

Abstract
MAP kinase phosphatases (MKPs) have crucial roles in regulating the signaling activity of MAP kinases and are potential targets for drug discovery against human diseases. These enzymes contain a catalytic domain (CD) as well as a binding domain (BD) that help recognize the target MAP kinase. We report here the crystal structures at up to 2.2 Å resolution of the BD and CD of human MKP5 and compare them to the known structures from other MKPs. Dramatic structural differences are observed between the BD of MKP5 and that of MKP3 determined previously by NMR. In particular, the cluster of positively charged residues that is important for MAP kinase binding is located in completely different positions in the two structures, with a distance of 25 Å between them. Moreover, this cluster is α-helical in MKP5, while it forms a loop followed by a β-strand in MKP3. These large structural differences could be associated with the distinct substrate preferences of these phosphatases, but further studies are needed to confirm this. The CD of MKP5 is observed in an active conformation, and two loops in the active site have backbone shifts of up to 5 Å relative to the inactive CDs from other MKPs.
Keywords: domains and motifs, exon/intron relationship, enzymes, active sites, structure
Mitogen-activated protein kinases (MAP kinases) have crucial roles in a large number of cellular signaling processes (Davis 2000; Chang and Karin 2001; Pearson et al. 2001; Johnson and Lapadat 2002). These enzymes, including ERK, p38, and JNK, are activated in response to a variety of signals, through phosphorylation of the Thr and Tyr residues in the Thr-X-Tyr motif in the activation loop.
MAP kinase phosphatases (MKPs) are important regulators of MAP kinase signaling, by dephosphorylating activated MAP kinases (Camps et al. 1999; Theodosiou and Ashworth 2002; Farooq and Zhou 2004; Martin et al. 2005; Dickinson and Keyse 2006). These enzymes are interesting targets for drug discovery against many human diseases. MKP5-deficient mice have increased JNK activity and elevated production of proinflammatory cytokines during innate immune responses (Zhang et al. 2004; Dickinson and Keyse 2006). Mice lacking MKP1 have elevated p38 activity and a hyperresponsive innate immune system (Dickinson and Keyse 2006). They are resistant to diet-induced obesity, as MKP1 also regulates JNK and ERK, but become glucose intolerant on a high fat diet (Dickinson and Keyse 2006).
MKPs belong to the larger family of dual specificity protein phosphatases (DUSPs or DSPs), as they can remove the phosphate group on both Thr and Tyr residues in MAP kinases. MKPs contain a phosphatase domain (or catalytic domain [CD]) in their C termini, and a MAP kinase binding domain (BD) at their N termini (Fig. 1A). MKP5 is unique among the MKPs in possessing an additional segment at the N terminus (Fig. 1A), although the function of this segment is not known. The CDs of the MKPs are highly conserved, with the catalytic Cys residue located in the phosphatase motif HCXXGXXR(S/T) (Fig. 1B). In comparison, the sequence conservation of the BDs is weaker (Fig. 1B), and this sequence diversity may help define the distinct substrate preferences of the MKPs as well as their different localizations in the cell. For example, MKP3 prefers ERK as the substrate, and MKP5 prefers p38 and JNK, while MKP1 can regulate all three MAP kinases. A cluster of positively charged residues in the BD mediates interactions with the target MAP kinases (Farooq et al. 2001; Tanoue et al. 2002; Dickinson and Keyse 2006; Zhou et al. 2006).
Figure 1.

Sequence alignment of human MKPs. (A) Domain organization of human MKP5. The MAP kinase binding domain (BD) and the catalytic domain (CD) are indicated. (B) Sequence alignment of human MKP5, MKP3, and MKP1. The secondary structure elements in MKP5 (S.S.) are labeled, and those in the NMR structure of the BD of MKP3 (Farooq et al. 2001) are shown at the bottom of the alignment. The start of the BD and CD are indicated in red. Residues not observed in our structures are shown in italic, and residues identical between MKP5 and MKP1 or MKP3 are shown in magenta.
We report here the crystal structures of the MAP kinase BD and the CD of human MKP5 and compare them to the known structures from other MKPs (Stewart et al. 1999; Farooq et al. 2001, 2003; Farooq and Zhou 2004; Jeong et al. 2006). We observed dramatic structural differences between the BDs of MKP5 and MKP3, and these differences may be the molecular basis for the different substrate preferences of these enzymes.
Results and Discussion
Structure determination
The crystal structure of the MAP kinase BD (residues 148–287) of human MKP5 has been determined at 2.2 Å resolution by the selenomethionyl single-wavelength anomalous diffraction (SAD) method (Hendrickson 1991). The crystal structure of the CD (residues 315–482) of human MKP5 has been determined at 2.8 Å resolution by the selenomethionyl multiple-wavelength anomalous diffraction (MAD) method (Hendrickson 1991).
The refined structures have excellent agreement with the crystallographic data and the expected bond lengths, bond angles, and other geometric parameters (Table 1). The majority of the residues are in the most favored region of the Ramachandran plot, and none of the residues are in the disallowed region. The two molecules of BD in the asymmetric unit have essentially the same conformation, with RMSD of 0.46 Å among their 130 equivalent Cα atoms. Both the BD and the CD are monomeric in solution, based on our static light scattering studies (data not shown).
Table 1.
Summary of crystallographic information

Structure of the MAP kinase binding domain
The structure of the MAP kinase BD of human MKP5 contains a central, fully parallel five-stranded β-sheet with helices on both faces (Fig. 2A). In addition, there is a small, two-stranded β-sheet involving residues 186–188 and 284–287 at the C terminus of the domain. The first residue observed in our structure (Lys148) is located just prior to the first β-strand. An additional nine N-terminal residues (139–147) in our expression construct appear to be disordered, as we did not observe any electron density for them. In addition, residues 162–171, in the loop connecting the first helix with the second β-strand (Fig. 2A), are also disordered.
Figure 2.

Structure of the MAP kinase binding domain of MKP5. (A) Schematic representation of the structure of the binding domain of MKP5. (B) Structural overlay of the MKP5 BD (in color) with the catalytic domain of bovine rhodanese (in gray) (Ploegman et al. 1978b). The catalytic Cys residue of rhodanese is shown in black and indicated with the red star. The equivalent residue in MKP5 is an Asp. (C) Structural overlay of the MKP5 BD with Cdc25A (in gray) (Fauman et al. 1998). Produced with Ribbons (Carson 1987).
The structure of BD is remarkably similar to that of sulfurtransferases (rhodaneses) (Bordo and Bork 2002). The closest homologs include bovine liver rhodanese (Fig. 2B; Ploegman et al. 1978a,b), Escherichia coli GlpE (Spallarossa et al. 2001), Azotobacter vinelandii rhodanese (Bordo et al. 2000), Leishmania major 3-mercaptopyruvate sulfurtransferase (Alphey et al. 2003), Thermus thermophilus single-domain rhodanese (Hattori et al. 2006), and Wolinella succinogenes polysulfide-sulfurtransferase (Lin et al. 2004). Interestingly, Cdc25A, a dual specificity phosphatase that has an important role in cell cycle control, also has the same backbone fold (Fig. 2C; Fauman et al. 1998). The RMSD is ∼2.3 Å for roughly 100 equivalent Cα atoms between the BD and these other structures, but the sequence identity is only about 20%, calculated with the program Dali (Holm and Sander 1993).
While the overall structures of the BD and these sulfurtransferases and Cdc25A are similar, there are also significant differences among them. Most importantly, these other enzymes contain a catalytic cysteine residue, located just after strand β4 (Fig. 2B,C). In BD, however, this residue is replaced by an Asp (Fig. 2A), and therefore the BD cannot have the same catalytic activity. Several of the rhodaneses contain two structurally homologous domains, but only one of them is catalytically active (Ploegman et al. 1978a,b; Bordo et al. 2000; Alphey et al. 2003). The equivalent residue in the inactive domain of these enzymes has also been mutated. In fact, it is an Asp in bovine rhodanese (Ploegman et al. 1978a,b).
Besides the catalytic residue itself, the loop containing this residue, connecting strand β4 and helix α4, is much more extended in the structure of BD (Fig. 2A), whereas it is much shorter in the sulfurtransferases and Cdc25A (Fig. 2B,C). In addition, three small helices are in the crossover connection between β3 and β4 in BD (Fig. 2A), whereas only one helix is present here in the other enzymes (Fig. 2B,C).
Large structural differences with the BD of MKP3
The structure of the BD from MKP3 has been determined in solution by NMR (Farooq et al. 2001). The two BD sequences share 28% identity (Fig. 1B). However, there are dramatic differences in many of the surface loops and helices between the two structures (Fig. 3A), even though their central β-sheets have a similar conformation (Fig. 3B). As a consequence, the molecular surfaces of the two domains are entirely different (Fig. 3C,D). Most remarkably, the cluster of positively charged residues that is important for interacting with the target MAP kinases has completely different conformations in the two structures, separated by ∼25 Å (Fig. 3B). Residues Leu63–Arg–Arg65 in MKP3 are located in a loop followed by a small β-strand, whereas their sequence equivalents (Arg202–Arg–Arg204) in MKP5 are located in a helix (α3′), on the opposite face of the central β-sheet (Fig. 3A). Therefore, these motifs are located at completely different positions on the surface of these domains (Fig. 3B,C). For the structure of the MKP5 BD reported here, 86.6% of the residues are in the “most-favored” regions, 12.9% in the “additional allowed” regions, 0.4% in the “generously allowed” regions, and 0% in the “disallowed” regions of the Ramachandran plot (Laskowski et al. 1993). For the structure of the MKP3 BD (Farooq et al. 2001), the four regions have 46.7%, 40.7%, 10.4%, and 2.2% of the residues, respectively. Of the 18 residues in the MKP3 BD structure in the generously allowed and disallowed regions, four are located near this positively charged cluster. If these large structural differences are substantiated by further studies, they may be associated with the distinct substrate preferences of the two enzymes.
Figure 3.
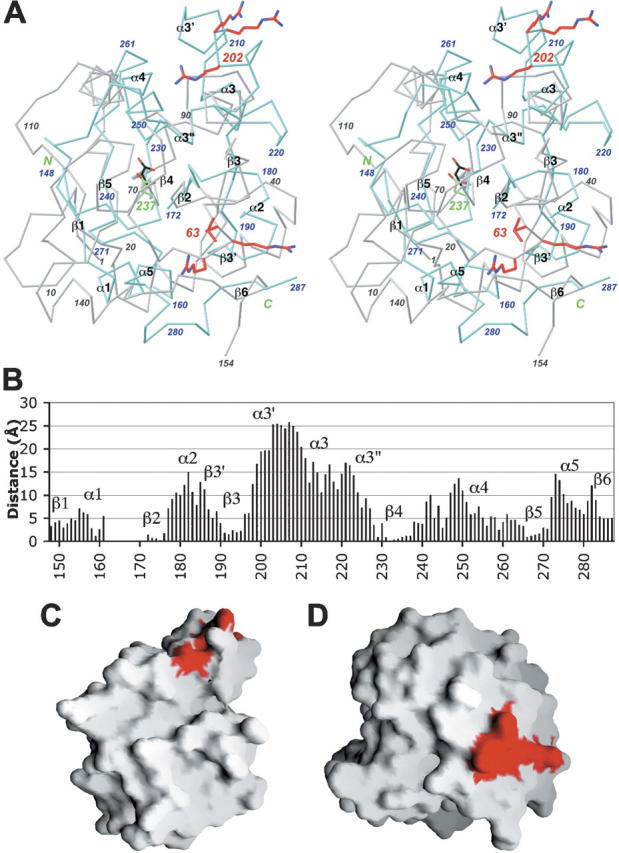
Large structural differences with the BD of MKP3. (A) Structural overlay of the MKP5 BD (in color) and the MKP3 BD (in gray) (Farooq et al. 2001). The positively charge cluster important for MAP kinase interactions is shown in red in both structures. Produced with Ribbons (Carson 1987). (B) Plot of the distance between equivalent Cα atoms in the structures of MKP5 BD and MKP3 BD. (C) Molecular surface of the MKP5 BD. (D) Molecular surface of the MKP3 BD, viewed in the same orientation as panel (C). Produced with Grasp (Nicholls et al. 1991).
Structure of the catalytic domain of MKP5
The structure of CD of MKP5 contains a central, mostly parallel five-stranded β-sheet that is surrounded by helices (Fig. 4A). Close structural homologs of this CD include the dual-specificity protein phosphatase VHR (Yuvaniyama et al. 1996), the CDK-interacting protein phosphatase KAP (Song et al. 2001), the PTEN tumor suppressor (Lee et al. 1999), and the tyrosine phosphatase SHP-2 (Hof et al. 1998). The RMSD is ∼2.5 Å for ∼115 equivalent Cα atoms, and the sequence identity varies from 10% to 35%, as calculated by the program Dali (Holm and Sander 1993). In addition, our structure is essentially identical to that reported recently for this domain (Jeong et al. 2006), with RMSD of ∼0.37 Å between 145 equivalent Cα atoms. An exception is the extended loop at the C terminus of the domain (Fig. 4A), residues 465–482, which are absent from the expression construct for the other structure (Jeong et al. 2006).
Figure 4.
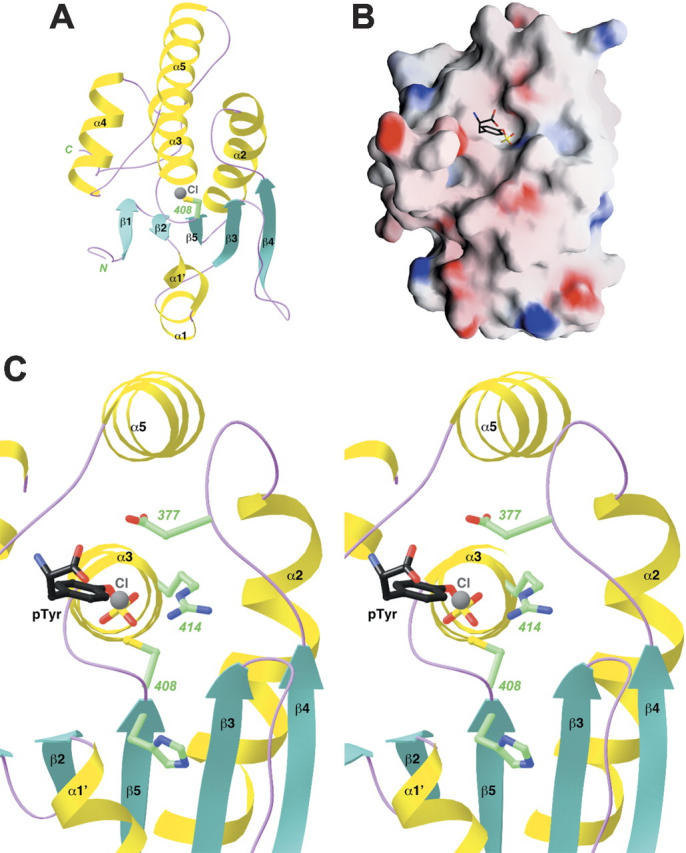
Structure of the catalytic domain of MKP5. (A) Schematic representation of the structure of the catalytic domain of MKP5. The catalytic Cys408 residue is shown in green. A chloride ion bound in the active site is shown in gray. (B) Molecular surface of the MKP5 catalytic domain. The binding mode of phosphotyrosine to PTP1B is shown (Puius et al. 1997). (C) A closeup of the active site region of the CD of MKP5. The side chains of residues His407, Cys408, Arg414, and Asp377 are shown in green. The chloride ion is shown in gray, and the phosphotyrosine (pTyr) observed in the structure of PTP1B is shown in black (Puius et al. 1997). Panels (A) and (C) were produced with Ribbons (Carson 1987) and panel (B) with Grasp (Nicholls et al. 1991).
The catalytic residue, Cys408, is located just after strand β5 and also near the N-terminal end of helix α3 (Fig. 4A). This side chain is probably activated by the expected stabilization of the thiolate by the dipole moment of this helix. The active site is positioned at the bottom of a shallow depression on the surface of the enzyme (Fig. 4B). We also observed the binding of a chloride ion in the active site of CD. It is located 3.3 Å from the Cys408 side chain and has ionic interactions with the side chain of Arg414 (Fig. 4C). In addition, the chloride ion is situated at the N-terminal end of helix α3 and should also have favorable interactions with the dipole moment of this helix. This chloride ion is a mimic for the phosphate group of the substrate, as revealed by a comparison with the structure of PTP1B in complex with phosphotyrosine (Fig. 4C; Puius et al. 1997). The strictly conserved Arg414 side chain helps recognize the phosphate group, and the Asp377 residue, in the β4–α2 loop (Fig. 4C), protonates the leaving group (MAP kinase after dephosphorylation) and then functions as the general base for the hydrolysis of the phosphocysteine intermediate.
Most MKPs have low phosphatase activity on their own and become activated upon binding the target MAP kinase. Our preliminary data show that the CD of MKP5 actually possesses phosphatase activity and can hydrolyze the p-nitrophenolphosphate (pNPP) model substrate. There are large structural differences between the MKP5 CD and the CD from other MKPs (Fig. 5), which have a distorted active site and are probably in an inactive state (Stewart et al. 1999; Farooq et al. 2003). Most importantly, the β5–α3 loop, which interacts with the phosphate group of the substrate, and the β4–α2 loop, which contains the catalytic Asp residue, have large structural differences in the catalytic domain of Pyst1 (Fig. 5; Stewart et al. 1999). In comparison, our studies suggest that the MKP5 CD may be in an active conformation on its own, and a similar conclusion was reached from a different study on this CD (Jeong et al. 2006). DUSP5, an ERK-specific MKP, also appears to be active in the absence of MAPK binding (Mandl et al. 2005), and the structure of the CD of this phosphatase confirms that it is in the active conformation (Jeong et al. 2007).
Figure 5.

Structural comparison between the catalytic domains of MKP5 and Pyst1. The CD of MKP5 is shown in color, and that of Pyst1 in gray (Stewart et al. 1999). The catalytic Cys408 and Asp377 residues of MKP5 are shown in green. Produced with Ribbons (Carson 1987).
In summary, we have determined the crystal structures of the MAP kinase binding domain and the catalytic domain of human MKP5. Both structures show large differences from that of BD and CD of other MKPs. The structural differences for the BD may help define the substrate preferences of these enzymes, and differences in the active site region of the CD may be the molecular basis for its constitutive activity.
Materials and Methods
Protein expression and purification
The MAP kinase binding domain (residues 139–287) and the catalytic domain (residues 305–482) of human MKP5 were subcloned into the pET26b vector (Novagen) and overexpressed in E. coli at 20°C. The recombinant proteins contain a hexa-histidine tag at the C terminus. After cell lysis, the soluble proteins were purified by nickel-agarose and gel-filtration chromatography. The binding domain was concentrated to 25 mg/mL in a buffer containing 20 mM Tris (pH 7.4), 500 mM NaCl, 5 mM DTT, and 5% (v/v) glycerol and stored at −80°C. The catalytic domain was concentrated to 45 mg/mL in a buffer containing 20 mM Tris (pH 7.4), 150 mM NaCl, 5 mM DTT, and 5% (v/v) glycerol. The C-terminal His-tag was not removed for crystallization.
The selenomethionyl protein of the binding domain was produced in DL41(DE3) cells, grown in defined LeMaster media supplemented with selenomethionine (Hendrickson et al. 1990). The selenomethionyl protein of the catalytic domain was produced in a minimum medium (M9) supplemented with several specific amino acids to inhibit the endogenous biosynthesis of methionine (Doublie et al. 1996). The samples were purified following the same protocol as that for the native proteins.
Protein crystallization
Crystals of the MAP kinase binding domain were prepared by the sitting-drop vapor diffusion method at 4°C. Initial crystallization conditions for the wild-type protein were identified by sparse-matrix screening with commercial kits (Hampton Research). The reservoir solution contained 100 mM sodium acetate (pH 4.5) and 2.3–2.5 M ammonium acetate. The protein was at a concentration of 12 mg/mL. Plate-shaped crystals appeared in about 4–5 d and grew to full size in 2 wk. They were cryo-protected with a solution containing 100 mM sodium acetate (pH 4.5), 2.3 M ammonium acetate, and 35% (v/v) glycerol and flash-frozen in liquid propane for data collection at 100 K.
Crystals of the MKP5 catalytic domain were prepared by the sitting-drop vapor diffusion method at 4°C. The reservoir solution contained 100 mM Bis-tris (pH 6.0) and 2.7–3.0 M sodium chloride. The protein was at a concentration of 15 mg/mL (diluted 1:2 with 30 mM Tris at pH 7.4 and 3 mM DTT from the original stock). Bipyramid-shaped crystals appeared overnight and grew to full size in 3–4 d. They were cryo-protected with the reservoir solution supplemented with 30% (v/v) ethylene glycol and flash-frozen in liquid propane for data collection at 100 K.
Crystals of the binding domain belong to space group P21, with cell parameters of a = 49.3 Å, b = 52.2 Å, c = 62.3 Å, and β = 89.4°. There are two molecules of the binding domain in the asymmetric unit. Crystals of the catalytic domain belong to space group P3221, with cell parameters of a = b = 92.5 Å and c = 78.1 Å. There is one molecule of the catalytic domain in the asymmetric unit.
Data collection and processing
X-ray diffraction data were collected at 100 K on an ADSC CCD at the X4A beamline of Brookhaven National Laboratory. The diffraction images were processed and scaled with the HKL package (Otwinowski and Minor 1997). A selenomethionyl single-wavelength anomalous diffraction (SAD) data set to 2.2 Å resolution was collected for the binding domain, while a selenomethionyl multiple-wavelength anomalous diffraction (MAD) data set to 2.9 Å resolution was collected for the catalytic domain. Structure refinement of the catalytic domain was carried out against a data set collected on the native protein, to 2.8 Å resolution. The data processing statistics are summarized in Table 1.
Structure determination and refinement
For the binding domain, the locations of six Se atoms were determined with the program SnB (Weeks and Miller 1999). Reflection phases to 2.2 Å resolution were calculated based on the SAD data and improved with the program SOLVE/RESOLVE (Terwilliger 2003), which also automatically located about 40% of the residues in the molecule. For the catalytic domain, the locations of five Se atoms were determined based on the MAD data, and reflection phases to 2.9 Å resolution were calculated and improved with the program SOLVE/RESOLVE (Terwilliger 2003).
For both structures, the full atomic model was built into the electron density with the program O (Jones et al. 1991). The structure refinement was carried out with the program CNS (Brunger et al. 1998). The statistics on the structure refinement are summarized in Table 1.
Acknowledgments
We thank Randy Abramowitz and John Schwanof for setting up the X4A beamline at the NSLS. This research is supported in part by a grant from the NIH to L.T.
Footnotes
Reprint requests to: Liang Tong, Department of Biological Sciences, Columbia University, 1212 Amserdam Ave., New York, NY 10027, USA; e-mail: ltong@columbia.edu; fax: (212) 865-8246.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.062712807.
References
- Alphey M.S., Williams, R.A.M., Mottram, J.C., Coombs, G.H., and Hunter, W.N. 2003. The crystal structure of Leishmania major 3-mercaptopyruvate sulfurtransferase. J. Biol. Chem. 278: 48219–48227. [DOI] [PubMed] [Google Scholar]
- Bordo D. and Bork, P. 2002. The rhodanese/Cdc25 phosphatase superfamily. Sequence-structure-function relations. EMBO Rep. 3: 741–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordo D., Deriu, D., Colnaghi, R., Carpen, A., Pagani, S., and Bolognesi, M. 2000. The crystal structure of a sulfurtransferase from Azotobacter vinelandii highlights the evolutionary relationship between the rhodanese and phosphatase enzyme families. J. Mol. Biol. 298: 691–704. [DOI] [PubMed] [Google Scholar]
- Brunger A.T., Adams, P.D., Clore, G.M., DeLano, W.L., Gros, P., Grosse-Kunstleve, R.W., Jiang, J.-S., Kuszewski, J., Nilges, M., Pannu, N.S., et al. 1998. Crystallography & NMR System: A new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 54: 905–921. [DOI] [PubMed] [Google Scholar]
- Camps M., Nichols, A., and Arkinstall, S. 1999. Dual specificity phosphatases: A gene family for control of MAP kinase function. FASEB J. 14: 6–16. [PubMed] [Google Scholar]
- Carson M. 1987. Ribbon models of macromolecules. J. Mol. Graph. 5: 103–106. [Google Scholar]
- Chang L. and Karin, M. 2001. Mammalian MAP kinase signaling cascades. Nature 410: 37–40. [DOI] [PubMed] [Google Scholar]
- Davis R.J. 2000. Signal transduction by the JNK group of MAP kinases. Cell 103: 239–252. [DOI] [PubMed] [Google Scholar]
- Dickinson R.J. and Keyse, S.M. 2006. Diverse physiological functions for dual-specificity MAP kinase phosphatases. J. Cell Sci. 119: 4607–4615. [DOI] [PubMed] [Google Scholar]
- Doublie S., Kapp, U., Aberg, A., Brown, K., Strub, K., and Cusack, S. 1996. Crystallization and preliminary X-ray analysis of the 9 kDa protein of the mouse signal recognition particle and the selenomethionyl-SRP9. FEBS Lett. 384: 219–221. [DOI] [PubMed] [Google Scholar]
- Farooq A. and Zhou, M.-M. 2004. Structure of regulation of MAPK phosphatases. Cell. Signal. 16: 769–779. [DOI] [PubMed] [Google Scholar]
- Farooq A., Chaturvedi, G., Mujtaba, S., Plotnikova, O., Zeng, L., Dhalluin, C., Ashton, R., and Zhou, M.-M. 2001. Solution structure of ERK2 binding domain of MAPK phosphate MKP-3: Structural insights into MKP-2 activation by ERK2. Mol. Cell 7: 387–399. [DOI] [PubMed] [Google Scholar]
- Farooq A., Plotnikova, O., Chaturvedi, G., Yan, S., Zeng, L., Zhang, Q., and Zhou, M.-M. 2003. Solution structure of the MAPK phosphatase PAC-1 catalytic domain. Insights into substrate-induced enzymatic activation of MKP. Structure 11: 155–164. [DOI] [PubMed] [Google Scholar]
- Fauman E.B., Cogswell, J.P., Lovejoy, B., Rocque, W.J., Holmes, W., Montana, V.G., Piwnica-Worms, H., Rink, M.J., and Saper, M.A. 1998. Crystal structure of the catalytic domain of the human cell cycle control phosphatase, Cdc25A. Cell 93: 617–625. [DOI] [PubMed] [Google Scholar]
- Hattori M., Mizohata, E., Tatsuguchi, A., Shibata, R., Kishishita, S., Murayama, K., Terada, T., Kuramitsu, S., Shirouzu, M., and Yokoyama, S. 2006. Crystal structure of the single-domain rhodanese homologue TTHA0613 from Thermus thermophilus HB8. Proteins 64: 284–287. [DOI] [PubMed] [Google Scholar]
- Hendrickson W.A. 1991. Determination of macromolecular structures from anomalous diffraction of synchrotron radiation. Science 254: 51–58. [DOI] [PubMed] [Google Scholar]
- Hendrickson W.A., Horton, J.R., and LeMaster, D.M. 1990. Selenomethionyl proteins produced for analysis by multiwavelength anomalous diffraction (MAD): A vehicle for direct determination of three-dimensional structure. EMBO J. 9: 1665–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hof P., Pluskey, S., Dhe-Paganon, S., Eck, M.J., and Shoelson, S.E. 1998. Crystal structure of the tyrosine phosphatase SHP-2. Cell 92: 441–450. [DOI] [PubMed] [Google Scholar]
- Holm L. and Sander, C. 1993. Protein structure comparison by alignment of distance matrices. J. Mol. Biol. 233: 123–138. [DOI] [PubMed] [Google Scholar]
- Jeong D.G., Yoon, T.-S., Kim, J.H., Shim, M.Y., Jung, S.-K., Son, J.H., Ryu, S.E., and Kim, S.J. 2006. Crystal structure of the catalytic domain of human MAP kinase phosphatase 5: Structural insight into constitutively active phosphatase. J. Mol. Biol. 360: 946–955. [DOI] [PubMed] [Google Scholar]
- Jeong D.G., Cho, Y.H., Yoon, T.-S., Kim, J.H., Ryu, S.E., and Kim, S.J. 2007. Crystal structure of the catalytic domain of human DUSP5, a dual specificity MAP kinase protein phosphatase. Proteins 66: 253–258. [DOI] [PubMed] [Google Scholar]
- Johnson G.L. and Lapadat, R. 2002. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science 298: 1911–1912. [DOI] [PubMed] [Google Scholar]
- Jones T.A., Zou, J.Y., Cowan, S.W., and Kjeldgaard, M. 1991. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A 47: 110–119. [DOI] [PubMed] [Google Scholar]
- Laskowski R.A., Macarthur, M.W., Moss, D.S., and Thornton, J.M. 1993. Procheck—A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26: 283–291. [Google Scholar]
- Lee J.-O., Yang, H., Georgescu, M.M., Di Cristofano, A., Maehama, T., Shi, Y., Dixon, J.E., Pandolfi, P., and Pavletich, N.P. 1999. Crystal structure of the PTEN tumor suppressor: Implications for its phosphoinositide phosphatase activity and membrane association. Cell 99: 323–334. [DOI] [PubMed] [Google Scholar]
- Lin Y.J., Dancea, F., Lohr, F., Klimmek, O., Pfeiffer-Marek, S., Nilges, M., Wienk, H., Kroger, A., and Ruterjans, H. 2004. Solution structure of the 30 kDa polysulfide-sulfur transferase homodimer from Wolinella succinogenes. Biochemistry 43: 1418–1424. [DOI] [PubMed] [Google Scholar]
- Mandl M., Slack, D.N., and Keyse, S.M. 2005. Specific inactivation and nuclear anchoring of extracellular signal-regulated kinase 2 by the inducible dual-specificity protein phosphatase DUSP5. Mol. Cell. Biol. 25: 1830–1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin H., Flandez, M., Nombela, C., and Molina, M. 2005. Protein phosphatases in MAPK signaling: We keep learning from yeast. Mol. Microbiol. 58: 6–16. [DOI] [PubMed] [Google Scholar]
- Nicholls A., Sharp, K.A., and Honig, B. 1991. Protein folding and association: Insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins 11: 281–296. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z. and Minor, W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276: 307–326. [DOI] [PubMed] [Google Scholar]
- Pearson G., Robinson, F., Beers Gibson, T., Xu, B.E., Karandikar, M., Berman, K., and Cobb, M.H. 2001. Mitogen-activated protein (MAP) kinase pathways: Regulation and physiological functions. Endocr. Rev. 22: 153–183. [DOI] [PubMed] [Google Scholar]
- Ploegman J.H., Drent, G., Kalk, K.H., and Hol, W.G.J. 1978a. Structure of bovine liver rhodanese. I. Structure determination at 2.5 Å resolution and a comparison of the conformation and sequence of its two domains. J. Mol. Biol. 123: 557–594. [PubMed] [Google Scholar]
- Ploegman J.H., Drent, G., Kalk, K.H., Hol, W.G.J., Heinrikson, R.L., Keim, P., Weng, L., and Russell, J. 1978b. The covalent and tertiary structure of bovine liver rhodanese. Nature 273: 124–129. [DOI] [PubMed] [Google Scholar]
- Puius Y.A., Zhao, Y., Sullivan, M., Lawrence, D.S., Almo, S.C., and Zhang, Z.-Y. 1997. Identification of a second aryl phosphate-binding site in protein-tyrosine phosphatase 1B: A paradigm for inhibitor design. Proc. Natl. Acad. Sci. 94: 13420–13425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song H., Hanlon, N., Brown, N.R., Noble, M.E.M., Johnson, L.N., and Barford, D. 2001. Phosphoprotein–protein interactions revealed by the crystal structure of kinase-associated phosphatase in complex with phosphoCDK2. Mol. Cell 7: 615–626. [DOI] [PubMed] [Google Scholar]
- Spallarossa A., Donahue, J.L., Larson, T.J., Bolognesi, M., and Bordo, D. 2001. Escherichia coli GlpE is a prototype sulfurtransferase for the single-domain rhodanese homology superfamily. Structure 9: 1117–1125. [DOI] [PubMed] [Google Scholar]
- Stewart A.E., Dowd, S., Keyse, S.M., and McDonald, N.Q. 1999. Crystal structure of the MAPK phosphatse PystI catalytic domain and implications for regulated activation. Nat. Struct. Biol. 6: 174–181. [DOI] [PubMed] [Google Scholar]
- Tanoue T., Yamamoto, T., and Nishida, E. 2002. Modular structure of a docking surface on MAPK phosphatase. J. Biol. Chem. 277: 22942–22949. [DOI] [PubMed] [Google Scholar]
- Terwilliger T.C. 2003. SOLVE and RESOLVE: Automated structure solution and density modification. Methods Enzymol. 374: 22–37. [DOI] [PubMed] [Google Scholar]
- Theodosiou A. and Ashworth, A. 2002. MAP kinase phosphatases. Genome Biol. 3: 3001–3010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weeks C.M. and Miller, R. 1999. The design and implementation of SnB v2.0. J. Appl. Crystallogr. 32: 120–124. [Google Scholar]
- Yuvaniyama J., Denu, J.M., Dixon, J.E., and Saper, M.A. 1996. Crystal structure of the dual specificity protein phosphatase VHR. Science 272: 1328–1331. [DOI] [PubMed] [Google Scholar]
- Zhang Y., Blattman, J.N., Kennedy, N.J., Duong, J., Nguyen, T., Wang, Y., Davis, R.J., Greenberg, P.D., Flavell, R.A., and Dong, C. 2004. Regulation of innate and adaptive immune responses by MAP kinase phosphatase 5. Nature 430: 793–797. [DOI] [PubMed] [Google Scholar]
- Zhou B., Zhang, J., Liu, S., Reddy, S., Wang, F., and Zhang, Z.-Y. 2006. Mapping ERK2-MKP3 binding interfaces by hydrogen/deuterium exchange mass spectrometry. J. Biol. Chem. 281: 38834–38844. [DOI] [PubMed] [Google Scholar]