

Abstract
PhyA from Selenomonas ruminantium (PhyAsr), is a bacterial protein tyrosine phosphatase (PTP)-like inositol polyphosphate phosphatase (IPPase) that is distantly related to known PTPs. PhyAsr has a second substrate binding site referred to as a standby site and the P-loop (HCX5R) has been observed in both open (inactive) and closed (active) conformations. Site-directed mutagenesis and kinetic and structural studies indicate PhyAsr follows a classical PTP mechanism of hydrolysis and has a broad specificity toward polyphosphorylated myo-inositol substrates, including phosphoinositides. Kinetic and molecular docking experiments demonstrate PhyAsr preferentially cleaves the 3-phosphate position of Ins P6 and will produce Ins(2)P via a highly ordered series of sequential dephosphorylations: D-Ins(1,2,4,5,6)P5, Ins(2,4,5,6)P4, D-Ins(2,4,5)P3, and D-Ins(2,4)P2. The data support a distributive enzyme mechanism and suggest the PhyAsr standby site is involved in the recruitment of substrate. Structural studies at physiological pH and high salt concentrations demonstrate the “closed” or active P-loop conformation can be induced in the absence of substrate. These results suggest PhyAsr should be reclassified as a D-3 myo-inositol hexakisphosphate phosphohydrolase and suggest the PhyAsr reaction mechanism is more similar to that of PTPs than previously suspected.
Keywords: inositol polyphosphate phosphatase, protein tyrosine phosphatase, phosphoinositide phosphatase, phytase, myo-inositol, P-loop, hydrolysis pathway
Protein tyrosine phosphatase (PTP) superfamily enzymes have been discovered in a range of prokaryotes, and most appear to serve roles that mimic their better-known eukaryotic counterparts as regulators of cellular function (Shi et al. 1998; Kennelly and Potts 1999). Some bacteria have adapted PTPs as “molecular missiles,” secreted into the infected host where they assist in the progression of infection (Bliska et al. 1991; Fu and Galan 1998; Bretz et al. 2003). The recently described PTP-like inositol polyphosphate phosphatase (IPPase) from Selenomonas ruminantium, PhyAsr, contains a PTP-like active site signature sequence (HCEAGVGR) but lacks significant primary sequence identity with known IPPases and PTPs (<20%). While its biological function is unclear, it is the first example of a PTP-like enzyme with activity toward myo-inositol hexakisphosphate (Ins P6, Fig. 1), one of the most abundant inositol polyphosphates (IPPs) in a majority of prokaryotic and eukaryotic cells (Sasakawa et al. 1995).
Figure 1.

The most energetically favorable conformation of myo-inositol hexakisphosphate, displaying 5 equatorial and 1 axial phosphate groups. Ins P6 is a myo compound with a plane of symmetry that crosses through C2 and C5, and two prochiral carbon pairs, C1/C3 and C4/C6. If the carbon ring is numbered counterclockwise, as shown by numbers inside the ring, assignment of a single substituent on C1 is 1D. Conversely, if the carbon ring is numbered clockwise (numbers outside the ring) the assignment is 1L (Loewus and Murthy 2000).
The X-ray crystallographic structure of PhyAsr (Chu et al. 2004) reveals a PTP-like fold and several novel catalytic properties have been inferred. In particular, it was suggested this enzyme is the first example of (1) a processive myo-inositol polyphosphatase (IPPase), (2) an IPPase that initiates hydrolysis at the 5-phosphate position of 2-equatorial Ins P6, and (3) a PTP-like enzyme that undergoes a significant substrate-induced P-loop conformation change. In the proposed mechanism, substrate moves between the catalytic site and a second substrate binding or standby site without being released as the successive intermediates are hydrolyzed. All previously characterized IPPases are distributive enzymes that release their products after each hydrolysis (Konietzny and Greiner 2002). These products may then act as substrates in further hydrolysis reactions. PTP and PTP-like enzymes frequently undergo substrate-induced conformational changes in the TrpProAsp(WPD)-loop that contains the conserved Asp general acid (Zhang 1998, 2003). In contrast, substantial substrate induced conformational changes in the P-loop, responsible for binding the scissile phosphate, have not been previously reported. Finally, preference for the 5-phosphate position of Ins P6 has only been reported in one other IPPase (Barrientos et al. 1994) and, to our knowledge, the 2-equatorial conformation of the substrate has not been observed in structures of IPPase complexes.
The catalytic mechanism of PTP superfamily enzymes has been extensively studied. The active site signature sequence, HC(X)5R, is required for activity (Zhang et al. 1994a; Zhang 1998, 2002, 2003) and follows a two-step, general acid–general base mechanism of dephosphorylation. The invariant Cys residue exists as a thiolate and catalysis involves the formation of a phosphocysteine intermediate (Cirri et al. 1993; Zhang et al. 1994b; Zhou et al. 1994). Main-chain amines and the guanidinium group of the conserved Arg coordinate the scissile phosphate in the catalytic site and stabilize the negative charge of the substrate (Barford et al. 1994; Zhang et al. 1994b) while an invariant Asp serves as the general acid (Zhang et al. 1994a; Jia et al. 1995; Lohse et al. 1997).
Given the movement of the P-loop, processive mechanism, and specificity for the 5-phosphate position of Ins P6, inferred from previous structural studies (Chu et al. 2004), we have investigated the catalytic mechanism using a combination of kinetic, site-directed mutant, and structural studies. We present experimental data that indicates PhyAsr follows a classical PTP mechanism of catalysis with a preference for the D-3-phosphate position of Ins P6. The preferred order in which individual phosphates are hydrolyzed from Ins P6 by the enzyme (dephosphorylation pathways) has been determined and is consistent with the sequential removal of phosphate groups. Finally, in the presence of high salt concentrations and pHs ≥ 6.5, the P-loop of PhyAsr is observed in the “closed” or active conformation in both the presence and absence of ligand.
Results
Catalytic mechanism
The catalytic properties of the recombinant wild-type PhyAsr toward the naturally occurring Ins P6 substrate have been determined at its pH optimum (Table 1). The rate of phosphate release can be saturated and the initial rates of reaction suggest a classical Michaelis-Menton enzyme mechanism. This is consistent with the kinetic properties of other PTP superfamily enzymes toward the commonly used phosphatase substrate p-nitrophenyl phosphate (pNPP) (Zhang et al. 1992; Guo et al. 2002). The apparent kcat and Km of PhyAsr for Ins P6 are 264 ± 19/s and 425 ± 28 μM, respectively. PhyAsr has some activity toward pNPP, phosphotyrosine, and phosphatidylinositol-(3,4,5)-trisphosphate (<1% relative).
Table 1.
Kinetic parameters for phytase activity of PhyAsr and various PhyAsr mutants

To characterize the PhyAsr catalytic mechanism and compare it with existing PTP enzymes, a series of site-directed mutants have been produced and subjected to kinetic and structural studies. In particular, the Cys252 nucleophile, Asp223 general acid, and the invariant Arg258 of the P-loop have been conservatively mutated to Ser, Asn, and Lys, respectively. The kinetic constants associated with these mutant enzymes and Cys252Ala are also shown in Table 1. The Cys252Ser and Cys252Ala mutants have no detectable IPPase activity in our assay, while the Asp223Asn and Arg258Lys mutant kcat/Km values are reduced by 30- and 1000–fold, respectively. The Asp223Asn Km decreases by more than 10-fold while the Arg258Lys Km is essentially unchanged. The effects of the Arg258Lys and both Cys252 mutants on PhyAsr activity are identical to those observed for equivalent mutations in PTPs (Zhang et al. 1994b; Flint et al. 1997). This suggests the P-loop movements previously observed in PhyAsr do not alter the roles of the invariant residues in the classical PTP-like catalytic mechanism. The decrease in Km for the Asp223Asn mutant is a novel feature of PhyAsr as equivalent mutations result in an increased Km for PTP1, Yersinia PTPase, and yeast low-molecular-weight PTPase (Wu and Zhang 1996; Lohse et al. 1997; Keng et al. 1999). The lower Km likely arises from favorable electrostatic interactions between Asn223 and Ins P6. This suggests the low pH optimum of wild-type PhyAsr is required to maintain Asp223 in its acid form and minimize unfavorable electrostatic interaction with the large net negative charge (−6 at pH 5) of Ins P6. This interpretation is supported by a shift in the pH optimum of Asp223Asn from 5 to 6 (Fig. 2). We speculate that a solvent molecule serves as the general acid in the Asp223Asn mutant and its higher pKa is responsible for the shift in pH optimum.
Figure 2.

The effect of pH on phytase activity of PhyAsr and the Asp223Asn mutant. Standard phosphatase assays were run over a pH range of 3.5–7. The overlapping buffer systems used were: 50 mM formate (pH 3.5–4), 50 mM sodium acetate (pH 4–6), and 50 mM imidazole (pH 6–7). The data are mean values ± standard deviation of three separate experiments. (Solid circles) Wild type; (open circles) D223N.
To further confirm that Cys252 is the nucleophile, PhyAsr was chemically modified using iodoacetic acid (IAA). A 60-min incubation with IAA resulted in a 96% loss of enzyme activity due to a single alkylation event (Supplemental material S1), as verified by mass spectrometry. Further, Ins P6 provides concentration-dependent protection against inactivation of the wild-type enzyme caused by IAA (Supplemental material S2).
X-ray crystallographic structures
Attempts to grow crystals of the PhyAsr mutants under the conditions used by Chu et al. (2004) were unsuccessful. A crystallization screen identified high salt conditions that yield crystals that diffract to a higher resolution than the previously published structure (1.8 Å). Despite the differences in the crystallization conditions, the space group, unit cell, crystal packing, and crystal contacts are nearly identical to those of the previously reported crystals (Chu et al. 2004).
In PTPs, the WPD-loop contains the conserved Asp general acid and undergoes a well-characterized conformational change during catalysis (Zhang 1998, 2003). The equivalent loop in PhyAsr contains an equivalent Asp (223) but cannot undergo a conformational change because of steric clashes that would result from contacts with the partial β-barrel domain, which is absent in other PTPs. Instead, the P-loop of PhyAsr has been observed in two distinct conformations: open (ligand free) and closed (ligand bound) (Chu et al. 2004). To determine whether our mutations affect the overall fold, or P-loop conformation of PhyAsr, the X-ray structures of each active site mutant have been determined and are shown in Figure 3. The structure of the Cys252Ser mutant has a sulfate ion bound in the active site, whereas the Asp223Asn and Arg258Lys structures are ligand free. Least-squares superposition of the main chain atoms of these structures with wild-type PhyAsr (Protein Data Bank [PDB] accession no. 1U24; Chu et al. 2004) yield root mean square deviations (RMSDs) in the range of 0.49–0.55 Å, indicating the overall structure of the protein is not disrupted by these active site mutations. The P-loop residues (251–259) of the three mutants have virtually identical conformations and can be superimposed with RMSDs of 0.13 (Asp223Asn onto Cys252Ser) and 0.19 Å (Arg258Lys onto Cys252Ser). Consequently, movement in the P-loop between an open, catalytically inactive form and a closed, catalytically active form on ligand binding is not observed in these structures (Fig. 3). Furthermore, least-squares superposition of the P-loops in our mutant structures and in the PhyAsr–hexasulfate complex (PDB accession no. 1U26; closed or active conformation) yield RMSDs below 0.30 Å. The equivalent superpositions between our mutants and 1U24 (open or inactive conformation) have RMSDs greater than 1.00 Å. Consequently, each of our mutant structures have P-loop conformations equivalent to the ligand-bound, closed conformation reported for 1U26. This is the same P-loop conformation observed in multiple PTP X-ray structures both in the presence and absence of ligand (Burke and Zhang 1998).
Figure 3.

Structures of three active site mutants of PhyAsr. (A) Cys252Ser; (B) Asp223Asn; (C) Arg258Lys; and (D) least-squares superposition of the Cys252Ser mutant structure (yellow loop) with the apo structure (1U24) (gray loop) previously solved (Chu et al. 2004). The critical active site residues are shown as sticks. Least-squares superposition of the P-loop main-chain atoms (residues 251–259) of the Asp223Asn and Arg258Lys mutants, 1U26, and the 1U24 with P-loop of the Cys252Ser mutant results in RMSDs of 0.13, 0.19, 0.11, and 1.15 Å, respectively. Oxygens are shown in red, nitrogens are shown in blue, and sulfur is shown in yellow. Figure was generated with PyMOL (Delano 2002).
While the P-loop remains in the catalytically competent conformation in each of our mutants, there are subtle differences in the positions of active site residues in the Arg258Lys structure. Arg258 forms a conserved salt bridge with Glu136 that orients the guanidinium relative to the scissile phosphate. The replacement of Arg with Lys results in a shorter side chain and the Lys258 salt bridge with Glu136 is lengthened by 0.88 Å. To compensate for both the size and a small movement in the Lys258 side chain, the Cγ of Asp223 shifts by 0.95 Å toward the Lys258 Nξ and the Asp223 carboxylate fills the volume vacated as a result of the mutation.
Substrate specificity
The presence of a second Ins P6 binding site (Chu et al. 2004) has led to the suggestion PhyAsr processively removes phosphate groups from Ins P6. To test this hypothesis and to determine the order of phosphate removal, the products of prolonged incubation of PhyAsr and Ins P6 were analyzed using high-performance ion-pair chromatography (HPIC). Purified PhyAsr was incubated with excess Ins P6 for 30 and 90 min prior to stopping the reaction. HPIC profiles of the Ins P6 hydrolysis products are shown in Figure 4. After 30 min of incubation, all Ins P6 has been hydrolyzed to lower-order IPPs. This is not consistent with a processive mechanism that hydrolyzes Ins P6 to a lower IPP product without releasing the intermediates. Additionally, D/L-Ins(1,2,4,5,6)P5 was the only detectable Ins P5 product. Shorter incubation times with Ins P6 were tested in an effort to detect other Ins P5 products; however, the only detectable Ins P5 product within the sensitivity limits of the HPIC is the D/L-Ins(1,2,4,5,6)P5.
Figure 4.

High-performance ion-pair chromatography (HPIC) analysis of the hydrolysis products of IPPs by PhyAsr. (A) Reference sample containing a mixture of IPP standards (≤6 phosphates). The source and identity of the unmarked standard peaks is indicated in Skoglund et al. (1998). Peaks: (1) D/L-Ins(1,2,4,5,6)P5; (2) Ins(2,4,5,6)P4; (3) D/L-Ins(1,2,5,6)P4; (4) D/L-Ins(1,2,4,6)P4; (5) D/L-Ins(1,4,5)P3; D/L-Ins(2,4,5)P3; (6) D/L-Ins(1,2,6)P3, Ins(1,2,3)P3; (7) D/L-Ins(1,2,4)P3; (8) D/L-Ins(2,4)P2; (9) D/L-Ins(1,2)P2, Ins(2,5)P2, D/L-Ins(4,5)P2. (B) PhyAsr incubated with Ins P6 for 30 min. (C) PhyAsr incubated with Ins P6 for 90 min. Peaks representative of major pathway products are labeled accordingly.
In addition to D/L-Ins(1,2,4,5,6)P5, the remaining major products present after 30 min of incubation are Ins(2,4,5,6)P4 and D/L-Ins(2,4,5)P3 and represent 80% of the total Ins P4 and Ins P3 products. Minor products (<10% of the total) formed are D/L-Ins(1,2,5,6)P4, D/L-Ins(1,2,4,6)P4, D/L-Ins(1,2,6)P3, and D/L-Ins(1,2,4)P3. Trace amounts of D/L-Ins(2,4)P2 and D/L-Ins(1,2)P2 are also formed. After 90 min of incubation, all of the Ins P5 and all but trace amounts of the major Ins P4 product had been hydrolyzed to the Ins P3 and Ins P2 products indicated above. Overall, the hydrolysis of Ins P6 to Ins P2 occurs in a largely ordered, sequential fashion despite the distributive nature of PhyAsr.
The kcat and Km for the enzymatic degradation of the myo-inositol phosphates present in our hydrolysis pathway were determined to aid in the elucidation of the hydrolysis pathway and preferred substrates of PhyAsr. The respective kinetic parameters are given in Table 2. To confirm the identity of the Ins P5 isomer, kinetic parameters were determined for the enzymatic hydrolysis of D-Ins(1,2,4,5,6)P5 and compared with those determined for the Ins P5 produced by PhyAsr. The kcat and Km for the hydrolysis of D-Ins(1,2,4,5,6)P5 are 295/s and 390 μM. This is virtually identical to the kcat (298/s) and Km (402 μM) for the hydrolysis of the Ins P5 produced by PhyAsr and is further proof the enzyme preferentially hydrolyzes the D-3-phosphate of Ins P6. Similarly, kinetic parameters were determined for D-Ins(2,4)P2 and D-Ins(2,6)P2, and compared with those for the Ins P2 generated by PhyAsr to confirm the isomer generated from Ins(2,4,5)P3. The results indicate that D-Ins(2,4)P2 is the major Ins P2 product. The results of gas chromatography-mass spectrometry analysis indicate that the end product of Ins P6 hydrolysis by PhyAsr is Ins(2)P. Thus, PhyAsr dephosphorylates Ins P6 via three independent pathways, the major pathway (80%) being Ins(1,2,3,4,5,6)P6, D-Ins(1,2,4,5,6)P5, Ins(2,4,5,6)P4, D-Ins(2,4,5)P3, and D-Ins(2,4)P2, and finally Ins(2)P (D-3,D-1,D-6,D-5,D-4; Fig. 5).
Table 2.
Kinetic parameters for enzymatic myo-inositol polyphosphate dephosphorylation by PhyAsr

Figure 5.
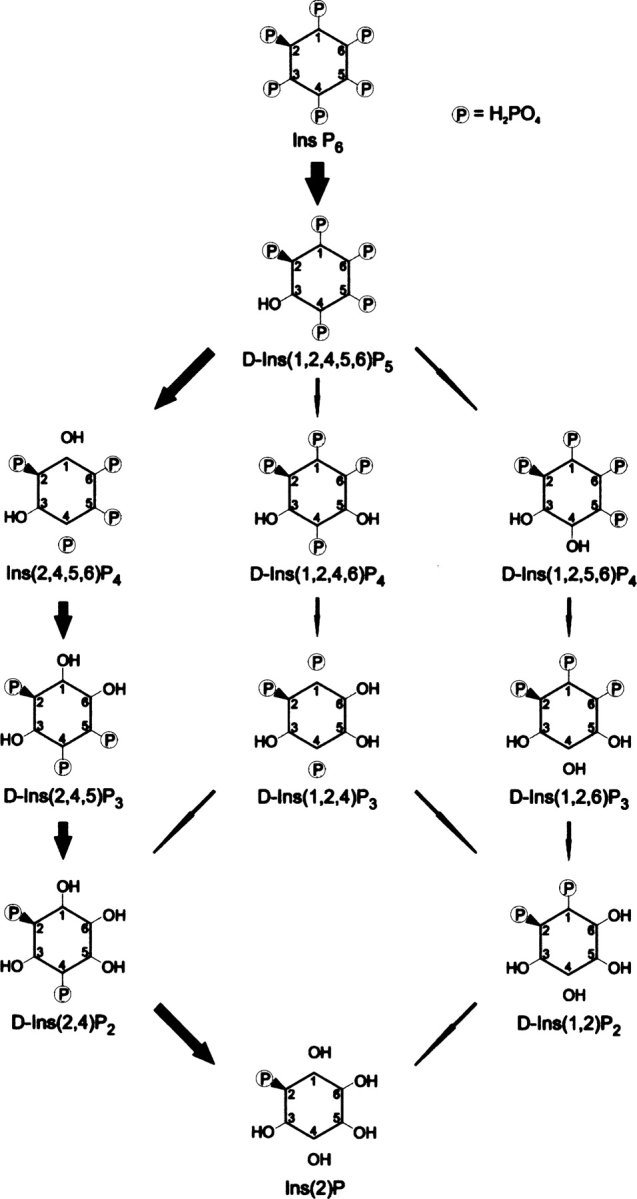
The dephosphorylation pathways of Ins P6 by PhyAsr as determined by high-performance ion-pair chromatography (HPIC) and kinetic analysis. Larger arrows indicate major pathway, smaller arrows indicate minor pathways. The major pathway accounts for ∼80% of degradation products.
The order of preference of PhyAsr for IPP substrates according to the kinetic parameters (Table 2), from highest to lowest, is Ins P5 > Ins P6 > Ins P4 > Ins P3 > Ins P2. This suggests a slight preference for a more highly phosphorylated substrate, likely because of the favorable electrostatic interactions between the positively charged binding pocket of the enzyme and the negative phosphate groups on the substrate.
Molecular docking
The demonstration that PhyAsr preferentially cleaves the D-3-phosphate contradicts the existing classification (5-phosphate preference) inferred from the PhyAsr–hexasulfate structure (1U26) at pH 7.5. Further, the HPIC results are not consistent with a processive mechanism utilizing a standby site. Given that PhyAsr is catalytically inactive at pH 7.5, molecular docking studies using the highest resolution PhyAsr structure (1.81 Å, PDB accession: 2B4P) and Ins P6 were performed to determine the potential effect of pH. The Ins P6 ligand was modeled with a charge of −6 and −9, corresponding to the charge of the ligand at the pH optimum of the enzyme (5.0) and at pH 7.5. In addition, the IPP ring conformation that predominates at pH 5.0 (D-2-axial) and pH 7.5 (D-2-equatorial) were also considered. Using default parameters, docking calculations consistently predict that Ins P6 binds with the 3-phosphate adjacent to the nucleophilic Cys252 (Table 3). The docking results further suggest the physiologically relevant Ins P6 6− ion binds more tightly to PhyAsr than the Ins P6 9− ion that exists at pHs (>7.5) where PhyAsr is inactive. Finally, there is a slight but consistent preference for D-2-axial over D-2-equatorial ring conformations among the lowest energy docking results. The preference for the D-2-axial ring conformation is supported by the position of the Asp223 general acid relative to the leaving group oxygen (Fig. 6). In 1U26, the hexasulfate ligand has a D-2-equatorial ring conformation and the bridging oxygen of the 5-phosphate is tilted away from the Asp223 carboxylate (4.0 Å contact with Oδ1). In the lowest energy docked structure, the Ins P6 ligand has a D-2-axial ring conformation and the bridging oxygen of the D-3-phosphate is directed toward the Asp223 carboxylate (3.2 Å contact with Oδ1). Taken together, the molecular docking results support the experimentally determined 3-phosphate preference and predict the physiologically relevant Ins P6 6− ion and D-2-axial ring conformations produce the lowest binding energies.
Table 3.
Summary of calculated binding energies for the molecular docking of Ins P6 with PhyAsr (accession no. 2B4P)
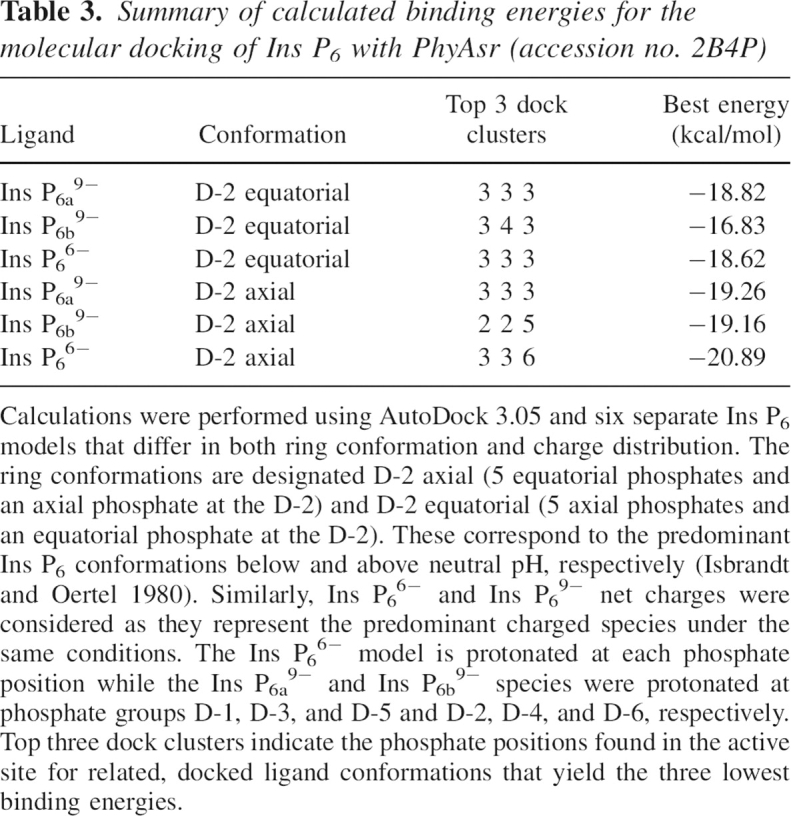
Figure 6.

The effect of binding an axial vs. an equatorial ligand in the active site of PhyAsr. PhyAsr (2B4P) with a 5-equatorial, 1-axial ligand docked into the active site was aligned with the 1U26. The inhibitor myo-inositol hexasulfate from 1U26 (Chu et al. 2004) is shown in yellow and the docked myo-inositol hexakisphosphate is shown in green. Asp223 is shown as sticks and colored cpk. Figure was generated with PyMOL (Delano 2002).
Discussion
The active site of PTP superfamily enzymes can undergo conformational rearrangements on substrate binding or as a regulatory mechanism. In PTPs, the main chain of the conserved WPD-loop undergoes significant conformational changes on substrate binding that bring the invariant Asp (general acid) within hydrogen bond distance of the leaving group (Zhang 1998, 2003). Several regulatory mechanisms have also been described that alter the main chain conformation of the active site signature sequence (HCX5R) or P-loop (Salmeen et al. 2003; van Montfort et al. 2003; Buhrman et al. 2005). More recently, the P-loop of PhyAsr has been observed in two distinct conformations with functional consequences: open (inactive) and closed (active) (Chu et al. 2004).
Isostructural and conservative mutations of the three PhyAsr active site residues that are invariant among PTP superfamily enzymes suggest the conformational differences observed in previous structural studies of PhyAsr do not affect the functional role of these residues. Each of the mutations (Cys252Ser, Asp223Asn, and Arg258Lys) produce comparable kinetic effects when compared with equivalent mutations in the mammalian receptor-like PTP LAR, the Yersinia PTP, and PTP1B (Cirri et al. 1993; Zhang et al. 1994a,c; Flint et al. 1997; Lohse et al. 1997; Orchiston et al. 2004). The sole exception is a 10-fold decrease in Km associated with the Asp223Asn mutation that is unique to PhyAsr. This difference is attributed to the large net charge (−6) associated with Ins P6 under physiological conditions and the polar, uncharged character of Asn223.
X-ray crystallographic structures of each PhyAsr mutant demonstrate the overall fold of the enzyme is unchanged in comparison with the wild-type enzyme. However, in contrast to previous suggestions that the P-loop undergoes a conformational change on substrate binding, the P-loop in each mutant structure adopts the closed or functional conformation in both the presence (Cys252Ser) and absence (Asp223Asn, Arg258Lys) of bound ligand. These differences cannot be due to differences in the crystalline lattice as each mutant structure has the same unit cell, space group, and crystalline contacts as 1U24 (open or inactive). Further, the Asp223Asn and Arg258Lys residues do not make contacts with P-loop residues in their respective structures and cannot account for the observed closed conformation. Finally, there are no conformational differences between our mutant structure and 1U24 that would prevent the P-loop from adopting the open conformation. This would suggest that factors other than substrate binding promote the functional conformation of the PhyAsr P-loop. Notable differences in the crystallization conditions of 1U24 and our mutant structures include pH and ionic strength. The higher pH of the mutant structures compared with 1U24 (4.6 vs. 6.5), the higher ionic strength (>2.0 M vs. ∼0.2 M), or both may stabilize the closed conformation.
All characterized IPPases remove phosphate from Ins Px substrates in a distributive fashion, such that each Ins Px − 1 and phosphate product are released from the enzyme and may act as a substrate in further hydrolysis reactions (Konietzny and Greiner 2002). It has been suggested that PhyAsr is processive and utilizes a standby site to bind and reorient substrate between successive hydrolysis reactions (Chu et al. 2004). Our PhyAsr-mediated Ins P6 hydrolysis reactions contain excess substrate and limiting enzyme. Hydrolysis is initiated at the D-3-phosphate position and all of the Ins P6 is converted to D-Ins(1,2,4,5,6)P5 and lower IPPs after relatively short periods of incubation. A limiting amount of a processive PhyAsr would not deplete all of the Ins P6 prior to the appearance of lower IPP end products. The D-Ins(1,2,4,5,6)P5 product is further hydrolyzed to Ins(2)P by one major (80%) and two minor pathways. The major pathway removes the phosphoryl groups in the order D-3, D-1, D-6, D-5, and finally D-4. These results provide experimental evidence that PhyAsr is a distributive enzyme and are not consistent with the standby site facilitating the processive degradation of Ins P6. While the functional role of the standby site is not readily apparent, we note the standby site does not significantly overlap the active site and suggest it may have a role recruiting additional substrate. This differs from the second substrate binding or affinity site present in PTP1B and PTPL1/FAP1 (Sun et al. 2003; Villa et al. 2005), which are structurally distinct and facilitate the binding of polypeptide substrates containing multiple phosphotyrosines.
Any enzyme that catalyzes the release of orthophosphate from Ins P6 can be referred to as a phytase (myo-inositol hexakisphosphate phosphohydrolase) (Mullaney and Ullah, 2003). Three structurally distinct classes of phytases have been kinetically and structurally characterized in the literature: histidine acid phosphatases; β-propeller phytases; and purple acid phosphatases. PhyAsr is the first example of a phytase with a PTP-like fold. On the basis of the position of the first phosphate hydrolyzed, three types of phytases are recognized by the Enzyme Nomenclature Committee of the International Union of Biochemistry, i.e., 3-phytases, which cut the D-3 phosphate (EC 3.1.3.8); 6-phytases, which cut the D-6 or L-6 phosphate (EC 3.1.3.26); and 5-phytase (EC 3.1.3.72) (Konietzny and Greiner 2002). Currently, PhyAsr is classified as a 5-phytase based upon the PhyAsr–hexasulfate structure (1U26) that shows the 5-sulfate adjacent to the invariant Cys nucleophile. The Ins P6 hydrolysis pathway studies demonstrate PhyAsr is a D-3-phytase under our experimental conditions. Of note, PhyAsr's closest structural homolog, the PTP-like phosphoinositide/-inositol phosphatase PTEN acts only on the D-3-phosphate position of its lipid substrate phosphatidylinositol (3,4,5)-trisphosphate and its IPP substrates Ins(1,3,4,5,6)P5 and Ins(1,3,4,5)P4 (Maehama and Dixon 1998; Caffrey et al. 2001). Additional evidence that PhyAsr is in fact a 3-phytase is provided by molecular docking studies that consistently predict Ins P6 binds to PhyAsr with the D-3-phosphate adjacent to the invariant thiolate under a wide variety of conditions. The docking studies also provide a rationale for some of the observed specificity of PhyAsr. In particular, the preferred Ins P6 ring conformation has five equatorial phosphate groups (D-2 axial) and corresponds to the lowest energy Ins P6 conformer at the pH optimum of the enzyme. This positions the leaving group within hydrogen bond distance of the invariant Asp223 (Fig. 6). This ring conformation also allows His224 to form a pair of hydrogen bonds between the two phosphate groups adjacent to the scissile phosphate. The bridging hydrogen bonds are optimal when the axial D-2 phosphate is adjacent to the phosphoryl group in the active site and explains the enzymes preference for hydrolyzing the D-3 phosphate and, subsequently, the D-1 phosphate.
Materials and Methods
Expression construct production
The region coding for the mature S. ruminantium IPPase (phyAsr; GeneBank accession number AF177214) was amplified from genomic DNA using polymerase chain reaction (PCR). PhyAsr forward and reverse primers included an NdeI site (CAT ATG) for cloning and a 5′ GC cap. The signal peptide sequence was determined using SignalP 3.0 (Nielsen et al. 1997; Bendtsen et al. 2004). PhyAsr numbering begins with 1 at the N terminus of the protein sequence found in GeneBank including the predicted signal peptide (first 27 residues). The PCR product was digested with NdeI and ligated into similarly digested pET28b vector (Novagen). Mutant proteins Cys252Ser, Cys252Ala, Arg258Lys, and Asp223Asn were prepared from phyAsr using the QuikChange Site-Directed Mutagenesis Kit (Stratagene) according to the manufacturer's instructions. Constructs were sequenced by automated cycle sequencing at the University of Calgary, Core DNA and Protein Services facilities. Sequence data were analyzed with the aid of SEQUENCHER version 4.0 (Gene Codes Corp.) and MacDNAsis version 3.2 (Hitachi Software Engineering Co., Ltd.).
Protein production and purification
Escherichia coli BL21 (DE3) cells (Novagen) were transformed with the phyAsr expression constructs. Protein expression was accomplished according to the instructions in the pET Systems Manual (Novagen) Protein overexpression was induced in cultures by adding IPTG to a final concentration of 1 mM. Incubation was continued overnight at 37°C.
Induced cells were harvested and resuspended in lysis buffer: 20 mM PO4 (pH 7), 300 mM NaCl, 1 mM β-mercaptoethanol (BME), 5% glycerol, and one Complete Mini, EDTA-free protease inhibitor tablet (Roche Applied Science). Cells were lysed with a Branson model 450 sonifier. Cell debris was removed by centrifugation at 20,000g. Recombinant 6×His tagged PhyAsr was purified to homogeneity by metal chelating affinity chromatography (Ni2+-NTA-agarose) according to the supplied protocol (Qiagen Corp). Protein was washed on the column with lysis buffer containing 15 mM imidazole and eluted with lysis buffer containing 400 mM imidazole. Purified protein was dialyzed overnight into 20 mM HEPES (pH 7), 300 mM NaCl, 0.1 mM EDTA, and 1 mM BME. The homogeneity of the purified protein was confirmed by 4/12% w/v SDS-polyacrylamide gel electrophoresis (Laemmli 1970) and Coomassie Brilliant Blue R-250 staining. Protein concentrations were determined using the extinction coefficient calculated by PROT-PARAM (Gasteiger et al. 2005).
Assay of enzymatic activity and quantification of the liberated phosphate
Activity measurements were carried out at 37°C. Enzyme reaction mixtures consisted of a 350-μL buffered substrate solution and 50 μL of a 25 nM enzyme solution. The buffered substrate solution contained 50 mM Na-acetate (pH 5) and 2 mM sodium phytate or a variable concentration (0.025–4 mM) of one of the individual IPPs used in our study. Ionic strength was held constant at 200 mM with the addition of NaCl. After the appropriate, empirically determined incubation period, the reactions were stopped and the liberated phosphate was quantified. Preliminary characterization, pH versus rate, and alkylation studies were done using the ammonium molybdate method previously described (Yanke et al. 1998). A 750-μL aliquot of 5% (w/v) trichloroacetic acid was added to stop the reaction, followed by the addition of 750 μL of phosphomolybdate coloring reagent. The coloring reagent was prepared by the addition of 4 volumes 1.5% (w/v) ammonium molybdate solution in 5.5% (v/v) sulfuric acid to 1 volume 2.7% (w/v) ferrous sulfate solution. Liberated inorganic phosphate was measured as A700 on the spectrophotometer. For kinetic studies we used a modified Heinonen and Lahti method, which was better suited to the range of substrate concentrations involved (Heinonen and Lahti 1981). A 1.5-mL aliquot of a freshly prepared solution of acetone/5 N H2SO4/10 mM ammonium molybdate (2:1:1 v/v/v) was added to the assay mixture for stopping and detection, followed by 100 μl of 1.0 M citric acid. Any cloudiness was removed by centrifugation prior to measurement of the absorbance at 355 nm.
To quantify the released phosphate, a calibration curve was produced for each quantification method over a range of 5–600 nmol phosphate/2 mL reaction mixture. Activity (U) was expressed as μmol phosphate liberated per min. Blanks were run by addition of the stop solution to the assay mixture prior to addition of the enzyme solution. The steady-state kinetic constants (Km, kcat) for the hydrolysis of Ins P6 and its derivatives by PhyAsr were calculated from regressional analysis (Sigma-plot 8.0; Systat Software Inc.) of Lineweaver-Burk plots of the data.
Crystallization
Recombinant 6×His tagged PhyAsr was polished by cation exchange (Macro-Prep High S) and size exclusion chromatography. Initial crystallization conditions were identified using the Hampton Research Crystal Screen (Hampton Research). Crystals were obtained in 100 mM sodium cacodylate (pH 6.5), 1.35 M ammonium sulfate, 0.80 M sodium chloride, and 1% 1,6-hexanediol by sitting-drop vapor diffusion. Drops were mixed using 2 μL of 30 mg/mL protein with 2 μL of mother liquor. Data were collected at the Advanced Light Source (ALS) beamline 8.3.1 for crystals of PhyAsr Cys252Ser and Asp223Asn. Data were collected from the Arg258Lys mutant using an FR591 Bruker-Nonius rotating anode X-ray generator (45 kV, 130 mA) and a Kappa CCD detector. Monochromatic Cu Kα radiation was generated using osmic mirrors. Data were integrated and scaled using HKL 2000 (Otwinowski and Minor 1997), the structures were determined by molecular replacement with aMoRe (Navaza 1994), and the CCP4 suite of programs (Collaborative Computational Project, Number 4 1994). Structure refinement of all three mutants was carried out using CNS 1.0 (Brunger et al. 1998). Statistics for the data collection and the refinement of PhyAsr Cys252Ser, Asp223Asn, and Arg258lys are shown in Table 4.
Table 4.
Data collection and refinement statistics for the X-ray crystallographic structures of the Cys252Ser, Asp223Asn, and Arg258Lys active site mutants of PhyAsr

Preparation of individual myo-inositol phosphate isomers
D-Ins(1,2,4,5,6)P5, D-Ins(1,2,5,6)P4, and Ins(2,4,5,6)P4 were obtained as described previously (Greiner et al. 2002a,b). For the production of IPP isomers generated by PhyAsr, sodium phytate (2.5 mmol) was incubated at 37°C in a mixture containing 50 mM NH4-acetate (pH 5.0) and 10 U of the purified enzyme in a final volume of 500 mL. After an incubation period of 30 min, the reaction was stopped by heat treatment (95°C, 10 min). The incubation mixture was lyophilized and the dry residues were dissolved in 10 mL of 1.0 M NH4-formate (pH 2.5). The solution was loaded onto a Q-Sepharose column (2.6 × 90 cm) equilibrated with 1.0 M NH4-formate (pH 2.5) at a flow rate of 2.5 mL/min. The column was washed with 500 mL of 1.0 M NH4-formate (pH 2.5); the bound IPPs were eluted with a linear gradient from 1.0 M to 1.4 M NH4-formate (pH 2.5) (1000 mL) at 2.5 mL/min. Fractions of 10 mL were collected. From even-numbered tubes, 100-μL aliquots were lyophilized. The residues were dissolved in 3 N H2SO4 and incubated for 90 min at 165°C to hydrolyze the eluted IPPs completely. The liberated phosphate was measured as previously described. The content of the fraction tubes corresponding to the individual IPPs were pooled and lyophilized until only a dry residue remained. The residue was dissolved in 10 mL of water. Lyophilization and redissolving were repeated twice. IPP concentration was determined by HPIC on Ultrasep ES 100 RP18 from Bischoff Chromatography (Sandberg and Ahderinne 1986). The purity of the IPP preparation was determined on an HPIC system (Skoglund et al. 1998). D-Ins(2,4,5)P3 and D-Ins(2,4)P3 were obtained in the same ways by using the phytate-degrading enzyme from Klebsiella terrigena.
Identification of enzymatically formed hydrolysis products
The enzymatic reaction was started at 37°C by addition of 50 μL of a suitable diluted solution of PhyAsr to the incubation mixtures (2 U/mL). The incubation mixture consisted of 1250 μL 0.1 M sodium acetate buffer (pH 5.0) containing 3.125 μmol sodium phytate or one of the purified individual lower IPP esters. From the incubation mixture, 100-μL samples were removed periodically and the reaction was stopped by heat treatment (95°C, 10 min). Heat-treated samples (50 μL) were resolved on an HPIC system using a Carbo Pac PA-100 (4 × 250 mm) analytical column from Dionex and a gradient of 5%–98% HCl (0.5 M, 0.8 mL/min) (Skoglund et al. 1998). The eluants were mixed in a post-column reactor with 0.1% Fe(NO3)3 in a 2% HClO4 solution (0.4 mL/min) (Phillippy and Bland 1988). The combined flow rate was 1.2 mL/min.
Identification of the myo-inositol monophosphate isomer
Myo-inositol monophosphates were produced by incubation of 1.0 U of PhyAsr with a limiting amount (0.1 μmol) of the individual IPP ester (Ins[1,2,3,4,5,6]P6, D-Ins[1,2,3,5,6]P5, D-Ins[1,2,5,6]P4, Ins[2,4,5,6]P40) in a final volume of 500 μL of 50 mM NH4-formate. After lyophilization, the residues were dissolved in 500 μL of a solution of pyridine:bis(trimethylsilyl)trifluoroacetamide (1:1 v/v) and incubated at room temperature for 24 h. The silylated products were injected at 270°C into a gas chromatograph coupled with a mass spectrometer. The stationary phase was methylsilicon in a fused silica column (0.25 mm × 15 m). Helium was used as the carrier gas at a flow rate of 0.5 m/s. The following heating program was used for the column: 100°C to 340°C, rate increase: 4°C/min. Ionization was performed by electron impact at 70 eV and 250°C.
Enzyme modification
Alkylation of PhyAsr was carried out in 20 mM HEPES (pH 7) containing 10 mM freshly prepared iodoacetic acid (IAA) and 1 M guanidine at room temperature in the dark. The modification reaction was initiated with the addition of 1 nmol of enzyme to the reaction mixture (final volume of 500 μL). A control reaction was prepared in the same way except iodoacetate was omitted. At 10-min intervals, 50-μL aliquots were withdrawn and added to 700 μL of 0.05 M NaAc (pH 5), which was assayed as described previously. The modification reaction was repeated in the presence of a range of sodium phytate concentrations. The percentage of residual phytase activity was calculated relative to the control. For samples analyzed by mass spectrometry, excess IAA was quenched with addition of 10× molar excess dithiothreitol.
Mass spectrometry
Mass analysis was performed on both PhyAsr and the alkylation product using Matrix Assisted Laser Desorption/Ionization Time-of-Flight (MALDI-TOF) at the McGill University proteomics lab. The theoretical accuracy of this instrument is 1 for every 2000 Da, or, 0.05% of the total mass of PhyAsr. Masses were obtained for the modified and unmodified proteins followed by tryptic digestion and tandem mass spectrometry (MS/MS) analysis.
Molecular docking
AutoDock 3.05 (Morris et al. 1998) was used to search for, identify, and calculate the interaction energy of the optimal Ins P6 complex with PhyAsr. The Ins P6 ligand used for these calculations was obtained from the 2.05 Å E. coli phytase structure (PDB accession: 1DKQ). The PhyAsr X-ray structure used as the target is a locally determined 1.8 Å structure (PDB accession: 2B4P). Electrostatic potentials assigned to PhyAsr are consistent with a neutral pH and constant throughout the calculations. Several ligand ring-conformations and charge distributions were considered. These include the two “chair” conformations (D-2 axial phosphate and D-2 equatorial phosphate), and net charges of −6 (hydrogen on the 2, 4, and 6 or 1, 3, and 5 phosphates), and −9. The C–O and P–O bonds of Ins P6 were allowed to freely rotate during the Monte Carlo simulated annealing. Finally, the docked ligand conformations were grouped or clustered based upon their pairwise all atom RMS deviation (0.5 Å cutoff).
Accession numbers
The nucleotide sequence for phyAsr has been deposited in the GenBank database under GenBank accession number AF177214. The amino acid sequence of this protein can be accessed through NCBI Protein Database under NCBI accession number AAQ13669. The atomic coordinates for the original crystal structure of this protein showing the P-loop in an “open” and “closed” conformation are available in the Research Collaboratory for Structural Bioinformatics Protein Databank under PDB No.1U24 and 1U26, respectively. Structure factors and coordinates for Cys252Ser, Asp223Asn, and Arg258Lys mutants have been deposited to the same Protein Databank under PDB No. 2B4U, 2B4P, and 2B4O, respectively.
Electronic supplemental material
Supplement 1
Time course of PhyAsr inactivation by 10 mM iodoacetate (IAA). Residual activity is plotted as a function of time incubated with IAA relative to a control reaction.
Supplement 2
The effect of varying concentrations of Ins P6 on the inactivation of PhyAsr by IAA. The modification reaction was repeated in the presence of 0, 2, 4, and 6 mM sodium phytate. Average activity of the protected and unprotected, modified enzyme relative to an unalkylated control for three separate experiments is presented.
Acknowledgments
Data was collected at beamline 8.3.1 at the ALS with the support of the Alberta Synchrotron Institute (ASI). The ASI synchrotron access program is supported by grants from the Alberta Science and Research Authority (ASRA) and the Alberta Heritage Foundation for Medical Research (AHFMR). R.J.G. is supported by the Alberta Ingenuity Fund (AIF) and the National Science and Engineering Research Council (NSERC). L.B.S. receives funding from NSERC. S.C.M. is funded by AHFMR, NSERC, and the Canadian Foundation for Innovation (CFI). Analysis of the isomers of the individual myo-inositol phosphate derivatives by N.-G. Carlsson, Chalmers University of Technology (Göteborg, Sweden), is gratefully acknowledged.
Footnotes
Supplemental material: see www.proteinscience.org
Reprint requests to: Steven C. Mosimann, Department of Chemistry and Biochemistry, University of Lethbridge, Lethbridge, Alberta T1K 3M4, Canada; e-mail: steven.mosimann@uleth.ca; fax: (403) 329-2057.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.062738307.
References
- Barford D., Flint, A.J., and Tonks, N.K. 1994. Crystal structure of human protein tyrosine phosphatase 1B. Science 263: 1397–1404. [PubMed] [Google Scholar]
- Barrientos L., Scott, J.J., and Murthy, P.P. 1994. Specificity of hydrolysis of phytic acid by alkaline phytase from lily pollen. Plant Physiol. 106: 1489–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendtsen J.D., Nielson, H., Von Heijne, G., and Brunak, S. 2004. Improved prediction of signal peptides: SignalP 3.0. J. Mol. Biol. 340: 783–795. [DOI] [PubMed] [Google Scholar]
- Bliska J., Guan, K., Dixon, J., and Falkow, S. 1991. Tyrosine phosphate hydrolysis of host proteins by an essential Yersinia virulence determinant. Proc. Natl. Acad. Sci. 88: 1187–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bretz J.R., Mock, N.M., Charity, J.C., Zeyad, S., Baker, C.J., and Hutcheson, S.W. 2003. A translocated protein tyrosine phosphatase of Pseudomonas syringae pv. tomato DC3000 modulates plant defence response to infection. Mol. Microbiol. 49: 389–400. [DOI] [PubMed] [Google Scholar]
- Brunger A.T., Adams, P.D., Clore, G.M., Delano, W.L., Gros, P., Grosse-Kunstleve, R.W., Jiang, J.-S., Kuszewski, J., Nilges, N., Pannu, N.S., et al. 1998. Crystallography and NMR systems (CNS): A new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 54: 905–921. [DOI] [PubMed] [Google Scholar]
- Buhrman G., Parker, B., Sohn, J., Rudolph, J., and Mattos, C. 2005. Structural mechanism of oxidative regulation of the phosphatase Cdc25B via an intramolecular disulfide bond. Biochemistry 44: 5307–5316. [DOI] [PubMed] [Google Scholar]
- Burke T.R. and Zhang, Z.Y. 1998. Protein-tyrosine phosphatases: Structure, mechanism, and inhibitor discovery. Biopolymers 47: 225–241. [DOI] [PubMed] [Google Scholar]
- Caffrey J.J., Darden, T., Wenk, M.R., and Shears, S.B. 2001. Expanding coincident signaling by PTEN through its inositol 1,3,4,5,6-pentakisphosphate 3-phosphatase activity. FEBS Lett. 499: 6–10. [DOI] [PubMed] [Google Scholar]
- Chu H.M., Guo, R.T., Lin, T.W., Chou, C.C., Shr, H.L., Lai, H.L., Tang, T.Y., Cheng, K.J., Selinger, B.L., and Wang, A.H.J. 2004. Structures of Selenomonas ruminantium phytase in complex with persulfated phytate: DSP phytase fold and mechanism for sequential substrate hydrolysis. Structure 12: 2015–2024. [DOI] [PubMed] [Google Scholar]
- Cirri P., Chiarugi, P., Camici, G., Manao, G., Raugei, G., Cappugi, G., and Ramponi, G. 1993. The role of Cys12, Cys17 and Arg18 in the catalytic mechanism of low-M(r) cytosolic phosphotyrosine protein phosphatase. Eur. J. Biochem. 214: 647–657. [DOI] [PubMed] [Google Scholar]
- Collaborative Computational Project, Number 4 1994. The CCP4 suite: Programs for protein crystallography. Acta. Cryst D50: 760–763. [DOI] [PubMed] [Google Scholar]
- Delano W.L. 2002. The PyMOL molecular graphics system. DeLano Scientific, San Carlos, CA.
- Flint A.J., Tiganis, T., Barford, D., and Tonks, N.K. 1997. Development of “substrate-trapping” mutants to identify physiological substrates of protein tyrosine phosphatases. Proc. Natl. Acad. Sci. 94: 1680–1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y. and Galan, J.E. 1998. The Salmonella typhimurium tyrosine phosphatase SptP is translocated into host cells and disrupts the actin cytoskeleton. Mol. Microbiol. 27: 359–368. [DOI] [PubMed] [Google Scholar]
- Gasteiger E., Hoogland, C., Gattiker, A., Duvaud, S., Wilkins, M.R., Appel, R.D., and Bairoch, A. 2005. Protein identification and analysis tools on the ExPASy server. In The proteomics protocols handbook (ed. J.M. Walker), pp. 571–607. Humana Press, Totowa, NJ.
- Greiner R., Alminger, M.L., Carlsson, N.G., Muzquiz, M., Burbano, C., Cuadrado, C., Pedrosa, M.M., and Goyoaga, C. 2002a. Pathway of dephosphorylation of myo-inositol hexakisphosphate by phytases of legume seeds. J. Agric. Food Chem. 50: 6865–6870. [DOI] [PubMed] [Google Scholar]
- Greiner R., Farouk, A., Alminger, M.L., and Carlsson, N.G. 2002b. The pathway of dephosphorylation of myo-inositol hexakisphosphate by phytate-degrading enzymes of different Bacillus spp. Can. J. Microbiol. 48: 986–994. [DOI] [PubMed] [Google Scholar]
- Guo X.L., Kui, S., Wang, F., Lawrence, D.S., and Zhang, Z.Y. 2002. Probing the molecular basis for potent and selective protein-tyrosine phosphatase 1B inhibition. J. Biol. Chem. 277: 41014–41022. [DOI] [PubMed] [Google Scholar]
- Heinonen J.K. and Lahti, R.J. 1981. A new and convenient colorimetric determination of inorganic orthophosphate and its application to the assay of inorganic pyrophosphatase. Anal. Biochem. 113: 313–317. [DOI] [PubMed] [Google Scholar]
- Isbrandt L.R. and Oertel, R.P. 1980. Conformational states of myo-inositol hexakis(phosphate) in aqueous solution. A 13C NMR, 31P NMR and Raman spectroscopy investigation. J. Am. Chem. Soc. 102: 3144–3148. [Google Scholar]
- Jia Z., Barford, D., Flint, A.J., and Tonks, N.K. 1995. Structural basis for phosphotyrosine peptide recognition by protein tyrosine phosphatase 1B. Science 268: 1754–1758. [DOI] [PubMed] [Google Scholar]
- Keng Y.-F., Wu, L., and Zhang, Z.-Y. 1999. Probing the function of the conserved tryptophan in the flexible loop of the Yersinia protein–tyrosine phosphatase. Eur. J. Biochem. 259: 809–814. [DOI] [PubMed] [Google Scholar]
- Kennelly P.J. and Potts, M. 1999. Life among the primitives: Protein O-phosphatases in prokaryotes. Front. Biosci. 4: d372–d385. [DOI] [PubMed] [Google Scholar]
- Konietzny U. and Greiner, R. 2002. Molecular and catalytic properties of phytate-degrading enzymes (phytases). Int. J. Food Sci. Technol. 37: 791–812. [Google Scholar]
- Laemmli U.K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227: 680–685. [DOI] [PubMed] [Google Scholar]
- Loewus F.A. and Murthy, P.P.N. 2000. Myo-inositol metabolism in plants. Plant Sci. 150: 1–19. [Google Scholar]
- Lohse L.D., Denu, M.J., Santoro, N., and Dixon, E.J. 1997. Roles of aspartic acid-181 and serine-222 in intermediate formation and hydrolysis of the mammalian protein tyrosine phosphatase PTP1B. Biochemistry 36: 4568–4575. [DOI] [PubMed] [Google Scholar]
- Maehama T. and Dixon, J.E. 1998. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J. Biol. Chem. 273: 13375–13378. [DOI] [PubMed] [Google Scholar]
- Morris G.M., Goodsell, D.S., Halliday, R.S., Huey, R., Hart, W.E., Belew, R.K., and Olson, A.J. 1998. Automated docking using a Lamarckian genetic algorithm and empirical binding free energy function. J. Comput. Chem. 19: 1639–1662. [Google Scholar]
- Mullaney E.J. and Ullah, A.H.J. 2003. The term phytase comprises several different classes of enzymes. Biochem. Biophys. Res. Commun. 312: 179–184. [DOI] [PubMed] [Google Scholar]
- Navaza J. 1994. aMoRe: An automated package for molecular replacement. Acta Crystallogr. A 50: 157–163. [Google Scholar]
- Nielsen H., Engelbrecht, J., Brunak, S., and Von Heijne, G. 1997. Identification of prokaryotic and eukaryotic signal peptides and prediction of their cleavage sites. Protein Eng. 10: 1–6. [DOI] [PubMed] [Google Scholar]
- Orchiston E.A., Bennett, D., Leslie, N.R., Clarke, R.G., Winward, L., Downes, C.P., and Safrany, S.T. 2004. PTEN M-CBR3, a versatile and selective regulator of inositol 1,3,4,5,6-pentakisphosphate (Ins(1,3,4,5,6)P-5). Evidence for Ins(1,3,4,5,6)P-5 as a proliferative signal. J. Biol. Chem. 279: 1116–1122. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z. and Minor, W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276: 307–326. [DOI] [PubMed] [Google Scholar]
- Phillippy B.Q. and Bland, J.M. 1988. Gradient ion chromatography of inositol phosphates. Anal. Biochem. 175: 162–166. [DOI] [PubMed] [Google Scholar]
- Salmeen A., Andersen, J.N., Myers, M.P., Meng, T.-C., Hinks, J.A., Tonks, N.K., and Barford, D. 2003. Redox regulation of protein tyrosine phosphatase 1B involves a sulphenyl-amide intermediate. Nature 423: 769–773. [DOI] [PubMed] [Google Scholar]
- Sandberg A.S. and Ahderinne, R. 1986. HPLC method for determination of inositol tri-, tetra-, penta-, and hexaphosphates in foods and intestinal contents. J. Food Sci. 51: 547–550. [Google Scholar]
- Sasakawa N., Sharif, M., and Hanley, M.R. 1995. Metabolism and biological activities of inositol pentakisphosphate and inositol hexakisphosphate. Biochem. Pharmacol. 50: 137–146. [DOI] [PubMed] [Google Scholar]
- Shi L., Potts, M., and Kennelly, P.J. 1998. The serine, threonine, and/or tyrosine-specific protein kinases and protein phosphatases of prokaryotic organisms: A family portrait. FEMS Microbiol. Rev. 22: 229–253. [DOI] [PubMed] [Google Scholar]
- Skoglund E., Carlsson, N.G., and Sandberg, A.S. 1998. High-performance chromatographic separation of inositol phosphate isomers on strong anion exchange columns. J. Agric. Food Chem. 46: 1877–1882. [Google Scholar]
- Sun J.P., Fedorov, A.A., Lee, S.Y., Guo, X.L., Shen, K., Lawrence, D.S., Alamo, S.C., and Zhang, Z.Y. 2003. Crystal structure of PTP1B complexed with a potent and selective bidentate inhibitor. J. Biol. Chem. 278: 12406–12414. [DOI] [PubMed] [Google Scholar]
- van Montfort R.L.M., Congreve, M., Tisi, D., Carr, R., and Jhoti, H. 2003. Oxidation state of the active-site cysteine in protein tyrosine phosphatase 1B. Nature 423: 773–777. [DOI] [PubMed] [Google Scholar]
- Villa F., Deak, M., Bloomberg, G.B., Alessi, D.R., and van Aalten, D.M. 2005. Crystal structure of the PTPL1/FAP-1 human tyrosine phosphatase mutated in colorectal cancer: Evidence for a second phosphotyrosine substrate recognition pocket. J. Biol. Chem. 280: 8180–8187. [DOI] [PubMed] [Google Scholar]
- Wu L. and Zhang, Z.Y. 1996. Probing the function of Asp128 in the low molecular weight protein-tyrosine phosphatase-catalyzed reaction. A pre-steady-state and steady-state kinetic investigation. Biochemistry 35: 5426–5434. [DOI] [PubMed] [Google Scholar]
- Yanke L.J., Bae, H.D., Selinger, L.B., and Cheng, K.J. 1998. Phytase activity of anaerobic ruminal bacteria. Microbiol 144: 1565–1573. [DOI] [PubMed] [Google Scholar]
- Zhang Z.Y. 1998. Protein-tyrosine phosphatases: Biological function, structural characteristics, and mechanism of catalysis. Crit. Rev. Biochem. Mol. Biol. 33: 1–52. [DOI] [PubMed] [Google Scholar]
- Zhang Z.Y. 2002. Protein tyrosine phosphatases: Structure and function, substrate specificity, and inhibitor development. Annu. Rev. Pharmacol. Toxicol. 42: 209–234. [DOI] [PubMed] [Google Scholar]
- Zhang Z.Y. 2003. Mechanistic studies on protein tyrosine phosphatases. Prog. Nucleic Acid Res. Mol. Biol. 73: 171–220. [DOI] [PubMed] [Google Scholar]
- Zhang Z., Clemens, J., Schubert, H., Stuckey, J., Fischer, M., Hume, D., Saper, M., and Dixon, J. 1992. Expression, purification, and physicochemical characterization of a recombinant Yersinia protein tyrosine phosphatase. J. Biol. Chem. 267: 23759–23766. [PubMed] [Google Scholar]
- Zhang Z., Wang, Y., and Dixon, J.E. 1994a. Dissecting the catalytic mechanism of protein-tyrosine phosphatases. Proc. Natl. Acad. Sci. 91: 1624–1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z.Y., Wang, Y., Wu, L., Fauman, B.E., Stuckey, A.J., Schubert, L.H., Saper, A.M., and Dixon, J.E. 1994b. The Cys(X)5Arg catalytic motif in phosphoester hydrolysis. Biochemistry 33: 15266–15270. [DOI] [PubMed] [Google Scholar]
- Zhou G., Denu, J.M., Wu, L., and Dixon, J.E. 1994. The catalytic role of Cys124 in the dual specificity phosphatase VHR. J. Biol. Chem. 269: 28084–28090. [PubMed] [Google Scholar]