

Abstract
12(S)-Hydroxyeicosatetraenoic acid (12-HETE) is one of the metabolites of arachidonic acid involved in pathological conditions associated with mitochondria and oxidative stress. The present study tested effects of 12-HETE on mitochondrial functions. In isolated rat heart mitochondria, 12-HETE increases intramitochondrial ionized calcium concentration that stimulates mitochondrial nitric oxide (NO) synthase (mtNOS) activity. mtNOS-derived NO causes mitochondrial dysfunctions by decreasing mitochondrial respiration and transmembrane potential. mtNOS-derived NO also produces peroxynitrite that induces release of cytochrome c and stimulates aggregation of mitochondria. Similarly, in HL-1 cardiac myocytes, 12-HETE increases intramitochondrial calcium and mitochondrial NO, and induces apoptosis. The present study suggests a novel mechanism for 12-HETE toxicity.
Keywords: 12-HETE, intramitochondrial ionized calcium, mtNOS, mitochondrial respiration, mitochondrial transmembrane potential, peroxynitrite, cytochrome c release, apoptosis
Introduction
12-Hydroxyeicosatetraenoic acid (12-HETE) is an arachidonic acid metabolite produced in lipoxygenase pathway. 12-HETE is involved in several oxidative stress-related pathological conditions including ischemia-reperfusion [1], hypertension [2,3], atherosclerosis [4], pancreas beta cells dysfunction [5] and diabetic nephropathy [6]. Moreover, pro-oxidant mediators such as 12-HETE along with increased NO synthesis may lead to progression of Alzheimer’s disease [7]. However, the exact role of 12-HETE in pathological conditions is yet to be established. Elevation of 12-HETE causes oxidative stress [4,8] and decrease in 12-HETE decreases oxidative stress [8]. Mitochondria are primary cellular sources of oxidative radicals and they are involved in oxidative stress-related conditions. While 12-HETE has been shown to interact with mitochondria [9,10], whether 12-HETE increases mitochondrial oxidant radicals is not fully understood.
The present study shows that in isolated rat heart mitochondria, 12-HETE increases intramitochondrial ionized calcium (Ca2+) concentration ([Ca2+]m) that stimulates mitochondrial nitric oxide (NO) synthase (mtNOS) activity. mtNOS-derived NO decreases mitochondrial respiration and transmembrane potential (ΔΨ). 12-HETE also increases mitochondrial peroxynitrite that induces cytochrome c (cyto c) release, tyrosine nitration of mitochondrial proteins, and aggregation of mitochondria. The present study also shows that in cardiac myocytes HL-1 cells, 12-HETE increases [Ca2+]m and mitochondrial NO, and induces apoptosis of those cells. Our study suggests a crucial role for [Ca2+]m and mtNOS in 12-HETE-induced apoptosis.
Material and Methods
Purification of rat heart mitochondria
Mitochondria were isolated from hearts of Sprague-Dawley rats by differential centrifugation and purified using Percoll (25%) gradient purification as described [11]. Purity of the mitochondria preparation was tested by measuring the cytochrome a content using ε605–630nm 12 mM−1 cm−1 [12]. Only mitochondria with less than 5% impurity were used in this study. Broken mitochondria were prepared by freezing the intact mitochondria in liquid nitrogen followed by thawing as described [13].
Cell culture
Cardiac myocyte cell line HL-1 (gift from Dr. Claycomb, LSUHSC, LA) were cultured in Claycomb medium (JRHBiosciences) supplemented with 10% fetal bovine serum, 0.1 mM norepinephrine, 2 mM L-glutamine, 100 U/ml penicillin, 100 U/ml streptomycin, and 0.25 μg/ml amphotericin B.
Treatment
Mitochondria were treated with 12-HETE (3 or 5 nM) for 20 min at room temperature. Control samples were treated with equal volume of the vehicle (ethanol) under the same conditions. In order to inhibit mtNOS activity, mitochondria were incubated with L-NMMA (150 μM) for 20 min on ice priory to 12-HETE treatment. Confocal microscopy studies used HL-1 cells treated with 12-HETE (3 nM) or equal volume of ethanol and cultured for 24 h. To measure apoptosis, HL-1 cells were treated with 12-HETE (3–20 nM) or equal volume of ethanol, cultured for 24 h and apoptotic cells were counted using trypan blue exclusion test.
Intramitochondrial ionized calcium
[Ca2+]m was measured by using the highly sensitive dual-wavelength excitation fluorometric assay recently established in our laboratory [11,14]. Briefly, mitochondria were loaded with fura-2/AM for 20 min at room temperature followed by twice wash. Fura-2/AM loaded mitochondria were excited at 352–362 nm and emission was collected at 510 nm.
Mitochondrial nitric oxide synthase activity
mtNOS activity was measured using the following three assays.
Oxyhemoglobin assay
Oxyhemoglobin assay was performed as described [12]. Broken mitochondria (30 μg) were added to a cuvette containing HEPES buffer (100 mM, pH 7.40), L-arginine (100 μM), tetrahydrobiopterin (10 μM), and Cu/Zn-superoxide dismutase (1 KU/ml). After reaching a steady state, oxyhemoglobin (4 μM) was added and ΔOD was detected at 401–420 nm. Quantitation was performed using ε401–420nm 100 mM−1 cm−1.
Citrulline radioassay
Mitochondria (1 mg) were supplemented with L-[3H]arginine (30,000–50,000 cpm) as described [15]. The exchange resin (Dowex) columns were prepared as described [16]. Samples were loaded on columns and the radioactivity of the effluents was determined.
Chemiluminescence assay
A sample of mitochondria (1 mg protein) was injected into the purge vessel containing vanadium chloride (0.8 % in 1M HCl), thermostated at 95 ºC and NO was measured using the NO chemiluminescence analyzer as described [14]. Standard curve was prepared by using NO saturated solution.
ΔΨ determination and mitochondrial oxygen consumption
K+-succinate (800 μM) supported ΔΨ was measured at 511–533 nm in the presence of safranin (10 μM) as described [16]. Mitochondria samples were suspended in a final volume of 1 ml HEPES buffer (100 mM, pH 7.40), respiration was supported by K+-succinate (800 μM) and the oxygen consumption was measured using a Clark-type oxygen electrode as described [16].
Mitochondria aggregation
Aggregation of mitochondria was tested as described [17]. Mitochondrial samples were placed on a Petroff-Hausser counting chamber (Hausser Scientific). 12-HETE or equal volume of vehicle was added and microphotographs were captured by a digital camera (Nikon Coolpix 4500, Japan).
Immunoblotting for tyrosine nitration and cytochrome c release
Mitochondrial proteins (25 μg) were separated on 10% gels, blotted onto nitrocellulose membranes, and probed with monoclonal anti-nitrotyrosine antibody (Alexis Biochemicals). For cyto c release, mitochondria were centrifuged at 100,000 xg for 10 min and the supernatant was re-centrifuged at 100,000 xg for 30 min. Cyto c was detected in the supernatant of the second centrifugation by Western blot using monoclonal anti-cytochrome c antibody (eBioscience) as described [14]
Confocal imaging
To measure [Ca2+]m, cells were loaded with Rhod2-AM (Molecular Probes; 5μM) simultaneously with mitotracker green (Molecular Probes; 200 nM) under 5% CO2 at 37 °C. To measure mitochondrial NO, cells were incubated with mitotracker red (Molecular Probes; 200 nM) and membrane permeable NO probe, 4,5 diaminofluorescein diacetate (DAF-2DA; Calbiochem, 5 μM) under 5% CO2 at 37 °C. Probed cells were permeabilized with digitonin (10 μM) to eliminate the cytosolic fraction of Rhod-2 and DAF.
The cover slips were mounted on glass slides with Vectashield (Vector Lab. Inc.) and images were captured with confocal microscope (Carl Zeiss) equipped with LSM 5 META software and 63x water objective. Fluorescent measurements were performed using multichannel detection of excitation of 488 nm line of an argon 543 nm line of HeNe1 and 633 nm lines of HeNe2 laser at room temperature. Fluorescence of mitotracker green and DAF were acquired using emission at 516 nm. Fluorescence of mitotracker red and Rhod-2 were acquired using emission at 579 nm. Images were obtained at 12 bit resolution by taking a single z-stack 1 μm steps. Merged images were obtained using LSM 5 software. Fluorescent intensities were measured using ImageJ (http://rsb.info.nih.gov/ij/).
Results
Mitochondrial ionized calcium concentration ([Ca2+]m) and mtNOS activity
It has been suggested that alteration of cellular calcium homeostasis contributes to cytotoxicity of 12-HETE. Fig. 1 shows that 12-HETE increased [Ca2+]m that was abolished with EGTA. Increase in [Ca2+]m was not sensitive to L-NMMA and L-NMMA did not interfere with the assay. mtNOS is [Ca2+]m-sensitive and elevation of [Ca2+]m stimulates mtNOS activity. Calculated Km value of mtNOS for Ca2+ is 23.5 μM [11]. Thus the present study tested whether 12-HETE stimulates mtNOS activity. Both oxyhemoglobin assay (Fig. 2a) and radioassay (Fig. 2b) show that 12-HETE increased mtNOS activity that was inhibited by NOS inhibitor L-NMMA. Increased mtNOS activity was also confirmed by chemiluminescence assay (Fig. 2c).
Fig. 1. 12-HETE and [Ca2+]m.
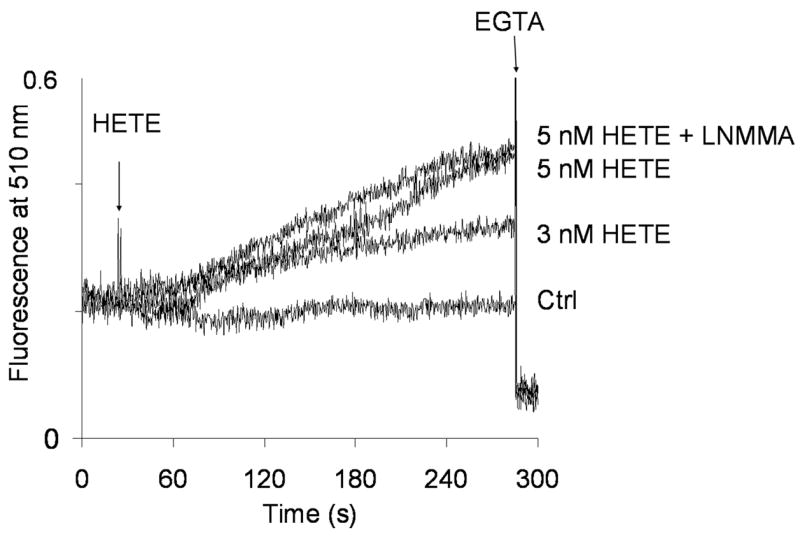
[Ca2+]m was measured real-time in isolated rat heart mitochondria using the highly sensitive fluorescent [Ca2+]m determination assay. Where indicated, 3 or 5 nM 12-HETE was added (HETE) in the presence or absence of L-NMMA (+LNMMA). Control (Ctr) was treated with same amount of vehicle. Representative of n=6.
Fig. 2. 12-HETE and mtNOS.

mtNOS activity was measured by using (a) Citrulline assay, (b) Oxyhemoglobin assay, and (c) Chemiluminescence assay where mitochondria were treated with 12-HETE (3 nM or 5 nM) or vehicle for control (Ctr). Where indicated, mtNOS was inhibited by L-NMMA (150 μM; +LNMMA). *Statistically different from Ctr (p<0.05), **Statistically different from HETE sample without L-NMMA (p<0.05).
Effect of 12-HETE on mitochondrial functions
The present study tested the effect of 12-HETE on mitochondrial oxygen consumption and ΔΨ. Fig. 3a shows that 12-HETE significantly decreased oxygen consumption and that this effect of 12-HETE was prevented when mtNOS was inhibited. The ΔΨ was dramatically lowered by 12-HETE (Fig 3b) and this affect of 12-HETE was also prevented when mtNOS was inhibited.
Fig. 3. Mitochondrial respiration and ΔΨ.

Succinate-supported mitochondrial respiration (a) and ΔΨ (b) were measured for mitochondria treated with 12-HETE (3 nM or 5 nM) or vehicle (Ctr). At the end of the measurements, ΔΨ was collapsed by uncoupling mitochondria with carbonyl cyanide m-chlorophenylhydrazone (CCCP; 1 μM). Where indicated, mtNOS was inhibited by L-NMMA (+LNMMA). Representative of n=6.
mtNOS-derived NO forms peroxynitrite that increases protein tyrosine nitration [14]. Fig 4a shows that 12-HETE increased protein tyrosine nitration. Increased mtNOS-derived peroxynitrite causes cyto c release from the mitochondria [16]. Fig. 4b shows 12-HETE induced release of cyto c from the mitochondria. mtNOS-derived peroxynitrite also causes aggregation of mitochondria [14]. Fig 4c shows that mitochondria treated with 12-HETE formed aggregates and that this effect of 12-HETE was prevented when mtNOS was inhibited.
Fig. 4. Mitochondrial aggregation, tyrosine nitration and cytochrome c release.

(a) Protein tyrosine nitration, and (b) cytochrome c release were detected in mitochondrial sample incubated with 12-HETE (3 nM or 5 nM) or vehicle (Ctr), in the absence or presence of L-NMMA (+LNMMA). Samples were subjected to SDS-PAGE and Western blot using anti-nitrotyrosine or anti-cytochrome c antibodies. (c) Aggregation of mitochondria was detected before treatment (T0) or 20 min after treatment (T20) with 12-HETE (3 nM or 5 nM) or vehicle (Ctr). Inhibition of mtNOS by L-NMMA (+LNMMA) prevented 12-HETE-induced aggregation. Typical aggregation is marked by arrows. Microphotographs are representative of 4–6 experiments.
Effect of 12-HETE on cardiac myocytes
In order to confirm our results obtain on isolated mitochondria, [Ca2+]m and mitochondrial NO were determinated for cardiac myocytes. Consistent with isolated mitochondria results, Fig. 5a shows that 12-HETE significantly increased [Ca2+]m in the mitochondria of cardiac myocytes. Likewise, results presented in Fig. 5b show that 12-HETE increased mitochondrial NO in cardiac myocytes (Fig. 5b). In order to confirm 12-HETE induced cytochrome c release and aggregation of isolated mitochondria, apoptosis was measured in HL-1 cells. Fig. 5c shows that 12-HETE induced apoptosis of HL-1 cells.
Fig. 5. 12-HETE and HL-1 cardiomyocytes [Ca2+]m, mitochondrial NO, and apoptosis.

(a) Control (Ctr) and 12-HETE treated (HETE) HL-1 cells were probed with, mitotracker green (Mito) and Rhod-2 (Calcium) and confocal images were acquired. Yellow areas in merge (Merge) indicate mitochondrial calcium. (b) Mitochondria of control (Ctr) and 12-HETE treated (HETE) HL-1 cells were visualized by mitotracker red (Mito) and NO by NO-sensitive probe DAF (NO). Yellow areas in merge (Merge) indicate mitochondrial NO. Fluorescence intensities are shown in right panels. (c) Apoptosis of HL-1 cells control (Ctr) or treated with 12-HETE (3–20 nM) is expressed as percent apoptotic cells. *Significantly different (p <0.05) from the Ctr. Images are representative of 4–6 experiments.
Discussion
12-HETE is an arachidonic acid metabolite that contributes to dysfunctions of various tissues [1–7]. It has been shown that 12-HETE alters Ca2+ homeostasis and that this effect of 12-HETE could be involved in its cytotoxicity [10,18,19]. Mitochondria play a crucial role in cellular Ca2+ homeostasis. The ΔΨ that renders mitochondrial inner membrane negatively charged is the driving force for mitochondria to take up large amounts of Ca2+ very rapidly. However, mitochondria maintain the [Ca2+]m very low by precipitating the [Ca2+]m to non-ionized Ca2+ stores known as matrix electron-dense granules [14,20]. Mitochondria maintain a dynamic intra-organelle Ca2+ homeostasis by continuously precipitating [Ca2+]m to the matrix granules and releasing [Ca2+]m from the granules. We and others have shown that conditions associated with oxidative stress increase [Ca2+]m [11,14,20–22] that initiates mitochondrial and cell injury [11,14]. Our laboratory recently introduced a sensitive dual-wavelength excitation fluorometric assay that allows real-time detection of [Ca2+]m under various conditions [11,14]. We used this assay and tested the effect of 12-HETE on [Ca2+]m. Fig. 1 shows that 12-HETE causes a dramatic increases in [Ca2+]m. This novel finding suggests that elevation of [Ca2+]m by 12-HETE contributes to alteration of cellular Ca2+ homeostasis that may stimulate oxidative stress [11,14,23]. Mitochondria produce NO via Ca2+-sensitive mtNOS [13,14,24-28]. Release of Ca2+ from matrix granules leading to increased [Ca2+]m stimulates mtNOS activity [11,14]. Results presented in Figs. 2a–c clearly show that 12-HETE increases mtNOS activity, and that this effect of 12-HETE was sensitive to L-NMMA. NO generated by mtNOS affects mitochondrial functions [29]. NO competes with oxygen for the oxygen binding site at complex IV and inhibits the activity of complex IV that results in decreased mitochondrial respiration and ΔΨ [13,29]. Fig. 3a shows that 12-HETE decreased oxygen consumption of mitochondria, and that this effect of 12-HETE was prevented when mtNOS was inhibited. Increased mtNOS-derived NO decreases ΔΨ [13]. Therefore, the present study tested the effect of 12-HETE on ΔΨ. Fig. 3b shows that 12-HETE dramatically decreased ΔΨ and that this effect of 12-HETE was partially prevented when mtNOS was inhibited. Mitochondria maintain [Ca2+]m in response to ΔΨ and decreased ΔΨ causes Ca2+ efflux from the mitochondria. Thus, elevated [Ca2+]m (Fig. 1) along with decreased ΔΨ (Fig. 3b) caused efflux of [Ca2+]m from mitochondria. This can contribute to 12-HETE-induced increased cellular Ca2+ reported by several groups [10, 18, 19].
mtNOS-derived NO readily and potently produces peroxynitrite [16]. Thus, we tested whether 12-HETE increases peroxynitrite in the mitochondria. Tyrosine nitration is one of the widely used and reliable biomarkers of peroxynitrite. Several studies have shown that elevated mitochondrial peroxynitrite increase tyrosine nitration of mitochondrial proteins [11,14,31,32]. Fig. 4a shows that 12-HETE increased tyrosine nitration of mitochondrial proteins, indicating that 12-HETE elevated peroxynitrite in the mitochondria. Release of cyto c from mitochondria is one of the key events during peroxynitrite-induced apoptosis [11,14,33]. Our previous studies showed that mtNOS stimulation induces cyto c release [11,14,16]. Fig. 4b shows that 12-HETE releases cyto c from the mitochondria. Increased protein tyrosine nitration together with cyto c release suggests a novel mechanism for 12-HETE-induced apoptosis reported by several studies [6,34].
Mitochondrial morphology is altered in various conditions. Under oxidative stress conditions and during apoptosis, mitochondria fuse and form aggregates [33]. Recently we showed that mtNOS-derived peroxynitrite causes aggregation of mitochondria [14]. Fig. 4c shows that 12-HETE stimulates mitochondrial aggregation that was prevented when mtNOS was inhibited. Those results further indicate a role for mtNOS in 12-HETE-induced mitochondrial oxidative stress. Several studies suggest crucial role of antioxidants in preventing oxidative stress induced by arachidonic acid metabolites [34,35]. Trolox that is a lipid-soluble antioxidant, and N-acetyl-L-cysteine that is the precursor of glutathione, decrease oxidative stress and protect neurons against toxicity caused by arachidonic acid metabolites [35].
Our studies on isolated mitochondria provided important insight into the molecular mechanism of 12-HETE toxicity. In order to verify isolated heart mitochondria results, the present study tested effect of 12-HETE on [Ca2+]m and mitochondrial NO, and apoptosis of HL-1 cardiac myocytes. Consistent with results on isolated mitochondria, Fig. 5 shows that 12-HETE at nanomolar concentration [36] increased [Ca2+]m and mitochondrial NO of those cells. Alteration of Ca2+ homeostasis contributes to pathological conditions including ischemia-reperfusion [11] and myocardial infarction [37], increase oxidative modification of cells components [23], and induces apoptosis [14,36]. Thus, our findings strongly suggest that 12-HETE increased [Ca2+]m and mitochondrial NO contributes to apoptosis induces by 12-HETE.
Taken together, our study suggests a novel mechanism for 12-HETE-induced mitochondrial dysfunction. Our study for the first time shows that 12-HETE increases [Ca2+]m and stimulates mtNOS activity, decreases respiration and ΔΨ, increases mitochondrial peroxynitrite, releases cyto c, and causes mitochondrial aggregation. Our studies also show for the first time that 12-HETE increases [Ca2+]m and mitochondria NO in HL-1 cardiac myocytes and cause apoptosis of those cells. Findings presented in the present study suggest a molecular mechanism underlying toxicity of 12-HETE that might be involved in pathological conditions associated with elevated 12-HETE, oxidative stress and mitochondrial dysfunction.
Acknowledgments
This work was supported by the National Institute on Aging (award AG023264-02) and American Heart Association (award 0562221B).
Abbreviations used in the text
- 12-HETE
2(S)-Hydroxyeicosatetraenoic acid
- NO
nitric oxide
- mtNOS
mitochondrial nitric oxide synthase
- ([Ca2+]m)
intramitochondrial ionized calcium concentration
- (ΔΨ)
mitochondrial transmembrane potential
- CCCP
carbonyl cyanide m-chlorophenylhydrazone
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kuzuya T, Hoshida S, Kim Y, Oe H, Hori M, Kamada T, Tada M. Cardiovasc Res. 1993;27:1056–1060. doi: 10.1093/cvr/27.6.1056. [DOI] [PubMed] [Google Scholar]
- 2.Gonzalez-Nunez D, Claria J, Rivera F, Poch E. Hypertension. 2001;37:334–338. doi: 10.1161/01.hyp.37.2.334. [DOI] [PubMed] [Google Scholar]
- 3.Chang WC, Su GW. Biochem Biophys Res Commun. 1985;127:642–648. doi: 10.1016/s0006-291x(85)80209-6. [DOI] [PubMed] [Google Scholar]
- 4.Natarajan R, Gerrity RG, Gu JL, Lanting L, Thomas L, Nadler JL. Diabetologia. 2002;45:125–133. doi: 10.1007/s125-002-8253-x. [DOI] [PubMed] [Google Scholar]
- 5.Prasad KM, Thimmalapura PR, Woode EA, Nadler JL. Biochem Biophys Res Commun. 2003;308:427–432. doi: 10.1016/s0006-291x(03)01418-9. [DOI] [PubMed] [Google Scholar]
- 6.Xu ZG, Li SL, Lanting L, Kim YS, Shanmugam N, Reddy MA, Natarajan R. Kidney Int. 2006;69:512–519. doi: 10.1038/sj.ki.5000137. [DOI] [PubMed] [Google Scholar]
- 7.Zhu X, Smith MA, Honda K, Aliev G, Moreira PI, Nunomura A, Casadesus G, Harris PL, Siedlak SL, Perry G. J Neurol Sci. 2007;257:240–246. doi: 10.1016/j.jns.2007.01.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pratico D, Zhukareva V, Yao Y, Uryu K, Funk CD, Lawson JA, Trojanowski JQ, Lee VM. Am J Pathol. 2004;164:1655–1662. doi: 10.1016/S0002-9440(10)63724-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gordon JA, Broekemeier KM, Spector AA, Pfeiffer DR. J Lipid Res. 1994;35:698–708. [PubMed] [Google Scholar]
- 10.Richter C, Frei B, Cerutti PA. Biochem Biophys Res Commun. 1987;143:609–616. doi: 10.1016/0006-291x(87)91397-0. [DOI] [PubMed] [Google Scholar]
- 11.Zenebe WJ, Nazarewicz RR, Parihar MS, Ghafourifar P. J Mol Cell Cardiol. 2007 doi: 10.1016/j.yjmcc.2007.05.019. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ghafourifar P, Asbury ML, Joshi SS, Kincaid ED. Meth Enzymol. 2005;396:424–444. doi: 10.1016/S0076-6879(05)96036-9. [DOI] [PubMed] [Google Scholar]
- 13.Ghafourifar P, Richter C. FEBS Lett. 1997;418:291–296. doi: 10.1016/s0014-5793(97)01397-5. [DOI] [PubMed] [Google Scholar]
- 14.Nazarewicz RR, Zenebe WJ, Parihar A, Larson SK, Alidema E, Choi J, Ghafourifar P. Cancer Res. 2007;67:1282–1290. doi: 10.1158/0008-5472.CAN-06-3099. [DOI] [PubMed] [Google Scholar]
- 15.Mayer B, Klatt P, Werner ER, Schmidt K. Neuropharmacology. 1994;33:1253–1259. doi: 10.1016/0028-3908(94)90024-8. [DOI] [PubMed] [Google Scholar]
- 16.Ghafourifar P, Schenk U, Klein SD, Richter C. J Biol Chem. 1999;274:31185–31188. doi: 10.1074/jbc.274.44.31185. [DOI] [PubMed] [Google Scholar]
- 17.Lemeshko VV, Solano S, Lopez LF, Rendon DA, Ghafourifar P, Gomez LA. Arch Biochem Biophys. 2003;412:176–185. doi: 10.1016/s0003-9861(03)00034-1. [DOI] [PubMed] [Google Scholar]
- 18.Sasaki M, Hori MT, Hino T, Golub MS, Tuck ML. Am J Hypertens. 1997;10:371–378. [PubMed] [Google Scholar]
- 19.Hasegawa G, Kumagai S, Yano M, Wang YG, Kobayashi Y, Saito Y. FEBS Lett. 2003;554:127–132. doi: 10.1016/s0014-5793(03)01128-1. [DOI] [PubMed] [Google Scholar]
- 20.Ashraf M, Bloor CM. Virchows Arch B Cell Pathol. 1976;22:287–297. doi: 10.1007/BF02889222. [DOI] [PubMed] [Google Scholar]
- 21.Coll KE, Joseph SK, Corkey BE, Williamson JR. Determination. J Biol Chem. 1982;257:8696–8704. [PubMed] [Google Scholar]
- 22.Pinton P, Leo S, Wieckowski MR, Di Benedetto G, Rizzuto R. J Cell Biol. 2004;165:223–232. doi: 10.1083/jcb.200311061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jain SK, Shohet SB. Biochim Biophys Acta. 1981;642:46–54. doi: 10.1016/0005-2736(81)90136-x. [DOI] [PubMed] [Google Scholar]
- 24.Dedkova EN, Ji X, Lipsius SL, Blatter LA. Am J Physiol, Cell Physiol. 2004;286:C406–C415. doi: 10.1152/ajpcell.00155.2003. [DOI] [PubMed] [Google Scholar]
- 25.Kanai AJ, Pearce LL, Clemens PR, Birder LA, VanBibber MM, Choi SY, de Groat WC, Peterson J. Proc Natl Acad Sci USA. 2001;98:14126–14131. doi: 10.1073/pnas.241380298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Boveris A, Valdez LB, Alvarez S, Zaobornyj T, Boveris AD, Navarro A. Antioxid Redox Signal. 2003;5:265–271. doi: 10.1089/152308603322110841. [DOI] [PubMed] [Google Scholar]
- 27.Arnaiz SL, Coronel MF, Boveris A. Nitric Oxide. 1999;3:235–243. doi: 10.1006/niox.1999.0229. [DOI] [PubMed] [Google Scholar]
- 28.Riobo NA, Melani M, Sanjuan N, Fiszman ML, Gravielle MC, Carreras MC, Cadenas E, Poderoso JJ. J Biol Chem. 2002;277:42447–42455. doi: 10.1074/jbc.M204580200. [DOI] [PubMed] [Google Scholar]
- 29.Ghafourifar P, Cadenas E. Trends Pharmacol Sci. 2005;26:190–195. doi: 10.1016/j.tips.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 30.Beltran B, Mathur A, Duchen MR, Erusalimsky JD, Moncada S. Proc Natl Acad Sci U S A. 2000;97:14602–14607. doi: 10.1073/pnas.97.26.14602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ischiropoulos H, Zhu L, Chen J, Tsai M, Martin JC, Smith CD, Beckman JS. Arch Biochem Biophys. 1992;298:431–437. doi: 10.1016/0003-9861(92)90431-u. [DOI] [PubMed] [Google Scholar]
- 32.Cruthirds DL, Novak L, Akhi KM, Sanders PW, Thompson JA, MacMillan-Crow LA. Arch Biochem Biophys. 2003;412:27–33. doi: 10.1016/s0003-9861(03)00039-0. [DOI] [PubMed] [Google Scholar]
- 33.Cereghetti GM, Scorrano L. Oncogene. 2006;25:4717–4724. doi: 10.1038/sj.onc.1209605. [DOI] [PubMed] [Google Scholar]
- 34.Kwon KJ, Jung YS, Lee SH, Moon CH, Baik EJ. J Neurosci Res. 2005;81:73–84. doi: 10.1002/jnr.20520. [DOI] [PubMed] [Google Scholar]
- 35.Canals S, Casarejos MJ, de Bernardo S, Rodriguez-Martin E, Mena AA. J Biol Chem. 2003;278:21542–21549. doi: 10.1074/jbc.M213174200. [DOI] [PubMed] [Google Scholar]
- 36.Liu B, Khan WA, Hannun YA, Timar J, Taylor JD, Lundy S, Butovich I, Honn KV. Proc Natl Acad Sci U S A. 1995;92:9323–9327. doi: 10.1073/pnas.92.20.9323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen X, Zhang X, Kubo H, Harris DM, Mills GD, Moyer J, Berretta R, Potts ST, Marsh JD, Houser SR. Circ Res. 2005;97:1009–1017. doi: 10.1161/01.RES.0000189270.72915.D1. [DOI] [PubMed] [Google Scholar]