

Abstract
The Nogo-66 receptor (NgR) plays a critical role in restricting axon regeneration in the central nervous system. This inhibitory action is in part mediated by a neuronal receptor complex containing p75NTR, a multifunctional receptor also well known to trigger cell death upon binding to neurotrophins such as NGF. In the present study, we show that Pep4 and NEP1–40, which are two peptides derived from the Nogo-66 sequence that modulate NgR-mediated neurite outgrowth inhibition, prevent NGF-stimulated p75NTR-dependent death of cultured embryonic motor neurons. They also confer protection on spinal cord motor neurons after neonatal sciatic nerve axotomy. These findings demonstrate an as-yet-unknown function of NgR in maintaining neuronal survival that may be relevant for motor neuron development and degeneration.
Keywords: NGF, NgR, amyotrophic lateral sclerosis, axotomy, reticulon
The glycosylphosphatidylinositol (GPI)-anchored Nogo-66 receptor (NgR) binds various structurally unrelated proteins: Nogo-A, myelin-associated glycoprotein, and oligodendrocyte-myelin glycoprotein. These myelin-derived proteins inhibit neurite outgrowth in vitro, and Nogo-A also prevents axonal regeneration in vivo after injury to the adult mammalian CNS (1). NgR forms a ternary receptor complex with the leucine-rich repeat transmembrane protein LINGO-1 (2) and either the neurotrophin receptor p75NTR (3, 4) or the orphan TNF receptor family member TAJ/TROY (5, 6). Downstream effectors of this receptor complex have not been completely elucidated but may include RhoA/Rho-kinase (7), PKC (8), and transactivation of the EGF receptor (9). Although the role of NgR in restricting the growth and repair of CNS axons has been clearly demonstrated (10–12), knockout mice for NgR display only subtle regeneration of tracts upon spinal cord trauma (13, 14), most probably because of compensatory mechanisms.
p75NTR is a versatile protein known to potentiate the trophic actions of Trk receptor tyrosine kinases but also, under particular conditions, to trigger apoptosis upon binding to neurotrophins such as NGF and BDNF or their related precursors (15). Embryonic motor neurons, which lack TrkA receptors, express p75NTR in vitro and undergo apoptosis when cocultured on reactive astrocytes secreting NGF or treated with exogenous NGF in the presence of low steady-state concentrations of NO (16, 17). This apoptotic activity strictly depends on p75NTR, because cultured motor neurons from p75NTR knockout mice do not respond to NGF (17). In vivo spinal motor neurons also express p75NTR during the embryonic period of naturally occurring cell death, but its expression gradually ends after birth. In contrast, adult motor neurons can reexpress p75NTR after axotomy (18) or under disease conditions such as ALS (17). This phenomenon, together with increased NGF levels, has indeed been involved in the motor neuron death characteristic of such conditions (17, 19, 20).
All of the above studies clearly indicate that p75NTR can trigger either neuronal death, when stimulated by NGF, or neurite outgrowth inhibition, when bound to NgR. The cross-talk between these two pathways has not been explored so far. In the present study, we asked whether NgR could regulate motor neuron survival by modulating p75NTR-induced cell death under conditions in which both receptor partners should appear coexpressed. The results presented herein provide compelling evidence that two peptides derived from the Nogo-66 sequence can prevent p75NTR-dependent cell death of embryonic cultured motor neurons. These peptides also conferred neuroprotection after neonate sciatic nerve axotomy.
Results
The NgR Pathway Is Expressed and Functional in Motor Neurons.
As a first step toward understanding the interactions between NgR and p75NTR pathways, we determined whether both receptors were coexpressed in our experimental paradigm. In embryonic day 16 (E16) mouse embryos, choline acetyl transferase (ChAT) immunoreactive motor neurons in the ventral horns of the spinal cord were also positive for NgR (Fig. 1A), and this staining persisted in neonate and adult spinal cord motor neurons [supporting information (SI) Fig. 6]. We also checked which components of the NgR receptor complex were present in cultures of purified embryonic motor neurons and NSC34 cells, a motor neuron-like cell line. NgR, LINGO-1, and p75NTR mRNAs were all expressed in these cultures; in turn, TAJ/TROY mRNA, present in whole spinal cord extracts, appeared undetectable in vitro (Fig. 1B). Moreover, all cultured motor neurons presented with both p75NTR and NgR immunoreactivity (Fig. 1C; two independent experiments were analyzed), thus indicating that NgR activation could involve p75NTR in this paradigm.
Fig. 1.

The Nogo-66/NgR pathway is expressed and functional in embryonic motor neurons. (A) Photomicrographs showing ChAT (green) and NgR (red) immunoreactivity in the lumbar spinal cord of embryonic (E16) rats. Note that ChAT and NgR mostly colocalize within the same motor neurons (merge), except for some few NgR-positive cells that are not stained for ChAT (asterisks). (Scale bar, 25 μm.) (B) RT-PCR showing the mRNA levels of the different constituents of the NgR signaling platform in cultured rat embryonic (E15) motor neurons (MNs), motor neuron-like NSC34 cells, and adult mouse spinal cord (SC). 18S rRNA levels are shown as internal control. (C) Photomicrographs showing p75NTR (green) and NgR (red) immunoreactivity in cultured rat embryonic (E15) motor neurons. (Scale bar, 25 μm.) (D) Total neurite length of cultured rat embryonic (E15) motor neurons treated for 24 h with Pep4 and/or NEP1–40 at 100 nM (+) or 1 μM (++). Motor neurons were cultured in the presence of GDNF. In some cases, motor neurons were pretreated with PI-PLC. Data are presented as percentages of total neurite length in the control condition (GDNF alone) (**, P < 0.01; ***, P < 0.001 vs. GDNF; #, P < 0.05 vs. Pep4; n = 3 independent experiments).
To ascertain whether NgR/LINGO-1/p75NTR signaling is functionally active in cultured embryonic motor neurons, we measured neurite outgrowth in response to Pep4, a peptide corresponding to residues 31–55 of Nogo-66 (21), and NEP1–40, another fragment of Nogo-66 that antagonizes Nogo-66-induced growth cone collapse (11). Total neurite length was reduced by ≈40% in the presence of 100 nM Pep4, and this effect was abolished by excess NEP1–40 (Fig. 1D). Pep4 was inefficient to reduce neurite length when motor neurons were phosphatidylinositol-specific phospholipase C (PI-PLC)-pretreated to cleave GPI-anchored proteins from the cell surface (22) (Fig. 1D). Together, these findings indicate that the NgR pathway leading to neurite outgrowth inhibition is functional in cultured embryonic motor neurons.
Nogo-66 Peptides Prevent p75NTR-Dependent Motor Neuron Death in Vitro.
We next turned to assess whether Nogo-66 peptides could affect motor neuron survival when added at the same doses as those found to regulate neurite outgrowth. Neither 100 nM Pep4 nor 100 nM NEP1–40 acted on motor neuron survival under normal culture conditions, i.e., in the presence of GDNF (Fig. 2A). These results are reminiscent of those found by Domeniconi et al. (23), who showed no proapoptotic effect of myelin-associated glycoprotein, a NgR ligand, or myelin extracts when treating cerebellar granule neurons under normal conditions. Pep4 and NEP1–40 did not alter the neuronal death observed upon GDNF deprivation (Fig. 2A), which suggests that the NgR pathway does not seem to interfere by itself with other mechanisms controlling survival, thus allowing testing of whether it can interact with p75NTR-induced cell death.
Fig. 2.

Pep4 and NEP1–40 prevent p75NTR-dependent motor neuron death. (A) Survival rates of cultured rat embryonic (E15) motor neurons treated for 24 h with 100 nM Pep4 or NEP1–40 in the presence (GDNF) or absence (none) of GDNF. Pep4 and NEP1–40 did not modify motor neuron survival in any condition (P > 0.05; n = 5 independent experiments). (B) Cultures were treated for 24 h with 100 nM Pep4 or NEP1–40 in the presence of NGF or NGF plus NOC-18 (NO generator) (*, P < 0.05 vs. control; #, P < 0.05 vs. NGF+NO alone; n = 5 independent experiments). (C) Rat embryonic (E15) motor neurons were cocultured on rat astrocyte monolayers and treated for 24 h with 100 nM Pep4 or NEP1–40 in the presence (NGF) or absence (none) of NGF (*, P < 0.05 vs. control; #, P < 0.05 vs. NGF; n = 3 independent experiments). (D) Cultures were treated for 24 h with 100 nM Pep4 or NEP1–40 and lumbar spinal cord lysates from symptomatic SOD1(G93A) or nontransgenic (WT) littermates in the presence of NOC-18 (NO generator) (*, P < 0.05 vs. G93A; #, P < 0.05 vs. G93A+NO; n = 3 independent experiments). (B–D) GDNF was present in all experimental conditions.
We have shown that 100 ng/ml NGF, in combination with low steady-state concentrations of NO (<50 nM), triggers the death of ≈45% of cultured embryonic motor neurons (17). In the present study, this effect was completely prevented by Pep4 or NEP1–40 administration (Fig. 2B). Identical results were obtained when NGF-treated motor neurons were cocultured with astrocytes as natural source of NO (17) (Fig. 2C). We had also shown in previous experiments that spinal cord extracts from SOD1(G93A) mice, a transgenic model for ALS, contain significant amounts of NGF-like activity and trigger p75NTR-dependent motor neuron death in the presence of low steady-state concentrations of NO (17). Here, extracts of ALS mouse spinal cord, but not from control littermates, decreased motor neuron survival by ≈40% under the described conditions, and this effect was prevented by adding Pep4 or NEP1–40 (Fig. 2D). Thus, Nogo-66 peptides have the ability to counteract the action of p75NTR in these several in vitro experimental paradigms and, surprisingly, both of them exhibit the same protection against p75NTR-mediated cell death, which clearly contrasts with their opposite effects on neurite outgrowth (Fig. 1D).
Neuroprotective Effects of Nogo-66 Peptides Are Mediated by NgR.
To ascertain whether the protective effects on cultured embryonic motor neurons were mediated directly by NgR, we first removed GPI-anchored proteins by PI-PLC treatment and looked at the effects of 100 nM Pep4 on p75NTR-dependent cell death. PI-PLC treatment did not modify motor neuron survival on its own but hampered Pep4 to restore neuronal survival in response to NGF (Fig. 3A). According to studies by Fournier et al. (12), the soluble form of NgR may bind to Nogo and prevent its binding to full-length Nogo receptors on the neuronal cell surface. Alternatively, it may also interact with surface-bound NgR and prevent receptor oligomerization or NgR interaction with the signal-transducing receptor subunit. We then challenged motor neuron cultures with NGF and NO in the presence of Nogo-66 peptides, soluble NgR, or both. Soluble NgR at 27 nM alone was unable to counteract NGF-mediated motor neuron death but completely prevented the protective actions of Pep4 and NEP1–40 (Fig. 3B). Altogether, the present data strongly suggest that Pep4 and NEP1–40 act on NgR signaling to avoid cell death.
Fig. 3.

The neuroprotective effect of Pep4 and NEP1–40 against p75NTR-dependent cell death is mediated by NgR. (A) Survival rates of cultured rat embryonic (E15) motor neurons treated for 24 h with 100 nM Pep4 with or without PI-PLC pretreatment in the presence of NGF plus NOC-18 (NO generator) (*, P < 0.05 vs. PIPLC alone; #, P < 0.05 vs. Pep4 plus NGF+NO; n = 3 independent experiments). (B) Cultures were treated for 24 h with 100 nM Pep4, 100 nM NEP1–40, and/or 27 nM soluble NgR in the presence or absence of NGF plus NOC-18 (NO generator) (*, P < 0.05 versus GDNF alone; #, P < 0.05 vs. corresponding control without NgR; n = 3 independent experiments). (C) Western blot showing NgR protein levels in NSC34 motor neuron-like cells after 24 h of transfection with antisense or corresponding scrambled NgR oligonucleotides 1 and 2. Actin immunoreactivity was assayed to ensure equal amounts of loaded proteins. (D) Survival rates of cultured rat embryonic (E15) motor neurons transfected with antisense or corresponding scrambled NgR oligonucleotides 1 and 2, and treated for 24 h with 100 nM Pep4 or NEP1–40 in the presence or absence of NGF plus NOC-18 (NO generator). (*, P < 0.05 vs. GDNF alone; #, P < 0.05 vs. corresponding control; three independent experiments). (A, B, and D) GDNF was present in all experimental conditions, unless otherwise indicated.
To further support this pharmacological evidence, we wished to knock down NgR expression in cultured embryonic motor neurons using an antisense oligonucleotide-based technique, as described (24). As a control of sequence specificity, we determined that transfection of NSC34 cells with two different NgR antisense oligonucleotides efficiently reduced NgR protein levels, compared with the corresponding nonspecific scrambled oligonucleotides (Fig. 3C). In the absence of Nogo-66 peptides, transfection of embryonic motor neuron cultures with either specific or scrambled NgR oligonucleotides did not modify the rate of cell death induced by NGF+NO (Fig. 3D), thus showing that NgR in its inactive form does not interfere with the death-promoting effect of p75NTR. When NgR expression was not compromised (i.e., in the presence of scrambled oligonucleotides), Pep4 and NEP1–40 reversed the toxic effect exerted by NGF+NO. In contrast, Nogo-66 peptides were completely ineffective in the presence of each one of the NgR antisense oligonucleotides (Fig. 3D). These findings, combined with the negative actions of PI-PLC and soluble NgR on the protective role of Pep4 and NEP1–40, clearly demonstrate that NgR is directly involved in the neuroprotection conferred by these peptides against p75NTR-mediated cell death.
Nogo-66 Peptides Antagonize p75-Dependent Motor Neuron Death in Vivo.
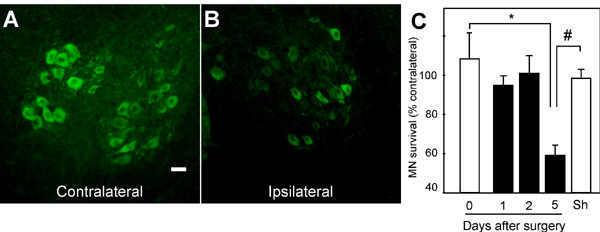
Neonatal motor neuron death after sciatic nerve axotomy has been typically described as depending on p75NTR activation (20, 25) and thus constitutes a very valuable model to study whether our Nogo-66 peptides are able to promote motor neuron survival in vivo. A time course of the neuronal loss observed in the transected side, compared with the contralateral one, showed 40% of motor neuron death within 5 days after axotomy (SI Fig. 7), which is consistent with other studies (26). Identical results were obtained when counting either ChAT-immunoreactive or large-sized toluidine blue-stained motor neurons. At this time point, a sharp up-regulation of several denervation-dependent genes, including nicotinic acetylcholine receptor α-subunit, myogenin and muscle-specific kinase, was found in the denervated hindlimb skeletal muscles of axotomized mice, thus ensuring the effectiveness of the nerve transection (Fig. 4I).
Fig. 4.

Pep4 and NEP1–40 prevent motor neuron death after sciatic nerve axotomy in neonate mice. (A–F) Photomicrographs showing ChAT immunoreactivity in the lumbar spinal cord of neonate mice. The sciatic nerve was axotomized and animals were treated during 5 days with vehicle (A and B), 11.6 mg/kg body mass Pep4 (C and D), or NEP1–40 (E and F). The sides contralateral (A, C, and E) and ipsilateral (B, D, and F) to the lesion are shown. (Scale bar, 25 μm.) (G and H) Quantification of the numbers of surviving motor neurons as shown in A–F after ChAT immunoreactivity (G) and toluidine blue staining (H). Data are presented as percentages of motor neurons in the corresponding side contralateral to the lesion [*, P < 0.05 vs. vehicle (Vh); n = 9–11 mice per group]. (I) Representative RT-PCR showing the mRNA levels of nicotinic acetylcholine receptor α-subunit (AchRα), myogenin (Myog), and muscle-specific kinase (Musk) in the hindlimb muscles contralateral (C) and ipsilateral (I) to the lesion. 18S rRNA levels are shown as internal control.
To determine whether s.c. injections of Nogo-66 peptides could be used in vivo, we biotinylated Pep4 and NEP1–40 and administered them to neonate animals (postnatal day 2) at 11.6 mg/kg body mass, a dose found to be efficient in other studies (27). Biotinylated peptides were then revealed using avidin-FITC (SI Fig. 8A). Upon biotinylated Pep4 or NEP1–40 injection, cells from the ventral horns of the spinal cord stained intensely, indicating that the Nogo-66 peptides could cross the blood–brain barrier easily. To determine whether these Nogo-66 peptides displayed neuroprotective potential in vivo after sciatic nerve axotomy, we administered daily Pep4 or NEP1–40 at the dose found to cross the blood–brain barrier. This dose did not modify the absolute number of motor neurons in the contralateral side of axotomized mice (SI Fig. 8B), thus attesting to its innocuity. In contrast, both Nogo-66 peptides abolished postaxotomy ipsilateral motor neuron loss compared with vehicle-treated animals (Fig. 4 A–F). This effect was confirmed twice by measuring ChAT-immunoreactive (Fig. 4G) and large-sized toluidine blue-stained motor neurons (Fig. 4H).
As a control of specificity, we used peptides corresponding to Pep4 and NEP1–40 in the Nogo-66-like domain of RTN3, which is another member of the reticulon family with very similar structural organization as Nogo but does not display any neurite outgrowth inhibitory properties (21). RTN3-derived Pep4 and NEP1–40 could not prevent NGF/p75NTR-dependent motor neuron death in vitro, nor rescue motor neurons from neonatal axotomy (Fig. 5). These findings further support the specific action of Nogo-derived Pep4 and NEP1–40 on the death-promoting effect of p75NTR.
Fig. 5.

RTN3-derived Pep4 and NEP1–40 do not prevent motor neuron death after p75NTR activation in vitro or sciatic nerve axotomy in neonate mice. (A) Survival rates of cultured rat embryonic (E15) motor neurons treated for 24 h with 100 nM Pep4 or NEP1–40 in the presence of NGF plus NOC-18 (NO generator). Pep4 and NEP1–40 were derived from the Nogo-66 sequence of Nogo or RTN3 (*, P < 0.05 vs. control; n = 3 independent experiments). (B) Photomicrographs showing ChAT immunoreactivity in the lumbar spinal cord of neonate mice. The sciatic nerve was axotomized and animals were treated during 5 days with vehicle, or 11.6 mg/kg body mass Pep4 or NEP1–40. The sides contralateral and ipsilateral to the lesion are shown. (Scale bar, 25 μm.) (C) Quantification of the numbers of surviving motor neurons as shown in B. Data are presented as percentages of motor neurons in the corresponding side contralateral to the lesion [*, P < 0.05 vs. vehicle (Vh); n = 5 mice per group].
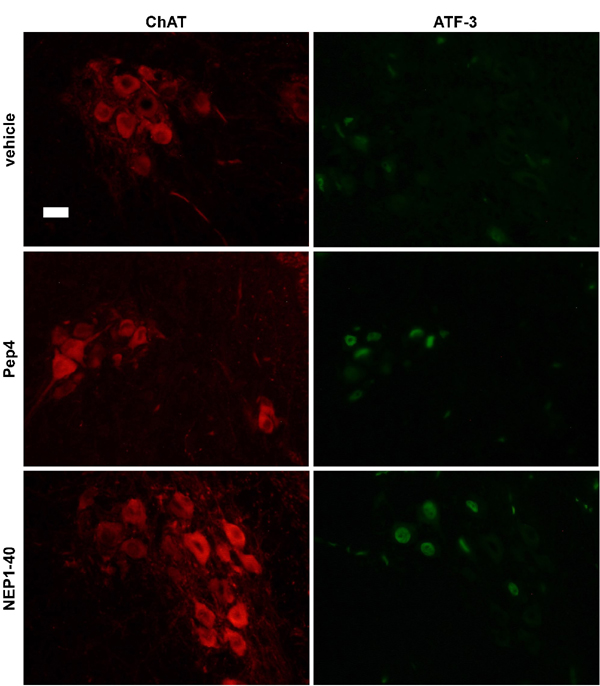
To gain further insight into the protective action of Pep4 and NEP1–40, we performed double immunolabeling using ChAT (to identify motor neurons) and ATF3 antibodies (SI Fig. 9). It has been recently shown that the up-regulation of the transcription factor ATF3 precedes motor neuron death after axotomy and in ALS (mutant SOD1) mice as a marker of the degenerative process (28). Under control conditions, ATF3 induction was detected 48 h after lesion, and all ATF3-positive motor neurons disappeared 3 days afterwards. In contrast, the number of ATF3-positive motor neurons stayed high in Pep4- and NEP1–40 treated mice. Because ATF3 up-regulation has been associated with increasing the intrinsic growth state of injured neurons (29), our findings suggest that the protection offered by the two Nogo-66 peptides maintains the number of alive motor neurons and hence allows the activation of regenerative efforts in the lesioned motor neurons.
Discussion
The evidence we put forward in this study shows a function of NgR in maintaining motor neuron survival in vitro and in vivo. Pep4 and NEP1–40, two peptides derived from the Nogo-66 sequence known to modulate NgR-mediated neurite outgrowth inhibition, are able to overcome the death-promoting effect of NGF on p75NTR-expressing motor neurons. They also significantly prevent motor neuron loss in neonate mouse spinal cord after sciatic nerve axotomy, an experimental paradigm characterized by p75NTR-dependent death of the injured motor neurons.
Pep4 activates the inhibitory function of NgR (7, 11, 21, 27), whereas NEP1–40 competes for binding of Nogo-66 to its receptor and hence promotes axonal regeneration (11, 27). In addition to these actions, we show here that Pep4 and, surprisingly, also NEP1–40 protected cultured embryonic motor neurons against NGF/p75NTR-induced cell death. The result of the treatment with NEP1–40 was clearly opposed to that found with soluble NgR, another well documented NgR antagonist (12) that triggered a massive increase in neurite outgrowth (data not shown) but had no effect on p75NTR-dependent motor neuron death. Thus, NEP1–40 does not always function as an antagonist for NgR, as previous studies had determined by its ability to prevent inhibition of neurite extension (11). Three sets of data attest to the specificity of the protective action exerted by NgR and Nogo-66 peptides. First, they could not rescue motor neurons from NGF/p75NTR-induced cell death when GPI-anchored NgR had been previously removed by PI-PLC treatment. Second, knocking down NgR expression using two different specific antisense oligonucleotides abolished the protection offered by Pep4 and NEP1–40. Finally, RTN3-derived Pep4 and NEP1–40 were also unable to prevent motor neuron death. In vivo, s.c.-administered Pep4 and NEP1–40 offered protection to neonate spinal cord motor neurons after sciatic nerve axotomy, whereas the two homolog peptides corresponding to Pep4 and NEP1–40 in the RTN3 sequence had no effect. Altogether, our findings strongly suggest that both Nogo-66 peptides exhibit a receptor-specific biological activity on their own under conditions where the death-promoting action of p75NTR would have killed the stressed motor neurons.
Many studies support the key role played by myelin NgR in restricting axonal growth after injury to the adult CNS (1). The results we present here may appear perplexing, because the same receptor that is limiting neurite extension can also promote neuronal survival. However, the nonpermissive role of NgR in axonal growth does not always function as such but can be modulated by intra- and extracellular signals (30, 31). In addition, it has been suggested that NgR could have a physiological role in the intact CNS unrelated to injury and regeneration (32). The embryonic expression of NgR observed in the present study, which is consistent with other reports (33, 34), suggested the neuroprotective effect of Pep4 and NEP1–40 could have been unperceived when focusing on the role of NgR in the adult. Our findings would thus favor the hypothesis that the major function of the p75NTR/NgR complex should involve, at least in embryonic and early postnatal life, the regulation of cell death.
The protection offered by NgR against p75NTR-induced cell death might also play an important role in motor neuron diseases. In ALS, the most common human adult motor neuron disease, reactive astrocytes produce NGF and surround degenerating motor neurons that express p75NTR. Most importantly, spinal cord lysates from symptomatic SOD1(G93A) mice, a transgenic model for ALS, stimulate motor neuron death, and this effect is prevented by NGF or p75NTR blocking antibodies (17). In this study, we show that Pep4 and NEP1–40 protect cultured motor neurons against the toxic effects of SOD1(G93A) spinal cord lysates, which prompt us to suggest that NgR could sustain, at least for a given period, the survival of motor neurons afflicted by ALS. In this regard, our recent work has shown that the genetic ablation of Nogo-A, one of the natural ligands of NgR, delays motor neuron loss in SOD1(G86R) mice, another model for ALS (35). These findings could seem paradoxical when compared with those presented in this report, because the absence of Nogo-A ligand in the knockout mice could prevent, at least in part, recruitment of p75NTR by Nogo-A-bound NgR and hence alleviate its death-promoting effect. One possible explanation of these results is that ablation of Nogo-A highly induces Nogo-B expression in the Nogo-A knockout mice (36, 37), therefore presumably leading to increased levels of Nogo-66 sequences necessary for the neuroprotective action of NgR.
Our present findings add an aspect to the spectrum of different biological effects mediated by p75NTR. We have demonstrated that in cells lacking TrkA receptors, such as motor neurons, the stimulation of NgR functions as a negative regulator of p75NTR-induced cell death. This study not only provides insight into a previously undescribed function for NgR in the CNS but also could have important implications for the design of therapeutic strategies to fight against motor neuron diseases using the numerous pharmacological agents targeting that receptor.
Methods
In Vitro Cell Counting.
Treatments of motor neuron and motor neuron/astrocyte cultures are described in SI Text. Total neurite length per motor neuron was determined by measuring the length of extensions longer than two cell bodies in 20–30 motor neurons per experimental condition using NIH Image version 1.62 software (National Institutes of Health). Motor neuron survival was assessed by directly counting all cells displaying intact neurites longer than 4 cells in diameter as described (17). Quantification was performed by an experiment-blinded observer.
Antisense Oligonucleotide Transfection.
Transfection with NgR antisense oligonucleotides was performed as described (24). Briefly, 5 μM HPLC-purified phosphorothioate antisense or scrambled oligonucleotide (Integrated DNA Technologies) was added to the cell suspension of purified embryonic motor neurons with repeated pipetting before seeding. Oligonucleotides were present during the time of culture. Using this technique, oligonucleotide uptake efficiency in embryonic motor neurons was >96% (24). Transfection of NSC34 cells was performed as described (38). Oligonucleotide sequences were as follows: NgR antisense 1, 5′-CTCCGGAGGACGCCCTCTTCAT-3′, NgR scrambled 1, 5′-ACCGATTTAGCGTCCGCCCTCG-3′, NgR antisense 2, 5′- CAGCCACAGGATGGTGAGATT -3′, and NgR scrambled 2, 5′- GACTAATCACTGAGTGAGGCG -3′.
Neonatal Sciatic Nerve Axotomy.
Animal manipulation followed current European Union regulations and was performed under the supervision of authorized investigators. Pups of the FVB/N strain younger than 5 days were hypothermically anesthesized, and the right sciatic nerve was exposed at the midthigh level and dissected sharply with microscissors. Muscles and skin incision were sutured, and animals were allowed to recover. Sham-operated mice served as control. Treatments consisted of daily s.c. injections with either vehicle (10% DMSO in PBS) or 11.6 mg/kg body mass Pep4 or NEP1–40, as reported (27). Five days after axotomy, spinal cords were processed for histological analysis, and hindlimb muscles were stored at −80°C until use. To perform motor neuron counts, lumbar spinal cord segments L3–L5 were cut into 10-μm cryostat sections, mounted in series of 10 consecutive slides, and stained with 1% toluidine blue in 5% sodium borate. Countings were performed at ×200 in the ventral horns of at least seven nonadjacent sections per animal. Special care was taken to compare anatomically matched sections between different animals. The cross-sectional areas of the stained cells were measured with NIH Image software, and only those cells with an area ≥1,000 μm2 were considered motor neurons (39). To verify this criterion, motor neurons on adjacent sections were immunostained for ChAT (SI Text). Counts of ChAT-positive motor neurons were performed by an independent experiment-blinded observer. Motor neuron numbers corresponding to the sciatic nerve transected side were expressed as a percentage of the contralateral side. Biotinylated Pep4 and NEP1–40 were obtained by using the EZ-link sulfo-NHS biotinylation kit (Pierce) following the manufacturer's intructions.
Immunohistochemistry, Western Blot Analysis, and RT-PCR.
Immunochemical and RT-PCR techniques are described in SI Text.
Statistical Analysis.
Data are expressed as means ± SEM. One-way ANOVA followed by the Newman–Keuls test was used to evaluate the significance of the effects on motor neuron survival and neurite length. The Kruskal–Wallis test, followed by Dunn's multiple comparison test, was used to assess for differences in the in vivo experiments. In all cases, P <0.05 was considered significant.
Supplementary Material
ACKNOWLEDGMENTS.
We thank Dr. Neil Cashman (University of British Columbia, Vancouver, BC, Canada) for providing the NSC34 cells. We acknowledge Mrs. A. Picchinenna and Mrs. M. J. Ruivo for excellent technical assistance. This work was supported by grants from Fondation pour la Recherche sur le Cerveau (L. Dupuis); Association Française contre les Myopathies, Biovalley, and Association pour la Recherche et le Développement de Moyens de Lutte contre les Maladies Neurodégénératives (J.P.L.); Association pour la Recherche sur la Sclérose Latérale Amyotrophique (J.P.L., L. Dupuis, and J.-L.G.d.A.); the PEDECIBA program (M.P.); PDT-Dinacyt 130/29 (L.B.); and Swiss National Science Foundation, the National Center of Competence in Research on Neural Plasticity and Repair, the Spinal Cord Consortium of the Christopher Reeve Foundation, and the European Union NeuroNetwork Project (M.E.S.). The collaboration between the laboratories of J.-P.L. and L.B. is supported by a grant from ECOS Sud (Foreign Affairs Ministry, France).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/cgi/content/full/0703842105/DC1.
References
- 1.Schwab ME. Curr Opin Neurobiol. 2004;14:118–124. doi: 10.1016/j.conb.2004.01.004. [DOI] [PubMed] [Google Scholar]
- 2.Mi S, Lee X, Shao Z, Thill G, Ji B, Relton J, Levesque M, Allaire N, Perrin S, Sands B, et al. Nat Neurosci. 2004;7:221–228. doi: 10.1038/nn1188. [DOI] [PubMed] [Google Scholar]
- 3.Wong ST, Henley JR, Kanning KC, Huang KH, Bothwell M, Poo MM. Nat Neurosci. 2002;5:1302–1308. doi: 10.1038/nn975. [DOI] [PubMed] [Google Scholar]
- 4.Wang KC, Kim JA, Sivasankaran R, Segal R, He Z. Nature. 2002;420:74–78. doi: 10.1038/nature01176. [DOI] [PubMed] [Google Scholar]
- 5.Shao Z, Browning JL, Lee X, Scott ML, Shulga-Morskaya S, Allaire N, Thill G, Levesque M, Sah D, McCoy JM, et al. Neuron. 2005;45:353–359. doi: 10.1016/j.neuron.2004.12.050. [DOI] [PubMed] [Google Scholar]
- 6.Park JB, Yiu G, Kaneko S, Wang J, Chang J, He XL, Garcia KC, He Z. Neuron. 2005;45:345–351. doi: 10.1016/j.neuron.2004.12.040. [DOI] [PubMed] [Google Scholar]
- 7.Yamashita T, Tohyama M. Nat Neurosci. 2003;6:461–467. doi: 10.1038/nn1045. [DOI] [PubMed] [Google Scholar]
- 8.Sivasankaran R, Pei J, Wang KC, Zhang YP, Shields CB, Xu XM, He Z. Nat Neurosci. 2004;7:261–268. doi: 10.1038/nn1193. [DOI] [PubMed] [Google Scholar]
- 9.Koprivica V, Cho KS, Park JB, Yiu G, Atwal J, Gore B, Kim JA, Lin E, Tessier-Lavigne M, Chen DF, He Z. Science. 2005;310:106–110. doi: 10.1126/science.1115462. [DOI] [PubMed] [Google Scholar]
- 10.Li S, Liu BP, Budel S, Li M, Ji B, Walus L, Li W, Jirik A, Rabacchi S, Choi E, et al. J Neurosci. 2004;24:10511–10520. doi: 10.1523/JNEUROSCI.2828-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.GrandPre T, Li S, Strittmatter SM. Nature. 2002;417:547–551. doi: 10.1038/417547a. [DOI] [PubMed] [Google Scholar]
- 12.Fournier AE, Gould GC, Liu BP, Strittmatter SM. J Neurosci. 2002;22:8876–8883. doi: 10.1523/JNEUROSCI.22-20-08876.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim JE, Liu BP, Park JH, Strittmatter SM. Neuron. 2004;44:439–451. doi: 10.1016/j.neuron.2004.10.015. [DOI] [PubMed] [Google Scholar]
- 14.Zheng B, Atwal J, Ho C, Case L, He XL, Garcia KC, Steward O, Tessier-Lavigne M. Proc Natl Acad Sci USA. 2005;102:1205–1210. doi: 10.1073/pnas.0409026102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nykjaer A, Willnow TE, Petersen CM. Curr Opin Neurobiol. 2005;15:49–57. doi: 10.1016/j.conb.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 16.Cassina P, Pehar M, Vargas MR, Castellanos R, Barbeito AG, Estevez AG, Thompson JA, Beckman JS, Barbeito L. J Neurochem. 2005;93:38–46. doi: 10.1111/j.1471-4159.2004.02984.x. [DOI] [PubMed] [Google Scholar]
- 17.Pehar M, Cassina P, Vargas MR, Castellanos R, Viera L, Beckman JS, Estevez AG, Barbeito L. J Neurochem. 2004;89:464–473. doi: 10.1111/j.1471-4159.2004.02357.x. [DOI] [PubMed] [Google Scholar]
- 18.Rende M, Giambanco I, Buratta M, Tonali P. J Comp Neurol. 1995;363:249–263. doi: 10.1002/cne.903630207. [DOI] [PubMed] [Google Scholar]
- 19.Turner BJ, Cheah IK, Macfarlane KJ, Lopes EC, Petratos S, Langford SJ, Cheema SS. J Neurochem. 2003;87:752–763. doi: 10.1046/j.1471-4159.2003.02053.x. [DOI] [PubMed] [Google Scholar]
- 20.Wiese S, Metzger F, Holtmann B, Sendtner M. Eur J Neurosci. 1999;11:1668–1676. doi: 10.1046/j.1460-9568.1999.00585.x. [DOI] [PubMed] [Google Scholar]
- 21.GrandPre T, Nakamura F, Vartanian T, Strittmatter SM. Nature. 2000;403:439–444. doi: 10.1038/35000226. [DOI] [PubMed] [Google Scholar]
- 22.Fournier AE, GrandPre T, Strittmatter SM. Nature. 2001;409:341–346. doi: 10.1038/35053072. [DOI] [PubMed] [Google Scholar]
- 23.Domeniconi M, Zampieri N, Spencer T, Hilaire M, Mellado W, Chao MV, Filbin MT. Neuron. 2005;46:849–855. doi: 10.1016/j.neuron.2005.05.029. [DOI] [PubMed] [Google Scholar]
- 24.Pehar M, Vargas MR, Robinson KM, Cassina P, England P, Beckman JS, Alzari PM, Barbeito L. Free Radic Biol Med. 2006;41:1632–1644. doi: 10.1016/j.freeradbiomed.2006.08.010. [DOI] [PubMed] [Google Scholar]
- 25.Lowry KS, Murray SS, Coulson EJ, Epa R, Bartlett PF, Barrett G, Cheema SS. J Neurosci Res. 2001;64:11–17. doi: 10.1002/jnr.1048. [DOI] [PubMed] [Google Scholar]
- 26.Sun W, Oppenheim RW. Mol Cell Neurosci. 2003;24:875–886. doi: 10.1016/s1044-7431(03)00219-7. [DOI] [PubMed] [Google Scholar]
- 27.Li S, Strittmatter SM. J Neurosci. 2003;23:4219–4227. doi: 10.1523/JNEUROSCI.23-10-04219.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vlug AS, Teuling E, Haasdijk ED, French P, Hoogenraad CC, Jaarsma D. Eur J Neurosci. 2005;22:1881–1894. doi: 10.1111/j.1460-9568.2005.04389.x. [DOI] [PubMed] [Google Scholar]
- 29.Seijffers R, Mills CD, Woolf CJ. J Neurosci. 2007;27:7911–7920. doi: 10.1523/JNEUROSCI.5313-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fischer D, He Z, Benowitz LI. J Neurosci. 2004;24:1646–1651. doi: 10.1523/JNEUROSCI.5119-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hasegawa Y, Fujitani M, Hata K, Tohyama M, Yamagishi S, Yamashita T. J Neurosci. 2004;24:6826–6832. doi: 10.1523/JNEUROSCI.1856-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hunt D, Coffin RS, Anderson PN. J Neurocytol. 2002;31:93–120. doi: 10.1023/a:1023941421781. [DOI] [PubMed] [Google Scholar]
- 33.Josephson A, Trifunovski A, Widmer HR, Widenfalk J, Olson L, Spenger C. J Comp Neurol. 2002;453:292–304. doi: 10.1002/cne.10408. [DOI] [PubMed] [Google Scholar]
- 34.Al Halabiah H, Delezoide AL, Cardona A, Moalic JM, Simonneau M. Gene Expr Patterns. 2005;5:561–568. doi: 10.1016/j.modgep.2004.10.010. [DOI] [PubMed] [Google Scholar]
- 35.Jokic N, Gonzalez de Aguilar JL, Dimou L, Lin S, Fergani A, Ruegg MA, Schwab ME, Dupuis L, Loeffler JP. EMBO Rep. 2006;7:1162–1167. doi: 10.1038/sj.embor.7400826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dimou L, Schnell L, Montani L, Duncan C, Simonen M, Schneider R, Liebscher T, Gullo M, Schwab ME. J Neurosci. 2006;26:5591–5603. doi: 10.1523/JNEUROSCI.1103-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Simonen M, Pedersen V, Weinmann O, Schnell L, Buss A, Ledermann B, Christ F, Sansig G, van der Putten H, Schwab ME. Neuron. 2003;38:201–211. doi: 10.1016/s0896-6273(03)00226-5. [DOI] [PubMed] [Google Scholar]
- 38.Trinh E, Boutillier AL, Loeffler JP. Mol Cell Neurosci. 2001;17:342–353. doi: 10.1006/mcne.2000.0928. [DOI] [PubMed] [Google Scholar]
- 39.Dupuis L, Oudart H, Rene F, Gonzalez de Aguilar JL, Loeffler JP. Proc Natl Acad Sci USA. 2004;101:11159–11164. doi: 10.1073/pnas.0402026101. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}