

Abstract
Intermediates along a protein's folding pathway can play an important role in its biology. Previous kinetics studies have revealed an early folding intermediate for T4 lysozyme, a small, well-characterized protein composed of an N-terminal and a C-terminal subdomain. Pulse-labeling hydrogen exchange studies suggest that residues from both subdomains contribute to the structure of this intermediate. On the other hand, equilibrium native state hydrogen experiments have revealed a high-energy, partially unfolded form of the protein that has an unstructured N-terminal subdomain and a structured C-terminal subdomain. To resolve this discrepancy between kinetics and equilibrium data, we performed detailed kinetics analyses of the folding and unfolding pathways of T4 lysozyme, as well as several point mutants and large-scale variants. The data support the argument for the presence of two distinct intermediates, one present on each side of the rate-limiting transition state barrier. The effects of circular permutation and site-specific mutations in the wild-type and circular permutant background, as well as a fragment containing just the C-terminal subdomain, support a model for the unfolding intermediate with an unfolded N-terminal and a folded C-terminal subdomain. Our results suggest that the partially unfolded form identified by native state hydrogen exchange resides on the folded side of the rate-limiting transition state and is, therefore, under most conditions, a “hidden” intermediate.
Keywords: T4 lysozyme, protein folding, intermediates, chevron plot
Partially folded forms of proteins are instrumental in a wide variety of important biological functions such as folding (Privalov 1996; Englander 2000; Galzitskaya et al. 2000), allostery (Kern and Zuiderweg 2003; Busenlehner and Armstrong 2005), catalysis (Schnell et al. 2004; Bae and Phillips 2006), protein translocation (Voos and Rottgers 2002), and signal transduction (Fink 2005; Veprintsev et al. 2006), as well as unwanted processes such as aggregation and formation of amyloid fibrils (Stefani and Dobson 2003; Jahn and Radford 2005). Understanding the structure, energetics, and dynamics of such intermediate states is thus a key problem in modern biochemistry. The rare and ephemeral nature of such species, however, makes them extremely difficult to study—with a lifetime often less than milliseconds, at equilibrium they typically only represent ∼1 in 106 molecules.
Experimental techniques using amide hydrogen exchange (HX) have proven extremely useful for investigating such elusive species. Transiently populated folding intermediates can be labeled by pulse-labeling HX studies, while native state HX (NSHX) experiments yield information about rarely populated forms at equilibrium with the native state during the experimental time period. For most proteins investigated by both the kinetic and equilibrium approaches, the two yield similar results, suggesting that they report on the same intermediate. The bacteriophage protein T4 lysozyme is one notable exception.
T4 lysozyme, T4L* (* refers to a cysteine-free variant) (Matsumura and Matthews 1989), is a small (164-residue) protein that displays cooperative unfolding behavior with an equilibrium denaturation profile well-modeled by a two-state transition. This simple picture, however, is complicated by structural and HX data. Although considered to be a single domain, T4 lysozyme is actually comprised of two structural lobes referred to as the N-terminal and C-terminal subdomains (see Fig. 1). NSHX has revealed a high-energy partially unfolded form that appears structured in only the C-terminal subdomain (Llinas et al. 1999), while pulse-labeling HX studies show protection factors that implicate an early intermediate with structure in both the N- and C-terminal subdomains. Hence for T4 lysozyme, the transient folding intermediate detected by pulse-labeling HX does not mirror the high-energy intermediate detected by NSHX.
Figure 1.
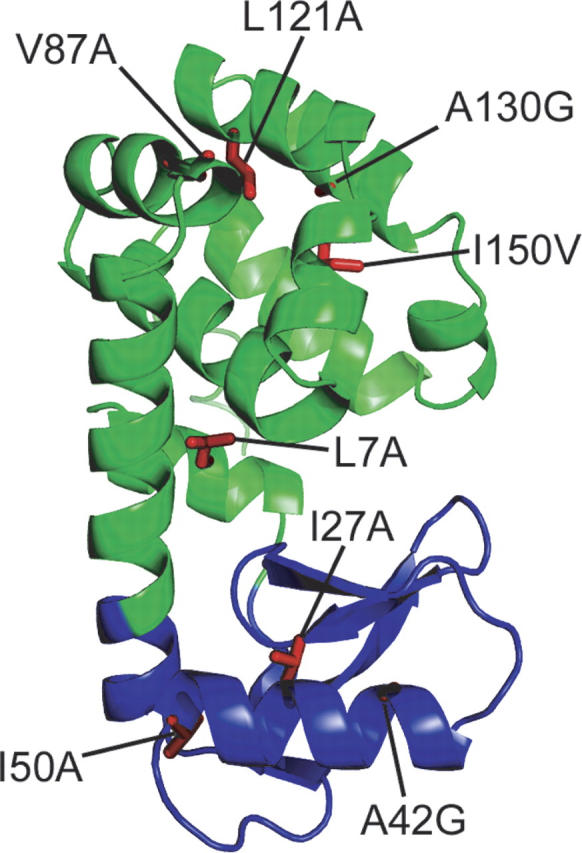
T4 lysozyme and the sites of mutations studied. (Green) The C-terminal subdomain and (blue) the N-terminal subdomain. Figure created using pdb 256L (Faber and Matthews 1990).
In the accompanying paper (Cellitti et al. 2007), we focused on structural and energetic characterizations of the partially unfolded form detected by NSHX. We demonstrated that a peptide model of this intermediate that contains residues from just the C-terminal subdomain forms a cooperatively folded native-like structure. Thus, residues in the N-terminal subdomain are not required to direct the folding of the C-terminal subdomain. We also demonstrated that in the full-length protein, there is cooperative coupling between the two lobes brought about, in part, by helix A. Although this helix resides sequentially at the N terminus of the protein, structurally it contributes to the C-terminal subdomain. Circular permutation (placing the N-terminal helix at the C terminus of the protein) diminishes the cooperativity between the subdomains, resulting in an even greater population of molecules with an independently folded C-terminal lobe.
Previous reports on the folding kinetics of T4L* have noted that the chevron plot (ln kobs vs. denaturant) displays the characteristic rollover in low denaturant suggestive of an early folding intermediate (Parker and Marqusee 1999). This kind of rollover can be modeled by either an on-pathway or an off-pathway early intermediate (U ⇆ I ⇆ N or I ⇆ U ⇆ N). For simplicity, the data for most proteins (including T4L*) are fit to the on-pathway obligatory folding intermediate model. Other refolding studies have also suggested the presence of a potential folding intermediate based on mutational analyses and detailed thermodynamic characterization of the refolding reaction (Desmadril and Yon 1984; Chen et al. 1989, 1992; Gassner et al. 2003).
Here, we use a protein engineering approach in conjunction with stopped-flow circular dichroism (CD) and chevron analyses to further characterize the transient intermediates populated during the folding and unfolding process of the protein. We find that the chevron plot of T4L* displays a rollover not only in the folding limb but also in the unfolding limb, a phenomenon that has been observed (Chen et al. 1992; Parker and Marqusee 1999) but never interpreted for this protein. By analyzing mutations in both structural subdomains of the protein (Fig. 1), in addition to the CP13* (Llinas and Marqusee 1998) and the LCTF variants introduced in the accompanying paper (Cellitti et al. 2007), we suggest that the intermediate probed by NSHX resides on the native state side of the rate-limiting transition state for folding and thereby represents an unfolding intermediate. Such intermediates, while difficult to identify, represent conformations easily sampled during excursions from the native state, and can play an important role in both folding and misfolding processes. Our conclusions also resolve a long-standing discrepancy between kinetics and equilibrium hydrogen exchange results for T4L* and highlight the importance of using multiple tools and approaches to characterize the energy landscape of a protein.
Results
Folding kinetics for T4L*
The unfolding/refolding process of T4L* was monitored as a function of urea concentration by following the change in CD signal as a function of time after rapid dilution with urea using a stop-flow mixing device. At all concentrations of urea, the observed data were fit to a single exponential process. A chevron plot derived from these data is shown in Figure 2. As seen previously (Chen et al. 1992; Parker and Marqusee 1999), the chevron plot for T4L* shows a “rollover” at low denaturant concentrations.
Figure 2.

Chevron plot of T4L*. The solid line is a fit using the three-state, on-pathway model.
At low denaturant concentrations, in the rollover region of the chevron plot, the amplitude of the observed phase does not account for the overall signal change of the reaction. Figure 3 shows how both these so-called burst-phase amplitudes and the final signals vary as a function of denaturant. Unlike the final signal, which reveals a cooperative unfolding transition, the burst-phase amplitudes change in a more-or-less linear fashion.
Figure 3.

Burst-phase amplitudes for T4L*. (A) Burst-phase amplitude at a final urea concentration of 0.6 M. The difference in signal between (○) the unfolded protein and (•) the initial signal upon refolding extrapolated to 0 time is the burst phase. (B) The burst phase as a function of urea. (•) Burst-phase amplitudes compared to (○) final amplitudes. (Solid line) A fit of final amplitudes to a two-state sigmoidal transition; (dashed line) an extrapolation of the unfolded baseline to low denaturant.
Folding of T4L* variants
Several site-specific variants were generated in both subdomains of T4L*: I27A, A42G, and I50A in the N-terminal subdomain and L7A, V87A, L121A, A130G, and I150V in the C-terminal subdomain. Mutations were designed to be minimally disruptive of structure and were placed at positions used as probes in HX studies. Five of the mutations were chosen based on their previous structural and thermodynamic characterizations (Xu et al. 1998), with the others chosen to provide more complete coverage of the protein. Most of the variants result in the removal of small hydrophobic side chains. All of the mutants and variants studied were soluble and well-behaved under the conditions we investigated; the resulting stabilities and kinetic parameters were independent of protein concentration. All the mutant proteins display apparent two-state behavior at equilibrium, and the resulting stabilities derived from the two-state fit are reported in Table 1. (Note that our resulting stabilities differ from those previously published [Xu et al. 1998], most likely because of differences in the pH and salt conditions.) The refolding/unfolding kinetics were monitored by CD in a manner similar to that described for T4L*, and the resulting chevron plots are shown in Figure 4.
Table 1.
Stabilities of T4L*, its mutants and variants

Figure 4.

Chevrons of T4L* mutants. (A) N-terminal mutations; (•) T4L*; (□) I27A; (▽) A42G; and (♦) I50A. (B) C-terminal mutations; (•) T4L*; (▴) L7A; (◊) V87A; ( ) L121A; (▾) A130G; and (×) I150V. (C) Large-scale variants; (•) T4L*; (○) CP13*; and (⊙) LCTF. (D) Mutants in the CP13* background; (□) I27A and (△) L7A in the context of the circular permutant. (•) T4L* and the mutations in the wild-type background, (▴) L7A and (▪) I27A, are shown for clarity.
) L121A; (▾) A130G; and (×) I150V. (C) Large-scale variants; (•) T4L*; (○) CP13*; and (⊙) LCTF. (D) Mutants in the CP13* background; (□) I27A and (△) L7A in the context of the circular permutant. (•) T4L* and the mutations in the wild-type background, (▴) L7A and (▪) I27A, are shown for clarity.
Chevron plots for the three variants containing point mutations in the N-terminal subdomain—I27A, A42G, and I50V—are shown in Figure 4A. These mutations all increase the rate of unfolding with minimal effects on the refolding rate. Thus, faster unfolding accounts for the decreased stability of these mutations. The other notable observation is the dramatic downward curvature in the unfolding limb of I27A, present to a lesser extent in the other two variants as well. These data were best fit to a four-state model, described in detail below.
Chevron plots for the five variants in the C-terminal subdomain are shown in Figure 4B. As with the N-terminal mutations, the folding limb is minimally affected. Three of the C-terminal mutations show only small effects on the unfolding limb. On the other hand, the unfolding rates of the two remaining variants, L7A and L121A, are markedly increased, and, perhaps most interestingly, their unfolding limbs appear to be entirely devoid of unfolding-limb curvature and were fit adequately using a three-state model (see below).
Refolding kinetics of the circular permutant CP13* were also monitored by stop-flow circular dichroism. CP13* has residues 1–12 grafted onto its C terminus using a six-amino-acid Ser(Gly)4Ala linker such that helix A is both sequentially and structurally associated with the rest of the C-terminal subdomain. As described in the accompanying paper (Cellitti et al. 2007), CP13* retains a wild-type-like structure with minimal change to the overall stability (Table 1). Compared to T4L*, however, the energetics of the two subdomains are less coupled in CP13* (as shown by NSHX in the accompanying paper [Cellitti et al. 2007]). The chevron plot for this variant is shown in Figure 4C (circles). Like the other variants presented above, the refolding limb is minimally affected by this permutation, and the destabilization appears to be reflected almost exclusively in the faster unfolding of the protein. It should also be noted that the unfolding limb of this permutation displays marked curvature.
LCTF (long C-terminal fragment), as described in the accompanying paper (Cellitti et al. 2007), was designed as a fragment modeling the partially unfolded form detected at equilibrium using NSHX. This fragment contains the C-terminal region of the protein, starting at residue 60, followed by the Ser(Gly)4Ala linker described for CP13*, and residues 1–12, which comprise helix A. Essentially, LCTF represents the C-terminal 98 residues of CP13* (the C-terminal structural subdomain). As demonstrated in the accompanying paper, LCTF forms a cooperatively folded domain (Table 1) with a structure that mirrors the C-terminal subdomain of the full-length protein (Cellitti et al. 2007). The chevron plot for the folding kinetics of LCTF is shown in Figure 4C; the plot looks remarkably like those for the full-length variants. The refolding rate appears very similar to that of the wild-type, CP13*, and the other variants, with the rollover at low denaturant concentrations. Unlike CP13*, however, LCTF shows no apparent curvature in its unfolding limb and was fit adequately to a three-state model (see below).
Finally, we evaluated the folding of the L7A and I27A mutations in the context of CP13* (Fig. 4D). Similar to mutations in the wild-type background, these mutations in CP13* have little effect on the folding limb and its rollover. On the other hand, the effects of these mutations on the unfolding limbs in the CP13* background are opposite from their effects in the wild-type background. While L7A eliminates unfolding limb curvature in the T4L* protein, the curvature remains in the CP13* background. Similarly, the greater unfolding limb curvature induced by I27A in T4L* is eliminated entirely when the mutation is in the CP13* context.
Analysis of the chevron plots
All of the kinetic data from this study were initially fit to a three-state model using Equations 1–5 (Baldwin 1996):
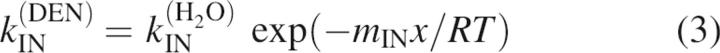 |
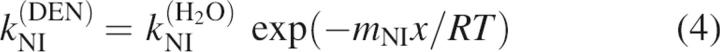 |
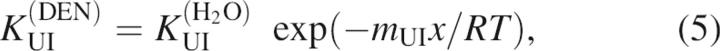 |
where kIN(DEN) and kNI(DEN) are the microscopic rate constants for folding and unfolding, respectively, kIN(H2O) and kNI(H2O) the rate constants in water, KUI the equilibrium constant between U and I, and fI the fraction of molecules present as I. mIN, mNI, and mUI are the denaturant dependencies of the two rate constants and equilibrium constant, respectively; x is the denaturant concentration, R the gas constant in kcal/mol·K, and T the temperature in K. Residuals from these fits, particularly from unfolding limb data, demonstrate the poorness of this model for all but three of the proteins. These fits and the residuals based on this model are shown in Figure 5A for T4L*, I27A, and L121A.
Figure 5.

Fits of T4L* and representative mutants. (•) T4L*; (○) I27A; and (▪) L121A. Residuals of all fits are inset. (A) Three-state fit. (B) Hammond behavior. (C) Four-state model.
Faced with rate profiles more complex than a simple three-state, on-pathway intermediate, alternative explanations for the curved chevron limbs were investigated. Deviations from linearity in chevron plots are typically attributed to one of two denaturant-dependent effects: Changes in the barriers (transition state) or changes in local minima, including any low-energy populated states on either side of the rate-limiting step. The two most common interpretations of curvature in the unfolding limb deal with effects on the folding barriers: (1) The moving transition state model, or Hammond behavior (Oliveberg 2001); and (2) the high-energy intermediate model (Sanchez and Kiefhaber 2003a,b,c; Scott and Clarke 2005). An alternate explanation is a four-state model that invokes both folding and unfolding intermediates. Reaction coordinate diagrams illustrating these models at high and low denaturant concentrations are shown in Figure 6.
Figure 6.

Energy diagrams representing the most common interpretations of complex unfolding and refolding kinetics. (Solid lines) Low (denaturant) conditions; (dotted lines) high (denaturant). (A) Transition state switching. The stability of the high-energy intermediate cannot be measured. (B) Hammond behavior. Transition state “sliding.” (C) Four-state model. The barriers between U and I and N and J cannot be measured.
The moving transition state model is based on the idea that the transition state in protein folding is a broad continuum of conformers referred to as the transition state ensemble (TSE) (Matouschek et al. 1995; Oliveberg et al. 1998; Otzen et al. 1999). In this broad continuum, one set of states will be rate limiting under a given set of conditions, but by changing solvent, that is, increasing the denaturant, or by altering the protein through mutation, the highest energy state in this continuum will “slide” along the TSE (Fig. 6B). In the absence of denaturant, the barrier will be closer on the reaction coordinate to the unfolded state, but with increasing denaturant, the transition state will become more native-like. Including a denaturant dependence term for this barrier is used to account for such Hammond behavior. This model has been successfully applied to chymotrypsin inhibitor 2 (Matouschek et al. 1995), barnase at different temperatures (Dalby et al. 1998), and most interestingly in the case of two structural homologs, U1A and S6 (Otzen et al. 1999). The revised rate equations are
and, as the derivative of ln k with respect to denaturant, the actual m-value becomes
This approach, however, did not adequately fit the data for T4L*. Fits and residuals for this model to T4L*, I27A, and L121A are shown in Figure 5B. Fitting the data to this model, modified slightly to include an on-pathway folding intermediate, resulted in rate constants that correspond to a dramatic overestimation of ΔGun. The existence of T4L* mutants that eliminate the curvature in the unfolding limb is also difficult to reconcile with this model.
The second model invoking effects on the transition state was developed by Kiefhaber and coworkers (Kiefhaber et al. 1992; Wagner and Kiefhaber 1999; Bachmann and Kiefhaber 2001; Sanchez and Kiefhaber 2003a,c) and has been applied to several proteins (Sanchez and Kiefhaber 2003b; Scott et al. 2004a,b; Scott and Clarke 2005). This model can be thought of as an extreme case of a single broad transition state in which there are two local high-energy maxima. The two transition states can be differentially affected by denaturant so that the first transition state (TS1) is rate limiting at low denaturant, while the second transition state (TS2) is rate limiting at high denaturant concentrations (Fig. 6A).
We have so far been unable to fit our data with this model. However, given the dramatic differences in the unfolding limb profiles among our variants, it is unlikely that the changes are due simply to effects on the transition state. In this model, a mutation that destabilizes the native state would also have to stabilize TS2 to prevent it from becoming rate limiting at high denaturant. Even if this were possible, the effect on TS2 would have to be so dramatic as to almost eliminate it. If not, the unfolding kinetics we observe in variants without curvature in the unfolding limb would be biphasic since there would be two significant barriers.
Finally, the data were fit using a four-state model with on-pathway folding and unfolding intermediates (Sauder et al. 1996; Fig. 6C; Scheme 1):
Figure .

This model is identical to the three-state model with the exception that a term for the fraction of protein present as an unfolding intermediate, J, must be included.
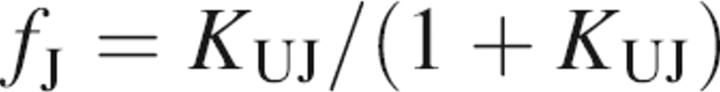 |
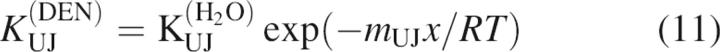 |
Figure 5C shows fits of the data from T4L*, I27A, and L121A to this model with the residuals inset. Variants such as L121A devoid of curvature in the unfolding limb can be fit with this model, but the resulting KNJ values indicate that J never becomes populated (Table 2). Thus, the simpler three-state on-pathway model was used for these variants. F-tests on the residuals of the three- and four-state fits confirm that for all but L121A, L7A, and LCTF, the four-state fit is significantly better (data not shown).
Table 2.
Kinetic parameters of T4L*, its mutants and variants

Based on the results from the above kinetics models, the stabilities of the intermediates and the activation energies of the major rate-limiting transition state were calculated using the following:
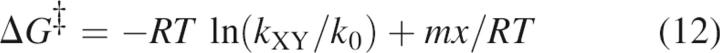 |
Although the absolute height of the energy barriers is not important, a value of k0 was set at 109 sec−1 (Bieri et al. 1999). Reaction-coordinate diagrams for T4L*, I27A, and L121A are shown at both 0 and 9 M urea in Figure 7.
Figure 7.
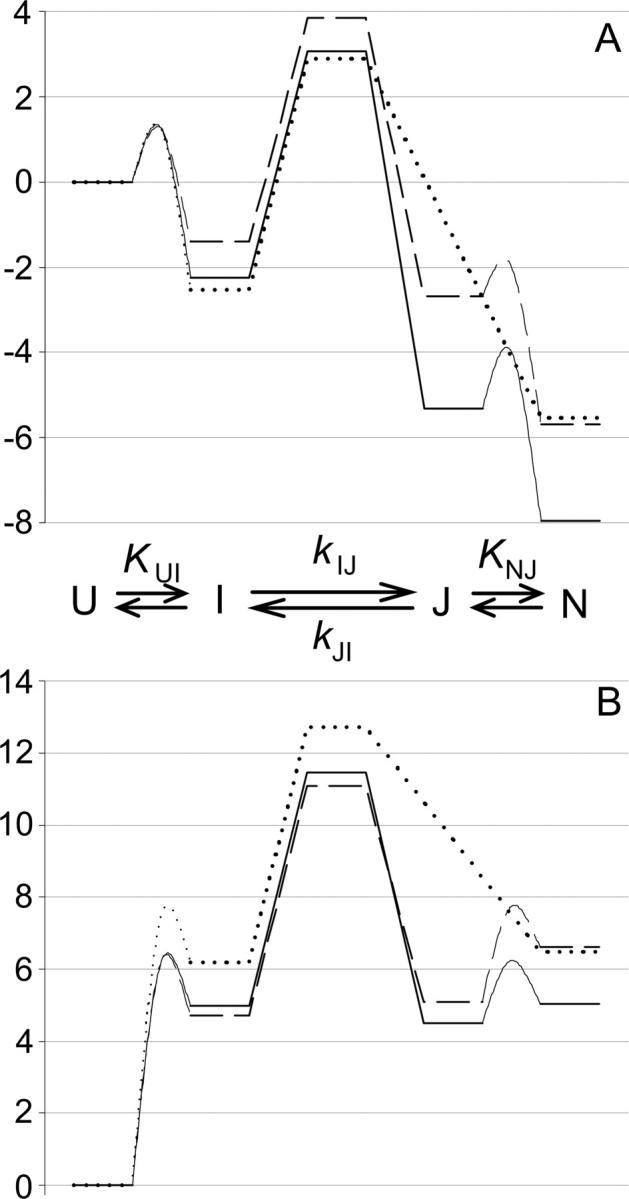
Reaction diagrams for (—) T4L*; (---) I27A; and (…) L121A. Diagrams are normalized to U, which is assigned 0. Barriers between U and I and N and J cannot be determined and were arbitrarily chosen. (A) 0 M urea. The native states of both mutants are destabilized, but only I27A is significantly destabilized in I and the transition state. (B) 9 M urea. J is more destabilized than N in I27A, making it a more populated state in this mutant than in T4L*.
Discussion
T4 lysozyme has been reported to fold through an early intermediate based on several observations: significant burst-phase amplitudes, rollover in the folding limb of the chevron plot, and pulse-labeling HX experiments that demonstrate a small amount of protection for a subset of amides. Based on the distribution of protected residues, both the N- and C-terminal subdomains are involved in this intermediate (Lu and Dahlquist 1992). In carrying out a detailed chevron analysis, we now find evidence for an intermediate on the folded side of the rate-limiting transition state, which for many of the variants studied here results in the population of an unfolding intermediate at high urea concentrations.
Many of our chevron plots revealed substantial curvature in the unfolding limb that cannot be accounted for by a simple three-state, on-pathway intermediate model. By invoking changes in the ground states, we have found a model with two intermediates, one populated upon folding (I) and one upon unfolding (J), that adequately explains our data. Single point mutations can either accentuate the population of the intermediate or eliminate it. A mutation that destabilizes N may also destabilize J; however, a destabilizing mutation that resides in a region not structured in J may actually stabilize J relative to N. Hence an increase in the relative curvature in the unfolding limb would be expected to correspond to changes in regions not involved in the structure and stability of the unfolding intermediate.
A comparison of the effects on folding between the different variants suggests that this unfolding intermediate corresponds to the partially unfolded form detected by NSHX that is structured only in the C-terminal domain. Mutations that specifically destabilize the N-terminal subdomain, such as I27A, stabilize this intermediate, resulting in greater rollover of the unfolding arm of the chevron plot, while mutations that destabilize the C-terminal subdomain either have no impact on the intermediate's formation because of equal effects on the native and high-energy states, as seen in V87A, A130G, and I150V, or they destabilize the intermediate to such an extent that it does not form at all, leading to chevron plots with linear unfolding arms for L7A and L121A. In both cases, it is the mutations with the largest effect on the free energy of unfolding that have the greatest impact on the intermediate's formation or disruption.
The chevron plots of the two large-scale variants of T4L*, CP13*, and LCTF, also support the idea that the unfolding intermediate resembles that detected by NSHX and modeled by LCTF. CP13*, shown by native state hydrogen exchange to specifically destabilize the N-terminal subdomain (see accompanying paper [Cellitti et al. 2007] in this issue), has a notably increased curvature in the unfolding limb of its chevron plot compared to T4L*, suggesting that the unfolding intermediate does not result from structure or stability in the N-terminal domain. In LCTF, the entire N-terminal subdomain is absent, as is any curvature of the chevron's unfolding limb. This too is consistent with the notion that the C-terminal region models the unfolding intermediate. Furthermore, the extrapolated folding rate of LCTF is similar to that of the intact protein, suggesting that for both variants folding may involve crossing a similar rate-limiting transition state. Hence, the kinetics profile of LCTF further supports the hypothesis that the intermediate containing an unfolded N-terminal domain and a folded C-terminal domain, as detected by NSHX, resides on the folded side of the rate-limiting transition state.
Mutations made in the context of the circular permutant also corroborate the idea that the two subdomains unfold sequentially. Combining the I27A mutation with the circular permutation, both of which specifically destabilize the N-terminal subdomain and increase the curvature of their chevrons’ unfolding limbs, leads to a chevron with no unfolding limb curvature. While this lack of curvature could imply that the intermediate is no longer formed, the low kinetic m-value of CP13*I27A (2.0 kcal/mol · M, lower than even LCTF) leads us to believe that this variant specifically destabilizes N relative to the intermediate. This mutant, like LCTF, appears to model the partially folded species seen in NSHX. On the other hand, L7A in the wild-type context destabilizes the C-terminal domain to such an extent that this intermediate is no longer populated. However, when combined with CP13*, the N-terminal subdomain is also destabilized, such that J is once again populated during unfolding, as seen by the curvature in the unfolding limb of its chevron.
Nature of the rollover and burst phase in the folding process
Burst phases are seen as hallmarks of intermediate formation, both during refolding and unfolding (Kuwajima et al. 1987; Bhuyan and Udgaonkar 1998; Fujiwara et al. 1999; Jamin et al. 1999). Additionally, if representative of rapid formation of a true intermediate, they should show a cooperative, sigmoidal denaturant dependence (Raschke and Marqusee 1997). While all of the proteins and variants investigated here show burst-phase amplitudes during refolding, their values show a linear, not sigmoidal, dependence on urea concentration (Fig. 3B). There are also some inconsistencies between the HX protection noted by Lu and Dahlquist in their pulse-labeling study (Lu and Dahlquist 1992) and the stability of I determined from our kinetic modeling (KUI). All of the variants we have studied imply an early intermediate with a stability on the order of 4 kcal/mol, while the protection factors suggest a much lower stability (∼2–3 kcal/mol). The basis for the difference is unclear. Nonetheless, our results certainly support an argument against a specific compact structure involving residues from both subdomains as it appears insensitive to mutation and/or the complete removal of the N-terminal subdomain such as in LCTF.
Conclusions
In sum, by examining the denaturant dependence of both folding and unfolding for a diverse array of variants of T4L*, we find evidence for an unfolding intermediate that seems to be structured in only the C-terminal subdomain. This intermediate resembles the species seen by NSHX (Llinas et al. 1999) and is evidence that the NSHX results from T4 lysozyme report on a species on the folded side of the rate-limiting barrier, rather than the unfolded side. NSHX detection of such species has been interpreted similarly by Bai and coworkers (Zhou et al. 2005) and called “hidden intermediates” because of the inability to detect them using kinetics methods. T4 lysozyme, however, appears to be a system in which these “hidden” intermediates are actually revealed by kinetics methods. Such high-energy intermediates on the folded side of the transition state barrier are likely to be as important as more commonly identified folding intermediates, if not more so, in the myriad biological functions mentioned earlier. The identification of such an intermediate species in T4L* via kinetic methods is an exciting step toward a more complete understanding of the role of intermediates on both sides of the transition state barrier in protein folding.
Materials and Methods
Site-directed mutagenesis
Mutants were created using the QuikChange method from Stratagene. Primers were purchased from Integrated DNA Technoligies. Mutations were chosen and primers were designed by Eric Nicholson.
Protein purification
All proteins were overexpressed and purified as previously described (Llinas and Marqusee 1998).
CD spectrometry and denaturation melts
Spectra and denaturation melts on all mutants were done as described for T4L* (Llinas and Marqusee 1998).
Kinetic folding and unfolding
Refolding was initiated by diluting unfolded protein at 4.0 mg/mL in urea (6.5 M for T4L*, 6.0 M for mutants) 1:11 (v/v) into refolding buffer (20 mM potassium phosphate at pH 6.0; 100 mM NaCl with low concentrations of urea) using an AVIV 202 stopped-flow CD spectrophotometer equipped with a delta mixer and a cuvette pathlength of 1 mm. The CD signal was followed over time at 222 nm. Unfolding experiments were performed by diluting folded protein in 1.0 M urea into high concentrations of urea. Folded and unfolded stock protein samples were equilibrated overnight. Each data point represents the averaged signal of at least seven individual shots. Experiments were performed at 25°C.
The observed signal for each concentration of urea described a single exponential (kobs) and was fit using SigmaPlot 8.0 for Windows (Systat Software Inc.) to the equation:
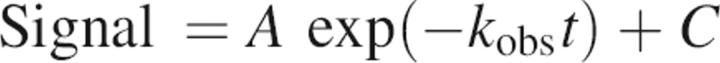 |
where C is the final signal, A the amplitude of the observable phase, and t is time. The initial points in the “rolled over” limbs of each variant describe the “burst phase” (the signal obtained in the undetected dead-time of the instrument, 18.1 msec), measured by
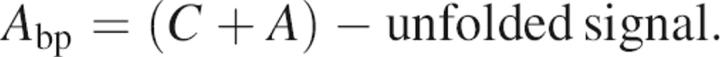 |
Note added in proof
Recently, Kato and coworkers (Kato et al. 2007) engineered a mimic of the equilibrium intermediate detected by hydrogen exchange by replacing residues 17–58 of the N-terminal subdomain with five glycines. This variant folded with the same rate as the wild-type protein leading the authors to conclude that the intermediate seen in native-state hydrogen exchange resides after the rate-limiting transition state.
Acknowledgments
We thank the entire Marqusee laboratory staff for help and discussions especially Eric Nicholson for initiating this study. This work was supported by an NIH grant (GM50945) to S.M.
Footnotes
Reprint requests to: Susan Marqusee, Department of Molecular and Cell Biology and QB3 Institute–Berkeley, 229 Hildebrand Hall, University of California, Berkeley, Berkeley, CA 94720-3206, USA; e-mail: marqusee@berkeley.edu; fax: (510) 643-9290.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.062632807.
References
- Bachmann A. and Kiefhaber, T. 2001. Apparent two-state tendamistat folding is a sequential process along a defined route. J. Mol. Biol. 306: 375–386. [DOI] [PubMed] [Google Scholar]
- Bae E. and Phillips Jr, G.N. 2006. Roles of static and dynamic domains in stability and catalysis of adenylate kinase. Proc. Natl. Acad. Sci. 103: 2132–2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldwin R.L. 1996. On-pathway versus off-pathway folding intermediates. Fold. Des. 1: R1–R8. [DOI] [PubMed] [Google Scholar]
- Bhuyan A.K. and Udgaonkar, J.B. 1998. Multiple kinetic intermediates accumulate during the unfolding of horse cytochrome c in the oxidized state. Biochemistry 37: 9147–9155. [DOI] [PubMed] [Google Scholar]
- Bieri O., Wirz, J., Hellrung, B., Schutkowski, M., Drewello, M., and Kiefhaber, T. 1999. The speed limit for protein folding measured by triplet–triplet energy transfer. Proc. Natl. Acad. Sci. 96: 9597–9601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busenlehner L.S. and Armstrong, R.N. 2005. Insights into enzyme structure and dynamics elucidated by amide H/D exchange mass spectrometry. Arch. Biochem. Biophys. 433: 34–46. [DOI] [PubMed] [Google Scholar]
- Cellitti J., Llinas, M., Echols, N., Shank, E.A., Gillespie, B., Crowder, S.M., Dahlquist, F.W., Alber, T., and Marqusee, S. 2007. Exploring subdomain cooperativity in T4 lysozyme. I. Structural and energetic studies of a circular permutant and protein fragment. Protein Sci (this issue). [DOI] [PMC free article] [PubMed]
- Chen B.L., Baase, W.A., and Schellman, J.A. 1989. Low-temperature unfolding of a mutant of phage T4 lysozyme. 2. Kinetic investigations. Biochemistry 28: 691–699. [DOI] [PubMed] [Google Scholar]
- Chen B.L., Baase, W.A., Nicholson, H., and Schellman, J.A. 1992. Folding kinetics of T4 lysozyme and nine mutants at 12 degrees C. Biochemistry 31: 1464–1476. [DOI] [PubMed] [Google Scholar]
- Dalby P.A., Oliveberg, M., and Fersht, A.R. 1998. Folding intermediates of wild-type and mutants of barnase. I. Use of φ-value analysis and m-values to probe the cooperative nature of the folding pre-equilibrium. J. Mol. Biol. 276: 625–646. [DOI] [PubMed] [Google Scholar]
- Desmadril M. and Yon, J.M. 1984. Evidence for intermediates during unfolding and refolding of a two-domain protein, phage T4 lysozyme: Equilibrium and kinetic studies. Biochemistry 23: 11–19. [DOI] [PubMed] [Google Scholar]
- Englander S.W. 2000. Protein folding intermediates and pathways studied by hydrogen exchange. Annu. Rev. Biophys. Biomol. Struct. 29: 213–238. [DOI] [PubMed] [Google Scholar]
- Faber H.R. and Matthews, B.W. 1990. A mutant T4 lysozyme displays five different crystal conformations. Nature. 348: 263–266. [DOI] [PubMed] [Google Scholar]
- Fink A.L. 2005. Natively unfolded proteins. Curr. Opin. Struct. Biol. 15: 35–41. [DOI] [PubMed] [Google Scholar]
- Fujiwara K., Arai, M., Shimizu, A., Ikeguchi, M., Kuwajima, K., and Sugai, S. 1999. Folding–unfolding equilibrium and kinetics of equine β-lactoglobulin: Equivalence between the equilibrium molten globule state and a burst-phase folding intermediate. Biochemistry 38: 4455–4463. [DOI] [PubMed] [Google Scholar]
- Galzitskaya O.V., Skoogarev, A.V., Ivankov, D.N., and Finkelstein, A.V. 2000. Folding nuclei in 3D protein structures. Pac. Symp. Biocomput. 2000: 131–142. [DOI] [PubMed] [Google Scholar]
- Gassner N.C., Baase, W.A., Mooers, B.H., Busam, R.D., Weaver, L.H., Lindstrom, J.D., Quillin, M.L., and Matthews, B.W. 2003. Multiple methionine substitutions are tolerated in T4 lysozyme and have coupled effects on folding and stability. Biophys. Chem. 100: 325–340. [DOI] [PubMed] [Google Scholar]
- Jahn T.R. and Radford, S.E. 2005. The Yin and Yang of protein folding. FEBS J. 272: 5962–5970. [DOI] [PubMed] [Google Scholar]
- Jamin M., Yeh, S.R., Rousseau, D.L., and Baldwin, R.L. 1999. Submillisecond unfolding kinetics of apomyoglobin and its pH 4 intermediate. J. Mol. Biol. 292: 731–740. [DOI] [PubMed] [Google Scholar]
- Kato H., Vu, N.D., Feng, H., Zhou, Z., and Bai, Y. 2007. The folding pathway of T4 lysozyme: An on-pathway hidden folding intermediate. J. Mol. Biol. 365: 881–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kern D. and Zuiderweg, E.R. 2003. The role of dynamics in allosteric regulation. Curr. Opin. Struct. Biol. 13: 748–757. [DOI] [PubMed] [Google Scholar]
- Kiefhaber T., Kohler, H.H., and Schmid, F.X. 1992. Kinetic coupling between protein folding and prolyl isomerization. I. Theoretical models. J. Mol. Biol. 224: 217–229. [DOI] [PubMed] [Google Scholar]
- Kuwajima K., Yamaya, H., Miwa, S., Sugai, S., and Nagamura, T. 1987. Rapid formation of secondary structure framework in protein folding studied by stopped-flow circular dichroism. FEBS Lett. 221: 115–118. [DOI] [PubMed] [Google Scholar]
- Llinas M. and Marqusee, S. 1998. Subdomain interactions as a determinant in the folding and stability of T4 lysozyme. Protein Sci. 7: 96–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llinas M., Gillespie, B., Dahlquist, F.W., and Marqusee, S. 1999. The energetics of T4 lysozyme reveal a hierarchy of conformations. Nat. Struct. Biol. 6: 1072–1078. [DOI] [PubMed] [Google Scholar]
- Lu J. and Dahlquist, F.W. 1992. Detection and characterization of an early folding intermediate of T4 lysozyme using pulsed hydrogen exchange and two-dimensional NMR. Biochemistry 31: 4749–4756. [DOI] [PubMed] [Google Scholar]
- Matouschek A., Otzen, D.E., Itzhaki, L.S., Jackson, S.E., and Fersht, A.R. 1995. Movement of the position of the transition state in protein folding. Biochemistry 34: 13656–13662. [DOI] [PubMed] [Google Scholar]
- Matsumura M. and Matthews, B.W. 1989. Control of enzyme activity by an engineered disulfide bond. Science 243: 792–794. [DOI] [PubMed] [Google Scholar]
- Oliveberg M. 2001. Characterisation of the transition states for protein folding: Towards a new level of mechanistic detail in protein engineering analysis. Curr. Opin. Struct. Biol. 11: 94–100. [DOI] [PubMed] [Google Scholar]
- Oliveberg M., Tan, Y.J., Silow, M., and Fersht, A.R. 1998. The changing nature of the protein folding transition state: Implications for the shape of the free-energy profile for folding. J. Mol. Biol. 277: 933–943. [DOI] [PubMed] [Google Scholar]
- Otzen D.E., Kristensen, O., Proctor, M., and Oliveberg, M. 1999. Structural changes in the transition state of protein folding: Alternative interpretations of curved chevron plots. Biochemistry 38: 6499–6511. [DOI] [PubMed] [Google Scholar]
- Parker M.J. and Marqusee, S. 1999. The cooperativity of burst phase reactions explored. J. Mol. Biol. 293: 1195–1210. [DOI] [PubMed] [Google Scholar]
- Privalov P.L. 1996. Intermediate states in protein folding. J. Mol. Biol. 258: 707–725. [DOI] [PubMed] [Google Scholar]
- Raschke T.M. and Marqusee, S. 1997. The kinetic folding intermediate of ribonuclease H resembles the acid molten globule and partially unfolded molecules detected under native conditions. Nat. Struct. Biol. 4: 298–304. [DOI] [PubMed] [Google Scholar]
- Sanchez I.E. and Kiefhaber, T. 2003a. Evidence for sequential barriers and obligatory intermediates in apparent two-state protein folding. J. Mol. Biol. 325: 367–376. [DOI] [PubMed] [Google Scholar]
- Sanchez I.E. and Kiefhaber, T. 2003b. Hammond behavior versus ground state effects in protein folding: Evidence for narrow free energy barriers and residual structure in unfolded states. J. Mol. Biol. 327: 867–884. [DOI] [PubMed] [Google Scholar]
- Sanchez I.E. and Kiefhaber, T. 2003c. Non-linear rate-equilibrium free energy relationships and Hammond behavior in protein folding. Biophys. Chem. 100: 397–407. [DOI] [PubMed] [Google Scholar]
- Sauder J.M., MacKenzie, N.E., and Roder, H. 1996. Kinetic mechanism of folding and unfolding of Rhodobacter capsulatus cytochrome c2. Biochemistry 35: 16852–16862. [DOI] [PubMed] [Google Scholar]
- Schnell J.R., Dyson, H.J., and Wright, P.E. 2004. Structure, dynamics, and catalytic function of dihydrofolate reductase. Annu. Rev. Biophys. Biomol. Struct. 33: 119–140. [DOI] [PubMed] [Google Scholar]
- Scott K.A. and Clarke, J. 2005. Spectrin R16: Broad energy barrier or sequential transition states? Protein Sci. 14: 1617–1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott K.A., Batey, S., Hooton, K.A., and Clarke, J. 2004a. The folding of spectrin domains I: Wild-type domains have the same stability but very different kinetic properties. J. Mol. Biol. 344: 195–205. [DOI] [PubMed] [Google Scholar]
- Scott K.A., Randles, L.G., and Clarke, J. 2004b. The folding of spectrin domains II: φ-value analysis of R16. J. Mol. Biol. 344: 207–221. [DOI] [PubMed] [Google Scholar]
- Stefani M. and Dobson, C.M. 2003. Protein aggregation and aggregate toxicity: New insights into protein folding, misfolding diseases and biological evolution. J. Mol. Med. 81: 678–699. [DOI] [PubMed] [Google Scholar]
- Veprintsev D.B., Freund, S.M., Andreeva, A., Rutledge, S.E., Tidow, H., Canadillas, J.M., Blair, C.M., and Fersht, A.R. 2006. Core domain interactions in full-length p53 in solution. Proc. Natl. Acad. Sci. 103: 2115–2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voos W. and Rottgers, K. 2002. Molecular chaperones as essential mediators of mitochondrial biogenesis. Biochim. Biophys. Acta 1592: 51–62. [DOI] [PubMed] [Google Scholar]
- Wagner C. and Kiefhaber, T. 1999. Intermediates can accelerate protein folding. Proc. Natl. Acad. Sci. 96: 6716–6721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J., Baase, W.A., Baldwin, E., and Matthews, B.W. 1998. The response of T4 lysozyme to large-to-small substitutions within the core and its relation to the hydrophobic effect. Protein Sci. 7: 158–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z., Huang, Y., and Bai, Y. 2005. An on-pathway hidden intermediate and the early rate-limiting transition state of Rd-apocytochrome b562 characterized by protein engineering. J. Mol. Biol. 352: 757–764. [DOI] [PubMed] [Google Scholar]