante, A. Fledel-Alon, S. Schmutz, M. M. Gray et al., 2005. Selective sweep mapping of genes with large phenotypic effects. Genome Res. 15: 1809–1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pool, J., V. DuMont, J. Mueller and C. Aquadro, 2006. A scan of molecular variation leads to the narrow localization of a selective sweep affecting both Afrotropical and cosmopolitan populations of Drosophila melanogaster. Genetics 172: 1093–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlötterer, C., 2002. Towards a molecular characterization of adaptation in local populations. Curr. Opin. Genet. Dev. 12: 683–687. [DOI] [PubMed] [Google Scholar]
- Schlötterer, C., 2003. Hitchhiking mapping—functional genomics from the population genetics perspective. Trends Genet. 19: 32–38. [DOI] [PubMed] [Google Scholar]
- Schöfl, G., and C. Schlötterer, 2006. Microsatellite variation and differentiation in African and non-African populations of Drosophila simulans. Mol. Ecol. 15: 3895–3905. [DOI] [PubMed] [Google Scholar]
- Shapiro, M. D., M. E. Marks, C. L. Peichel, B. K. Blackman, K. S. Nereng et al., 2004. Genetic and developmental basis of evolutionary pelvic reduction in threespine sticklebacks. Nature 428: 717–723. [DOI] [PubMed] [Google Scholar]
- Stinchcombe, J. R., and H. E. Hoekstra, 2007. Combining population genomics and quantitative genetics: finding the genes underlying ecologically important traits. Heredity (in press). [DOI] [PubMed]
- Storz, J. F., 2005. Using genome scans of DNA polymorphism to infer adaptive population divergence. Mol. Ecol. 14: 671–688. [DOI] [PubMed] [Google Scholar]
- Storz, J. F., and H. E. Hoekstra, 2007. The study of adaptation and speciation in the genomic era. J. Mammal. 88: 1–4. [Google Scholar]
- Storz, J. F., S. J. Sabatino, F. G. Hoffmann, E. J. Gering, H. Moriyama et al., 2007. The molecular basis of high-altitude adaptation in deer mice. PLoS Genet. 3: e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutter, N. B., C. D. Bustamante, K. Chase, M. M. Gray, K. Zhao et al., 2007. A single IGF1 allele is a major determinant of small size in dogs. Science 316: 112–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teshima, K. M., G. Coop and M. Przeworski, 2006. How reliable are empirical genomic scans for selective sweeps? Genome Res. 16: 702–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornton, K., and J. Jensen, 2007. Controlling the false-positive rate in multilocus genome scans for selection. Genetics 175: 737–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungerer, M. C., L. C. Johnson and M. A. Herman, 2007. Ecological genomics: understanding gene and genome function in the natural environment. Heredity (in press). [DOI] [PubMed]
- Vitarius, J. A., E. Sehayek and J. L. Breslow, 2006. Identification of quantitative trait loci affecting body composition in a mouse intercross. Proc. Natl. Acad. Sci. USA 103: 19860–19865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weir, B. S., and C. C. Cockerham, 1984. Estimating F-statistics for the analysis of population structure. Evolution 38: 1358–1370. [DOI] [PubMed] [Google Scholar]
- Wiehe, T., V. Nolte, D. Zivkovic and C. Schlötterer, 2007. Identification of selective sweeps using a dynamically adjusted number of linked microsatellites. Genetics 175: 207–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Q., F. Zhang, X. H. Chen, Y. Q. Wang, W. Q. Wang et al., 2007. Rapid evolution, genetic variations, and functional association of the human spermatogenesis-related gene NYD-SP12. J. Mol. Evol. 65: 154–161. [DOI] [PubMed] [Google Scholar]