

Abstract
The ability to determine conformational parameters of protein-folding landscapes is critical for understanding the link between conformation, function, and disease. Monitoring hydrogen exchange (HX) of labile protons at equilibrium enables direct extraction of thermodynamic or kinetic landscape parameters in two limiting extremes. Here, we establish a quantitative framework for relating HX behavior to landscape. We use this framework to demonstrate that the range of predicted global HX behavior for the majority of a set of characterized two-state proteins under near-native conditions does not readily span between both extremes. For most, stability may be quantitatively determined under physiological conditions, with semiquantitative boundaries on kinetics additionally determined using modest experimental perturbations to shift HX behavior. The framework and relationships derived in the simple context of two-state global folding highlight the importance of understanding HX across the entire continuum of behavior, in order to apply HX to map landscapes.
Keywords: hydrogen exchange, protein folding, two-state, conformational landscape, mass spectrometry
Folded proteins sample an ensemble of conformations in addition to the native state (Hvidt and Nielsen 1966; Karplus and McCammon 1983; Dill and Chan 1997). These alternative conformations may be involved in normal protein function, regulation, and degradation, as well as in diseases related to misfolding and aggregation (Kelly 1996; Dobson 1999). The distribution and transit of molecules among the different states is governed by the free-energy differences and barriers between them. This collection of states and barriers make up the folding landscape for a given protein. One approach to collecting information about a protein's landscape involves making measurements as a function of a controllable perturbant such as temperature or denaturant. These progressively unfold the protein, permitting estimation of landscape parameters by extrapolation to physiological conditions (Matthews and Hurle 1987). However, for the majority of proteins that depend on the assistance of chaperone networks to fold in the cell, such methods lead to aggregation (Clark 2004). Therefore, investigating the interplay between folding, function, and disease requires a method with the potential to extract landscape parameters for the protein of interest under near-native solution conditions.
Hydrogen-exchange detection of landscape
Hydrogen-deuterium exchange (HX) is a powerful method that has the capacity to measure directly landscape parameters of conformations sampled under physiological conditions (Englander et al. 1997). In recent years, the utility of hydrogen exchange has been greatly broadened by detection methods using mass spectrometry (MS) (Katta and Chait 1991; Hoofnagle et al. 2003; Eyles and Kaltashov 2004; Wales and Engen 2006). For example, the ability of HXMS to distinguish populations has been invaluable in resolving controversies of parallel protein-folding pathways (Miranker et al. 1993; Heidary et al. 1997; Tsui et al. 1999). The extreme sensitivity enabling investigations at low concentrations has enabled HXMS to probe amyloidogenic processes (Nettleton et al. 1998; Yao et al. 2005). In addition, dynamics of chaperone allostery (Rist et al. 2006), as well as assembly of complexes as large as the HIV capsid protein (Lanman and Prevelige Jr. 2004), have been investigated through HXMS. Changes in surface accessibility mapped by HXMS protection have been used to identify protein–protein interaction sites (Mandell et al. 1998; Woods Jr. and Hamuro 2001; Lee et al. 2004). The ability to monitor HXMS of individual proteins in complexes has provided new insights into substrates bound to chaperones (Robinson et al. 1994; Gross et al. 1996; Weber-Ban et al. 1999). Finally, HXMS has shown great promise for the possibility of monitoring protein stability and dynamics in vivo (Ghaemmaghami and Oas 2001; Engen et al. 2002).
HX is an established tool for mapping conformational landscapes of native proteins (Chamberlain and Marqusee 1997; Arrington and Robertson 2000b; Englander 2000; Woodward et al. 2004; Bai 2006; Bai et al. 2007). The transient sampling of alternative conformations by native proteins at equilibrium leads to an alteration of hydrogen-bonded and solvent-exposed structure. HX capitalizes on a subset of labile protein protons that can exchange with those of water when they are solvent accessible. At pH values above ∼4, this process is initiated by OH− abstraction of a protein proton, notably the amide NH (Eigen 1964). The rate of this process (kint) is governed predominantly by chemical considerations such as pH, temperature, and protein sequence. However, kint is only observed in short model peptides (as well as surface exposed amides), as protein structure protects the amide NH through burial and hydrogen bonding, slowing the rate of its exchange (Englander et al. 1997). The exchange of an amide site in a native protein occurs, therefore, with an observed rate constant (kHX), which is governed in part by chemistry and in part by the sampling of landscape features that result in exposure of the site.
Two regions of a landscape separated by a barrier can be distinguished if they have distinct HX properties. This was first modeled for a single amide site in terms of an equilibrium between two states with the sites inaccessible and accessible to solvent, respectively (Hvidt 1964). This equilibrium is governed by the rate constants for opening (ku) and closing (kf) linked to an exchange reaction occurring with kint only from the open state (Equation 1) (Hvidt 1964):
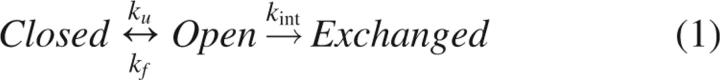 |
The closed and open states represent two points on a landscape, with the stability difference and heights of the barriers separating the closed and open states given by:
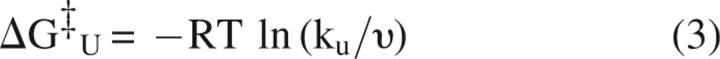 |
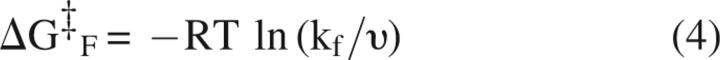 |
respectively, where R = the universal gas constant, T = temperature, υ = kbT/h, the rate of molecular vibration, kb = Boltzmann's constant, and h = Planck's constant (Matthews and Hurle 1987).
The landscape is effectively encoded into the apparent hydrogen-exchange rate kHX. kHX for a protected site is slowed relative to kint to an extent determined by ku and kf of the opening reaction exposing the site to solvent (Hvidt and Nielsen 1966):
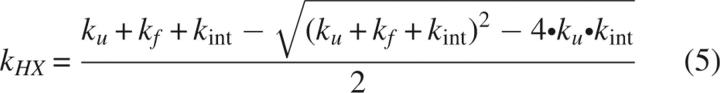 |
For a stable protein (kf >> ku), Equation 5 simplifies to (Hvidt and Nielsen 1966):
 |
Here, we focus on HX behavior arising from sites exposed only during the global unfolding equilibrium (N <-> U), with the long-term aim of extending our framework to exploit the broad range of equilibria detected by HX, including breathing from the native state, local unfolding of particular regions, transient population of intermediates, and intermolecular interfaces.
HX in limiting regimes directly extracts conformational parameters
Landscape parameters are readily decoded from kHX in two limiting extremes, where Equation 6 simplifies to allow direct extraction of either kinetics or thermodynamics. When kf << kint, Equation 6 reduces to:
 |
becoming first order, in what as known as the EX1 regime (Hvidt and Nielsen 1966). Measurements of kHX in EX1 have extracted the global ku for Protein L (Yi and Baker 1996), ubiquitin (Sivaraman et al. 2001), turkey ovomucoid third domain (Arrington and Robertson 1997), cytochrome c (Hoang et al. 2002), CD2.d1 (Cliff et al. 2004), and OspA (Yan et al. 2002). In the EX2 regime, kf >> kint, and Equation 6 reduces to a second order form (Hvidt and Nielsen 1966):
 |
Host—guest peptide studies have permitted calculation of kint for any amino acid sequence under a broad range of solution conditions (Bai et al. 1993). Global stability calculations for nearly a dozen proteins using ku/kf extracted from kHX measurements under EX2 conditions of residues, which exchange only through global unfolding yield ΔGU values that closely match those measured through calorimetry or chemical denaturation (Bai et al. 1994; Huyghues-Despointes et al. 1999). Furthermore, the powerful native-state HX approach has also identified and determined the stability of intermediates in many cases (Bai 2006). Thus, features of landscapes can be quantitatively mapped by HX in limiting regimes (Arrington and Robertson 2000b).
EX2 and EX1 behavior are distinguished experimentally through the profile in hydrogen exchange mass spectrometry or the pH dependence of kHX (Ferraro and Robertson 2004). The evolution of the mass profile as amide protons exchange with heavier solvent deuterons is exceptionally distinct in EX1 vs. EX2 (Miranker et al. 1993; Chung et al. 1997). HX approximated by EX1 gives a two (or more) peak mass profile, whereas EX2 yields only one. This distinction is trivially made, provided the number of sites exposed lead to a resolved separation in position between the unexchanged and exchanged peaks. For situations in which peaks are unresolved, statistical considerations of peak width can be used to distinguish regime (Arrington and Robertson 2000a; Weis et al. 2006). Regimes can additionally be distinguished by making multiple measurements as a function of pH, which changes kint in a defined manner (Hvidt 1964). In EX2, kHX scales linearly with changes in kint (Equation 8), while in EX1, kHX is independent of changes to kint (Equation 7) (Ferraro and Robertson 2004). Thus, either HXMS, or multiple measurements of kHX as a function of pH can be used to identify the HX regime.
Mapping full landscapes (i.e., ku and kf for a two-state protein) using HX requires shifting between both limiting regimes (Ferraro and Robertson 2004). Experimentally, solution conditions are altered to shift HX from EX2 toward EX1 by increasing kint and/or decreasing kf (Englander et al. 1972; Loh et al. 1996). The most common strategy is to affect kint using pH (Loh et al. 1996; Swint-Kruse and Robertson 1996; Arrington and Robertson 2000c; Simmons et al. 2003). Intrinsic exchange is base catalyzed above ∼pH 3, resulting in a log-linear dependence of kint on pH (Hvidt 1964; Englander et al. 1972). For several proteins, this has been exploited to successfully map both thermodynamics and kinetics by measuring kHX as a function of pH (Arrington and Robertson 1997, 2000c; Sivaraman et al. 2001; Rodriguez et al. 2002; Yan et al. 2002).
Alternatively, HX is shifted toward EX1 by slowing kf through adding denaturant (Equation 9) (Clarke and Fersht 1996; Loh et al. 1996; Deng and Smith 1998; Sivaraman et al. 2001; Simmons et al. 2003) or increasing temperature (Equation 10) (Loh et al. 1996; Swint-Kruse and Robertson 1996; Kim et al. 2002). These affect ku as well (Chen et al. 1989):
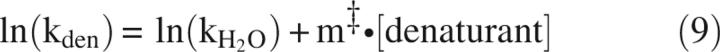 |
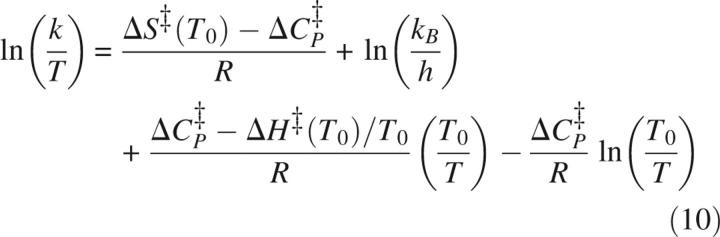 |
where kden and kH2O are ku or kf in the presence and absence of denaturant, respectively, m‡ is the slope of denaturant dependence of ln(ku) or ln(kf), k is ku or kf, T is temperature, and T0 is the reference temperature. ΔS‡ (T0) and ΔH‡(T0) are the activation entropy and enthalpy at T0, and ΔCp ‡ is heat-capacity change associated with reaching the transition state. In addition to affecting ku and kf, temperature influences kint: A 10-fold increase in kint is observed for every increase of ∼22°C (Englander et al. 1997). While there may be some effect of denaturant on kint, these effects are not large. This is evident in the observation that ΔGU calculated with Equations 2 and 8 using kHX values measured in denaturant and calculated values of kint yields values very close to ΔGU determined through other means (Huyghues-Despointes et al. 1999).
Perturbation of the landscape to shift HX behavior has important limitations. Multiple measurements as a function of denaturant or temperature are required to extrapolate kHX to native conditions. The growing recognition that landscapes affect in vivo function and disease makes landscape mapping an essential tool for cell biology and molecular medicine (Kelly 1996; Dobson 1999). However, the majority of proteins cannot be relied upon to fold correctly from a denatured state (Clark 2004). To fully exploit the potential of HX to characterize landscapes, it is necessary to investigate the extent to which landscape parameters may be extracted when solution conditions are limited to the near native. This requires a more quantitative understanding of the intersection between the influence of experimental tools on HX behavior and their impact on the landscape.
In this work, we assess the feasibility of using HX for measuring landscape parameters of proteins. While most proteins are large, with complex landscape topologies, small two-state proteins serve as models of protein domains. Because landscapes of many such model proteins are already well defined, we can assess the type of HX behavior experimentally accessible to them and determine the feasibility and difficulties with regard to using this technique on more complex proteins. We have therefore exploited an existing collection of thermodynamic and kinetic parameters for two-state proteins to evaluate the solution requirements for reaching both limiting regimes to extract both thermodynamics and kinetics from HX measurements. This is done by establishing a generalized framework for examining the interplay of the requirements for reaching the limiting regimes and the underlying conformational landscape. We then use the framework to probe the range of global HX behavior accessible under near-native conditions. Based on this analysis, we determine the extent to which the landscape of an uncharacterized two-state protein may be mapped through native-state HX.
Results
Quantitative framework for relating HX behavior to landscape
To quantitatively investigate the relationship between HX and conformational landscape, we first focused on a single two-state protein. The landscape of PWT spectrin SH3 has been extensively characterized, allowing us to model its global HX behavior and determine under which conditions kHX simplifies to reveal its thermodynamics or kinetics. ku and kf as a function of both denaturant and temperature have been measured for the PWT variant of spectrin SH3, which has a single Gly replacing the second and third residues of the wild-type protein (Martinez et al. 1999). Therefore, kHX for global HX may be calculated for a given condition using Equation 6 and the average kint calculated from its sequence at the matched pH and temperature.
kHX is readily interpreted in two limiting extremes where kf is either much larger than, or much smaller than kint. For PWT spectrin SH3 under physiological conditions (pH 6.5, 25°C), the reported kf is 4.6 s−1, (Martinez et al. 1999) and kint is 8.5 s−1. Obviously these are too close in value to fulfill either limiting condition. Instead, the global kHX that would be observed, (1.8 × 10−3 s−1 calculated using Equation 6) would lead to 15% error relative to the EX2 approximation (kHX,EX2 = 2.1 × 10−3 s−1), and 85% error relative to the EX1 approximation (kHX,EX1 = 1.2 × 10−2 s−1). Thus, to measure the landscape parameters accurately, it is necessary to shift PWT spectrin SH3′s HX from what has been termed the EXX regime (Xiao et al. 2005) into either limiting regime through changing kint or kf through experimental manipulations.
To determine and compare the extent of manipulations required to shift HX behavior between limiting regimes in a quantitative and general way, we define a parameter ρ:
As the transition between the limiting regimes is governed by the relative rates of refolding (kf) and the intrinsic exchange rate (kint), ρ quantifies HX behavior relative to the extremes. By systematically comparing kHX calculated with Equation 6 relative to the limiting approximations of EX1 (Equation 7) and EX2 (Equation 8) for a variety of ku, kf, and kint values over a broad range of ρ, we determined that for a stable protein (ku/kf < 0.01) kHX is within 5% of the EX2 approximation when ρ ≤ −1.3 (kf > 20•kint), and within 5% of the EX1 approximation when ρ ≥ 1.3 (kf < kint/20). PWT spectrin SH3′s physiological ρ calculated with Equation 11 is –0.7 (Fig. 1), indicating that ρ must be decreased by 0.6 to reach EX2, and increased by 2.0 to reach EX1.
Figure 1.

Global HX behavior for PWT spectrin SH3. Dependence of the apparent HX rate on ρ as a function of pH (solid), denaturant (dashed), or temperature (dotted). Gray shading specifies EXX regime. Black circle indicates HX under physiological conditions (23°C, pH 7). Changes in solution conditions, i.e., pH, T, or [denaturant] are indicated at the EXX boundaries and strongly EX1 (ρ = −1.3, 1.3, and 3.3, respectively).
Together, ρ and kHX setup a quantitative framework with which to effectively describe the HX behavior of unfolding events leading to exchange. ρ establishes a coordinate along which HX behavior may be characterized relative to the limiting extremes where kHX reports directly on landscape. As ρ increases from EX2 to EX1, the shift in relative landscape contributions (Equation 6) drives kHX along a coordinate between a minimum of (ku/kf)•kint to a maximum of ku. Thus, (ρ, kHX) may be used to characterize and compare the HX behavior of a given protein under different conditions (Fig. 1) or a set of proteins under the same conditions (Fig. 3A, below).
Figure 3.
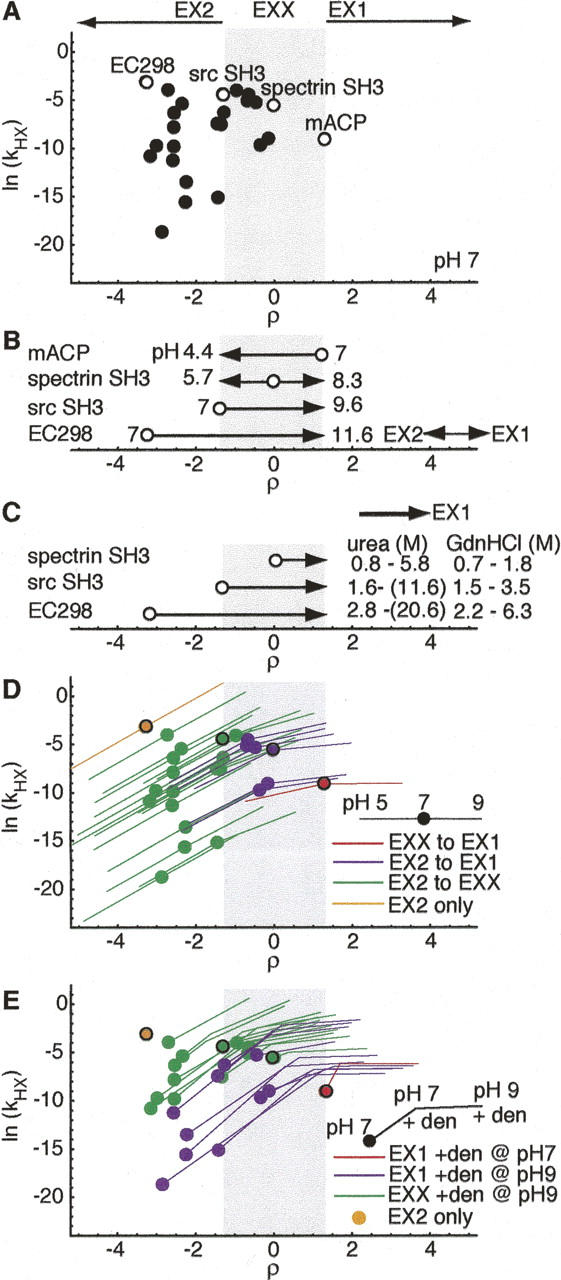
Global HX behavior for two-state proteins. (A) Distribution of HX behavior expected for stable two-state proteins with ku and kf characterized under standard conditions of 50 mM ionic strength, pH 7, 25°C (Maxwell et al. 2005). Circles denote global exchange of proteins from analysis set. Open circles indicate representative proteins spanning the range of observed HX behavior (ρ = −3.3, Eco298; ρ = 1.3, srcSH3; ρ = 0, specSH3; ρ = 1.3, mACP). (B) Calculated change in pH required to reach limiting regimes, relative to ρ positions at pH 7 (open circles) of representative proteins. (C) Calculated denaturant concentration required to shift HX from physiological ρ value to EX1. Concentration ranges calculated using maximum and minimum urea and GdnHCl m‡ f values reported for proteins in analysis set. (D) HX behavior accessible between pH 5 and pH 9. Circles indicate HX sampled at pH 7. Red indicates mACP, which samples only EX1, purple indicates proteins, which sample both EX2 and EX1, green indicates proteins, which sample EX2 and EXX, and orange indicates EC298, which samples only EX2 between pH 5 and pH 9. (E) HX behavior accessible upon addition of denaturant at modest levels (maintaining ku/kf = 0.01: ΔGU = 11.4 kJ/mol), at pH 7 and pH 9. Red indicates proteins that sample HX far enough into EX1 with denaturant addition at pH 7 to allow ku extrapolation, purple indicates proteins that sample HX far enough into EX1 with denaturant added at pH 9 to allow ku extrapolation, green indicates those that shift only to EXX with denaturant addition at pH 9, and orange indicates EC298, which is only 11.4 kJ/mol stable without denaturant.
The relationship between HX and landscape was first investigated for an individual protein by exploiting the known dependence of ku and kf for PWT spectrin SH3 on solution conditions. kHX can be calculated as a function of ρ using alterations to pH (affects kint), temperature (affects kint and kf), or denaturant (affects kf) (Fig. 1). pH has the greatest ability to shift ρ, in terms of range and direction, as seen by the magnitude of the X-axis change. We calculated the pH curve assuming that only kint, and neither ku nor kf, is affected by side-chain ionization. Given this assumption and the fact that both pH and ρ are based on a log10 scale, a one-unit pH change corresponds to a one-unit change in ρ. Thus, for PWT spectrin SH3, shifting between pH 3 and pH 11 covers seven ρ units. While the assumption that pH has no effect on landscape is unlikely to hold over such a wide range, it is reasonable to expect only minor perturbations over the range required to switch between EX2 and EX1 (pH 5.9–8.6). As such, the calculated change in kHX when using pH to shift ρ ranges from the limiting EX2 approximation (ku/kf)•kint to the limiting EX1 approximation (ku).
In contrast to pH, the primary effect of alterations in temperature and addition of denaturant is on landscape and not ρ. Increases in these two perturbants typically slow kf, resulting in a shift to larger values of ρ (i.e., toward EX1). For PWT spectrin SH3, addition of up to 9 M urea or increasing the temperature between 10°C and 100°C covers just over half of the ρ range accessible through pH (3.6 and 3.9 ρ units, respectively). Importantly, using these to shift HX behavior along the ρ axis changes ku and kf to such an extent (determined by Equations 9 or 10), that the protein is destabilized. This is reflected in the greater slopes of the ln kHX dependence on ρ for the temperature and denaturant curves, relative to the pH curve. Thus, while adding 5.1 M urea, or increasing temperature to 73°C is sufficient to reach the EX1 regime (where kHX = ku), still further measurements with increasing temperature or denaturant are needed to extrapolate ku back to physiological conditions.
The differential effects on PWT spectrin SH3′s landscape from shifting ρ to the same extent using either pH, temperature, or denaturant are illustrated in reaction coordinate form in Figure 2. For pH, ΔGU is maintained at –15 kJ/mol compared with ΔGU = 0 at standard state from EX2 to EX1 (Fig. 2B). Increasing temperature or denaturant to simply reach the EX1 boundary, however, leads to a completely destabilized protein (Fig. 2C,D). The ΔGU at 73°C = +1.4 kJ/mol, and ΔGU at 5.1 M urea = −0.4 kJ/mol. Continuing into EX1 to enable fitting of the dependence of ku on temperature or denaturant further destabilizes the protein (ΔGU at 88°C = +10.7 kJ/mol, and ΔGU at 7.6 M urea = +6.6 kJ/mol for ρ = 2.3). Thus, applying the (ρ, kHX) framework using the known landscape parameters of PWT spectrin SH3 illustrates both the relative effectiveness of the common methods for shifting HX (through the magnitude of the ρ change) and the relative consequences on landscape (through the magnitude and slope of the changes to ln[kHX]).
Figure 2.
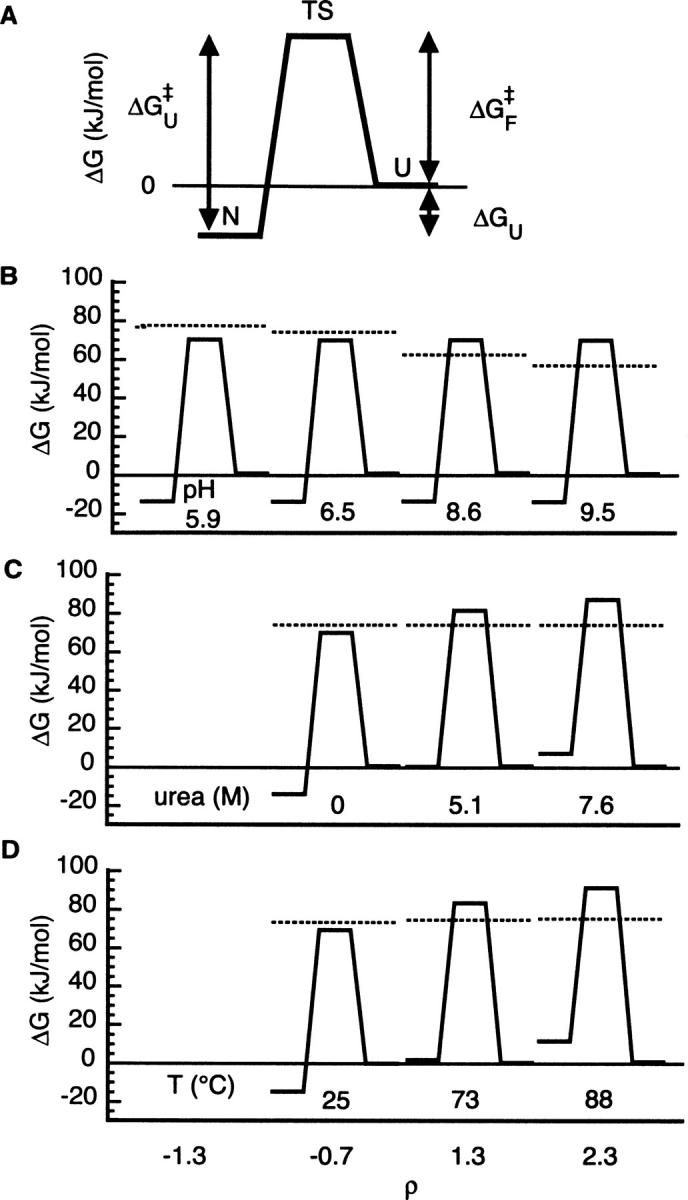
Landscape consequences of shifting PWT spectrin SH3 HX. (A) The conformational parameters of a two-state protein can be expressed in terms of the free energy landscape for unfolding from the native state (N) to the unfolded state (U) via a transition state ensemble (TS). The stability (ΔGU) and activation free energy barriers to unfolding (ΔG‡ U) and folding (ΔG‡ F) are calculated from ku, kf, and ku/kf using Equations 2, 3, and 4, respectively. Free energies are relative to U, arbitrarily set to 0. (B–D) Free energy landscape for PWT spectrin SH3 global unfolding calculated for ρ at the EX2 cutoff (ρ = −1.3), physiological (ρ = −0.5), EX1 cutoff (ρ = 1.3), and strongly EX1 (ρ = 3.3) conditions achieved using pH (B), denaturant (C), or temperature (D). Horizontal line across each landscape indicates apparent free energy barrier to intrinsic chemical exchange under matched conditions calculated from Equation 3 with kint in place of ku.
Distribution of HX for characterized two-state landscapes
We then applied this framework to a set of two-state proteins whose landscapes have been measured as a function of denaturant and extrapolated to standard conditions (25°C at pH 7, 50 mM buffer). We calculated the HX behavior for those 25 proteins from the 30 in the Maxwell collection (Maxwell et al. 2005), whose measurements were performed at pH 7, and that met our requirement of ku/kf ≥ 0.01, above which kHX vs. ρ is unchanging with stability. For each protein, the intrinsic exchange rate (from the unfolded conformation), kint, was calculated for each residue in the sequence at pH 7 (Bai et al. 1993), and the average kint was used in the calculations. The ρ values at pH 7 for global HX calculated using Equation 11 and the kHX values calculated and averaged using Equation 6 are listed in Table 1.
Table 1.
Expected HX behavior of two-state proteins under physiological conditions (pH 7, 25°C, 50 mM ionic strength)

Global HX under physiological conditions for characterized two-state proteins range from strongly EX2 up to, but not exceeding, the EX1 cutoff (Table 1; Fig. 3A). kint ranges between 1.7 and 4.1 s−1 for the set, while kf spans 8800 s−1 to 0.2 s−1. This corresponds to a total range of four ρ units (−3.3 to 1.3). The majority of the proteins (17 of 25) may be classified as EX2 exchangers. They range from EC298 as the most extreme EX2 exchanger (ρ = −3.3), with the fastest folding rate (8800 s−1), to Src SH3, at the EX2 cutoff (ρ = −1.3) with a folding rate of 78 s−1. The remaining characterized proteins fold too slowly to sample EX2. Only one, mACP, folds slowly enough to result in EX1 HX (kf = 0.2 s−1, ρ = 1.3). Strikingly, seven undergo HX in EXX (kf = 63–2.9 s−1). Thus, HX under physiological conditions can only readily extract quantitative landscape parameters for 2/3 of characterized proteins.
Figure 4.

Theoretical accessibility of two-state landscapes to HXMS mapping. (A) Calculated landscapes (solid lines) based on reported ku and kf for proteins spanning the spectrum of observed HX behavior under physiological conditions. (B) Landscape estimates (dashed lines) derived solely from kHX, ρest, and kint. Maximum and minimum estimates shown by double lines for semiquantitatively determined parameters. (C) Comparison of calculated and HX-derived parameters.
In EX2, the distribution in ln(kHX) directly reflects the variation in stability (|ΔGU| = 11.4–50 kJ/mol). The most stable protein of the set, CheW (ΔGU = −48 kJ/mol) has the slowest kHX. Notably, its ρ (−2.9) is very close to one of the least stable proteins of the set, Im7 (ρ = –2.7, ΔGU = −12 kJ/mol), whose kHX is four orders of magnitude faster. In EX1, the range in ln(kHX) values reflects the range in unfolding barrier heights, while in EXX, ln(kHX) values are a complex function of both stability and the unfolding barrier.
Shifting global HX behavior to reach limiting regimes
To determine the requirements for mapping landscapes, we calculated the theoretical range of experimental manipulation required to shift between the EX2 and EX1 regimes for proteins undergoing HX at representative ρ values. (Fig. 3B). Because it affects kint, pH shifts ρ uniformly (one unit pH change/one ρ unit) for all proteins. Therefore, an EXX exchanger exactly midway between the two limiting regimes at neutral pH like spectrin SH3 can span the 2.6 unit difference between the EX2 and EX1 cutoffs within the most reasonable pH range (5.7–8.3). (Note: In contrast to the PWT spectrin SH3 variant at pH 6.5 above, spectrin SH3 here refers to the wild-type protein at pH 7 in the standardized collection [Maxwell et al. 2005].) However, for those proteins undergoing global HX in a limiting regime at neutral pH, 2.6 reflects the minimum pH change required to reach the other limiting regime. The maximum required in our set is for the strongly EX2 protein, EC298, which nominally samples EX2 up to pH 9, and does not reach the EX1 cutoff until pH 11.6. Thus, for strongly EX2 proteins, increasing pH is effectively insufficient to shift HX into EX1.
Denaturant is always less effective than pH at shifting HX behavior. As demonstrated for PWT spectrin SH3 in Figure 2, denaturant affects all landscape parameters in a protein-specific matter. Using the reported m‡ f values for characterized two-state proteins, the denaturant concentration required to increase ρ by one unit ranges from 0.6 to 4.5 M urea and 0.6 to 1.4 M GdnHCl. Thus, modest concentrations are theoretically sufficient to shift ρ of an EXX exchanger to the EX1 cutoff (Fig. 3C). However, for proteins with m‡ f values from the lower end of the reported range, shifting ρ from EX2 to the EX1 cutoff would require urea in excess of the solubility limit. In addition, because denaturant affects the ku as well as kf, simply reaching the cutoff is not enough. The change in ρ must be yet larger to enable denaturant-dependent measurements of ku to the standard state. Thus, using denaturant alone to shift HX behavior is unlikely to enable extraction of ku except for a small subset of EXX exchangers.
Range of global HX behavior accessed under near-native conditions
An important advantage of HX is its potential to extract landscape parameters at equilibrium under conditions specifically related to function or disease and from aggregation-prone proteins whose landscapes are not accessible through traditional methods. We therefore investigated the extent to which parameters could be extracted from global HX if a more limited and native range of pH (pH 7 ± 2 units) and denaturant (ku/kf ≤ 0.01) are used to perturb the landscape (Table 2).
Table 2.
Range of HX behavior and landscape parameters extracted with mild experimental manipulations

Less than one-quarter of characterized proteins could sample both EX2 and EX1 for extraction of both thermodynamic and kinetic parameters within this pH range (Fig. 3D). Lowering pH to 5, the seven proteins in Table 1 exchanging in EXX at pH 7 can be switched into EX2, theoretically enabling the equilibrium constant to be measured. Six could be shifted into EX1 by adjusting pH to 9, additionally enabling extraction of the kinetic unfolding rate constant to completely characterize the landscape (purple symbols). On the other hand, the range of HX behavior for one EXX exchanger (GW1), and all except one of the EX2 exchangers at neutral pH, would only span EX2 and EXX with pH (green symbols), while mACP (red symbol), which samples EX1 HX at pH 7, would also only shift into EXX through pH manipulation. Furthermore, EC298, which samples the most extreme EX2 HX at pH 7 (orange), still samples EX2 at pH 9. Thus, for the majority of proteins, manipulating pH within a reasonable range from neutral shifts HX into EXX.
Denaturant alone is not effective in shifting HX to enable EX1 measurements, even for EXX exchangers, when restricted to amounts that only mildly destabilize the native protein. We determined the maximum denaturant concentration that could be added to each protein without destabilizing it beyond –11.4 kJ/mol, i.e., ku/kf ≤ 0.01, using Equation 9 (Table 2). We then calculated the change in ρ and kHX at that maximum denaturant concentration (Fig. 3E). HX must be shifted to ρ > 1.3 in order to make multiple EX1 measurements as a function of denaturant and extrapolate ku to 0 denaturant. Therefore, to classify HX as EX1 in the presence of denaturant, we required a minimum fivefold change in ku to be induced by the addition of denaturant between the concentration required to shift HX to ρ = 1.3 and the maximum concentration allowed without destabilizing the protein. Hence, even though addition of denaturant at pH 7 shifts HX of FKBP12, an EXX exchanger, and CheW, an EX2 exchanger, to ρ = 1.5 and 1.3, respectively (Table 2), only mACP HX is shifted far enough to enable the necessary measurements to qualify as EX1 under these conditions (Fig. 3E, red symbol).
Plainly, modest amounts of denaturant can theoretically enable ku measurements in combination with increased pH. By increasing pH from 7 to 9, ku measurements for 10 additional proteins could be extrapolated from EX1 HX measurements as a function of denaturant (Table 2; Fig. 3E, purple symbols). However, for those proteins exchanging in EXX at pH 7, the addition of denaturant at pH 9 is not an advantage over pH alone, as all except GW1 sample EX1 at pH 9 in the absence of denaturant (Fig. 3B). (Even with denaturant at pH 9, extrapolation of ku is not possible for GW1.) In contrast, addition of denaturant does shift HX to allow EX1 measurements at pH 9 for seven of the 17 proteins that still sample EX2 at pH 9. Thus, pH and denaturant in combination would enable measurement of ku for fewer than half of EX2 exchangers, assuming they are pH stable and would not be an advantage over pH alone for EXX exchangers.
Extracting landscape parameters from HX for uncharacterized two-state proteins
We next asked how closely the landscape of an uncharacterized protein (with only kint known from the sequence) would be determined from HX measurements in EX2, EXX, or EX1 regimes. Since HX regime can be classified through the HXMS profile (Miranker et al. 1993) or the pH dependence of kHX (Ferraro and Robertson 2004), ρ for any protein undergoing HX can be reasonably estimated as either ρest ≤ −1.3 (EX2), −1.3 < ρest < 1.3 (EXX), or ρest ≥ 1.3 (EX1). Obviously, for EX2 and EX1 HX, identification of the regime allows direct determination of either ku/kf or ku from the measured kHX.
In addition, assignment of the HX regime enables semiquantitative boundaries to be placed on kf and ku, regardless of regime. Because of the relationship between kint and kf in determining ρ, estimation of ρest enables calculation of the minimum and/or maximum possible value of kf using the known kint for the sequence. For EX2, kf ≥ 20•kint, in EXX 20•kint > kf > kint/20, and in EX1, kf ≤ kint/20. In addition, in EX2 and EXX, these kf limits may be used to determine boundaries on the possible value of ku relative to the measured kHX. In EX2, ku ≥ 20•kHX, and EXX, 20•kHX > ku > kHX. These limits on kf and ku can in turn be expressed in landscape terms using Equations 2–4, leading to semiquantitative estimates of ΔGU, ΔG‡ U, and ΔG‡ F.
To determine how fully the landscape of an unknown protein could be mapped, we compared the landscape information that would theoretically be gained from measurements of HX for four known proteins ranging in ρ from EX2 to the EX1 cutoff. For these proteins, we used only ρest, kHX, and the average kint calculated from the amino acid sequence to generate HX-derived landscape estimates. These are shown relative to the actual landscape calculated from the reported ku and kf values (Fig. 5).
Observation of global EX2 HX under physiological conditions would identify ρest ≤ −1.3 for EC298 and src SH3. Therefore, their stability (ΔGU = −11.4 and −14.0 kJ/mol, respectively) would be quantitatively extracted from their measured kHX (Fig. 5). In addition, this would set the upper limit for their folding barriers as ΔG‡ F, est ≤ 61.9 for Eco298 and ΔG‡ F, est ≤ 62.3 kJ/mol for src SH3. The upper estimate for the folding barriers also places an upper limit for the unfolding barriers, as ΔG‡ U = |ΔGU| + ΔG‡ F: ΔG‡ U, est ≤ 73.3 for Eco298 and ΔG‡ U,est ≤ 76.3 kJ/mol for src SH3. These estimates are reasonable for src SH3 (ΔG‡ F = 62.2 kJ/mol, ΔG‡ U = 76.1 kJ/mol), as its HX is right on the EX2 boundary, but that would not be known for an uncharacterized EX2 protein without further information to pinpoint ρ. As EC298 is the most extreme EX2 exchanger in the standardized set, its actual ΔG‡ F (50.5 kJ/mol) roughly sets the lower bound on the possible folding barrier for a protein with ρest ≤ −1.3, although some faster folding proteins, such as λ repressor (ΔG‡ F = 47.3 kJ/mol) (Maxwell et al. 2005) have been observed. Therefore, the range in folding and unfolding barrier estimates for an uncharacterized protein in EX2 is practically set at ∼15 kJ/mol.
On the other hand, an estimate of ρest ≥ 1.3 by observation of EX1, as for mACP (Fig. 5) would quantitatively determine the barrier to unfolding (ΔG‡ U = 95.3 kJ/mol), yield an estimate of the minimum possible folding barrier (ΔG‡ F,est ≥ 77 kJ/mol) and, therefore, the maximum possible stability (|ΔGU,est | ≤ 18.3 kJ/mol). For a protein with an actual ρ = 1.3, like mACP, these estimates are accurate (ΔG‡ F = 77 kJ/mol, ΔGU = −18.4 kJ/mol). Without knowing ρ, 0 is the effective boundary for minimum stability. Therefore, for an uncharacterized protein, the uncertainty range for the EX1 folding barrier and stability estimates is set by ΔGU,est.
For EXX proteins, while no parameter is exactly determined, upper and lower bounds for all of the parameters are obtained based on limits on ku and kf set by −1.3 ≤ ρest ≤ 1.3. Thus, for spectrin SH3, reasonable ranges for all of the parameters would be obtained from observing its HX in EXX: ΔG‡ F = 63.1–78.0 kJ/mol, ΔG‡ U = 78–84.3 kJ/mol, and ΔGU = 0 to −22.3 kJ/mol. The magnitude of those ranges hold for any protein with EXX exchange. Therefore, the uncertainty ranges for parameters estimated for an uncharacterized protein in EXX are 14.8, 7.5, and 22.4 kJ/mol for ΔG‡ F, ΔG‡ U, and ΔGU, respectively.
Limited pH changes can be used to refine the ρ estimate, greatly narrowing the uncertainty in landscape parameter determination. While only the EXX exchangers in our analysis would reach both limiting regimes within two units of pH 7, all of the proteins, except the extreme EX2 exchanger EC298, do switch into EXX (Fig. 3D). The EX2 to EXX transition could be identified, for example, by the change in width at the 50% exchange point (Miranker et al. 1996). By titrating pH until such a regime is observed, the average kint at that pH could be calculated from the sequence and used to estimate kf from the boundary ρ. Such a procedure could give estimates of kf within 10-fold, which would give ΔG‡ F and ΔG‡ U estimates accurate to within 6 kJ.
Discussion
We have developed a general theoretical framework to interrogate the landscape of proteins through HX. The approach capitalizes on qualitative use of HXMS to establish a semiquantitative localization of regime to a defined axis, ρ. Experimental manipulations shift HX into the regimes necessary to enable direct extraction of global landscape parameters. Using this approach, we can extrapolate that under physiological conditions, HX would not allow direct extraction of accurate landscape parameters for nearly one-third of globular proteins. However, it is only this group of proteins that can be manipulated by mild alteration of solution conditions to sample both limiting regimes, and thereby fully map their landscapes.
The optimal tool for shifting HX to limiting regimes is pH. This enables shifts across the broadest range of ρ, and does not rely on changing the landscape. Indeed, monitoring kHX as a function of pH by NMR has been applied successfully to extract both ku and kf through fitting the dependence to Equation 6 (Sivaraman et al. 2001) and through exploiting the range in kint values in the amino acid sequence to cover the range from EX2 to EX1 (Yan et al. 2002). Denaturant and temperature are far less effective at shifting ρ into EX1. This is consistent with observations that HX rates measured in 2 M denaturant experiments may be used to accurately determine ΔGU using the EX2 approximation (Equation 8) (Dai and Fitzgerald 2006). Only very stable proteins with steep dependencies of ln(kf) on denaturant and temperature (large m‡ f and ΔCp ‡ F), indicating very native-like transition states (Jackson 1998) sample EX1 without significant destabilization. In addition, multiple measurements of kHX beyond the EX1 boundary are required to extrapolate landscape parameters to a physiological standard state from added denaturant or increased temperature. Where possible, however, this approach provides additional structural information about the landscape, such as ΔCp ‡ U and m‡ U, which correlate with the relative solvent exposure of the transition state (Myers et al. 1995).
In this work, we have assumed that qualitative assessment of exchange regime can be performed by inspection of a hydrogen-exchange mass profile and/or the dependence of hydrogen exchange on pH. Given this, we have shown that simply knowing the sequence and the approximate value of ρ enables a reasonable estimate of global-folding landscape parameters even between the limiting regimes. While two-state behavior cannot be presumed for an uncharacterized system, it is reasonable to presume that a core of residues will exchange at the slowest rates and reflect only the global transition (Li and Woodward 1999; Ferraro and Robertson 2004). Thus, it is our expectation that at least a portion of the landscape of any globular protein can be readily characterized, even for biologically important systems that are particularly challenging experimentally.
HXMS on representative two-state proteins from the published set bear out our assumptions and conclusions. For example, the protein CI2 (ρ = −2.6 at pH 7), gives a characteristic EX2 mass profile with two phases in experiments at pH 6.8 (Xiao et al. 2005). Calculation of ΔGU from the kHX for the slow phase (corresponding to exchange of 36/65 residues) measured at 23°C is very close to that reported in Table 1 (ΔGU [HX] = −32.2 kJ/mol vs. ΔGU [calc.] = −39.8 kJ/mol). Furthermore, the solution pH must be raised above 10 in order to observe the EX1 profile. This is precisely what is predicted by the ρ framework, as CI2 HX is predicted to just reach the EX1 boundary (i.e., ρ = 1.3) at pH 10.
For protein L, an EX2 exchanger just on the cutoff at neutral pH (ρ = −1.3), a bimodal (i.e., EX1) HXMS profile corresponding to HX of 21/62 residues was observed at 60°C and pH 11 (Yi and Baker 1996). The reported kHX of 0.06 s−1 corresponds to ku, and the kf was estimated to be ∼0.3 s−1 based on the equilibrium constant from other experiments. Here, we calculate the average kint for protein L under those conditions to be kint = 5.9 × 105 s−1. These constants therefore correspond to ρ = 6.3, fully five ρ units above the EX1 cutoff. Importantly, the extreme pH used appears to have destabilized the landscape. ku and kf extrapolated from measurements in GdnHCl at 58°C, pH 7 (ku = 2.7 s−1, kf = 150 s−1) (Scalley and Baker 1997) differ significantly from those reported at pH 11, indicating that the four-unit increase in pH destabilizes the protein by 6.6 kJ/mol.
For most biologically relevant proteins, it is reasonable to assume that solution conditions will not be found that will allow for adequate sampling of both EX1 and EX2 regimes. Strong alteration of the landscape can be expected. We have shown here, for example, that even a model system like PWT spectrin SH3 appears strongly subject to landscape perturbation at denaturant and temperatures required to sample EX1, while high pH appears to destabilize protein L. Plainly, much milder perturbations are desirable to avoid landscape perturbation as well as aggregation. As we have shown, such limited experimental manipulations will primarily enable proteins to sample EXX.
In this work, we presumed a requirement for sampling at several points in the limiting regimes to extract full landscape information. However, landscape information is present, albeit convoluted, in the EXX regime. Recognition of this has driven the development of tools such as numerical simulation of protein HXMS (Arrington and Robertson 2000a; Xiao et al. 2005). The further development and refinement of such tools will ultimately be essential to unlocking the landscape information that is present in EXX. Coupling regime-independent extraction of landscape parameters from native HX with the applicability of MS to virtually any protein promises to greatly expand the range of protein landscapes accessible to experimental investigation.
Materials and Methods
Calculation of expected HX behavior
Our large analysis set (Table 1) consisted of 25/30 proteins from the previously published data set of Maxwell et al. (2005), whose ku and kf had been monitored as a function of denaturant exactly at pH 7, and which met the minimum stability requirement of ku/kf > 0.01. For all proteins analyzed, kint was calculated as described (Bai et al. 1993) at the appropriate pH and temperature for each residue and averaged over the sequence. kHX and ρ were calculated under physiological conditions (pH 6.5, 25°C for PWT spectrin SH3, and pH 7, 25°C, 50 mM ionic strength for Table 1 proteins) with Equations 6 and 11, using the appropriate kint and the reported ku and kf (Martinez et al. 1999; Maxwell et al. 2005) for PWT spectrin SH3 and Table 1 proteins, respectively.
Limiting values of ρ that define EX1 and EX2 were calculated as follows: kHX was calculated using Equation 6 and compared with the limiting approximations calculated with Equations 7 and 8 as a function of ρ. For stable proteins (ku/kf > 0.01), regardless of the individual values of ku, kf, or kint, kHX is within 5% of the EX1 approximation when ρ ≤ −1.3, and within 5% of the EX2 approximation when ρ ≥ −1.3.
Calculation of effect of solution conditions on ρ and kHX
Changes in kint with pH or temperature were calculated based on Bai et al. (1993). Changes in kf and ku based on denaturant were calculated with Equation 9 using the m‡ f and m‡ u reported (Maxwell et al. 2005). For PWT spectrin SH3, changes in kf and ku based on temperature were calculated using the reported ΔCp ‡ F and ΔCp ‡ U (Martinez et al. 1999). The corresponding changes in ρ and kHX were calculated as above using the calculated ku, kf, and kint values.
Acknowledgments
S.S.J. was supported by a Damon Runyon Cancer Foundation fellowship while this work was carried out. This work was supported, in part, by the National Institutes of Health, Grant No. DK54899. We acknowledge Drs. Stephanie Truhlar, Catherine Eakin, Erik Miller, and Judith Frydman for careful reading of the manuscript.
Footnotes
Reprint requests to: Andrew D. Miranker, Department of Molecular Biophysics and Biochemistry, Yale University, 260 Whitney Avenue, New Haven, CT 06520-8114, USA; e-mail: Andrew.Miranker@yale.edu; fax: (203) 432-5175.
Abbreviations: EX1, first order hydrogen exchange regime where Equation 7 holds; EX2, second order hydrogen exchange regime where Equation 8 holds; EXX, hydrogen exchange regime between EX2 and EX1; GdnHCl, guanidine hydrochloride; h, Planck's constant; HXMS, hydrogen exchange mass spectrometry; kb, Boltzmann's constant; kf, microscopic rate constant for folding; ku, microscopic rate constant for unfolding; kHX, rate constant for exchange of amide proton buried in N; kHX(EX1), rate constant for exchange when EX1 limiting approximation holds; kHX(EX2), rate constant for exchange when EX2 limiting approximation holds; kint, rate constant for intrinsic chemical exchange of amide proton; m‡ f, slope of denaturant dependence of ln(kf); m‡ u, slope of denaturant dependence of ln(ku); N, native state; R, universal gas constant; T, temperature; T0, reference temperature; U, unfolded ensemble; ΔCp ‡, heat capacity change upon unfolding to transition state ensemble; ΔG‡ F, activation free energy upon folding to transition state ensemble; ΔG‡ U, activation free energy upon unfolding to transition state ensemble; ΔGU, free energy change upon unfolding; ΔH‡(T0), activation enthalpy at T0; ΔS‡ (T0), activation entropy at T0; ρ, parameter characterizing HX relative to limiting approximations (Log10(kint/kf)); υ, rate of molecular vibration; kH20, rate constant in absence of denaturant.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.072994207.
References
- Arrington C.B. and Robertson, A.D. 1997. Microsecond protein folding kinetics from native-state hydrogen exchange. Biochemistry 36: 8686–8691. [DOI] [PubMed] [Google Scholar]
- Arrington C.B. and Robertson, A.D. 2000a. Correlated motions in native proteins from MS analysis of NH exchange: Evidence for a manifold of unfolding reactions in ovomucoid third domain. J. Mol. Biol. 300: 221–232. [DOI] [PubMed] [Google Scholar]
- Arrington C.B. and Robertson, A.D. 2000b. Kinetics and thermodynamics of conformational equilibria in native proteins by hydrogen exchange. Methods Enzymol. 323: 104–124. [DOI] [PubMed] [Google Scholar]
- Arrington C.B. and Robertson, A.D. 2000c. Microsecond to minute dynamics revealed by EX1-type hydrogen exchange at nearly every backbone hydrogen bond in a native protein. J. Mol. Biol. 296: 1307–1317. [DOI] [PubMed] [Google Scholar]
- Bai Y. 2006. Protein folding pathways studied by pulsed- and native-state hydrogen exchange. Chem. Rev. 106: 1757–1768. [DOI] [PubMed] [Google Scholar]
- Bai Y., Milne, J.S., Mayne, L., and Englander, S.W. 1993. Primary structure effects on peptide group hydrogen exchange. Proteins 17: 75–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai Y., Milne, J.S., Mayne, L., and Englander, S.W. 1994. Protein stability parameters measured by hydrogen exchange. Proteins 20: 4–14. [DOI] [PubMed] [Google Scholar]
- Bai Y., Feng, H., and Zhou, Z. 2007. Population and structure determination of hidden folding intermediates by native-state hydrogen exchange-directed protein engineering and nuclear magnetic resonance. Methods Mol. Biol. 350: 69–81. [DOI] [PubMed] [Google Scholar]
- Chamberlain A.K. and Marqusee, S. 1997. Touring the landscapes: Partially folded proteins examined by hydrogen exchange. Structure 5: 859–863. [DOI] [PubMed] [Google Scholar]
- Chen B.L., Baase, W.A., and Schellman, J.A. 1989. Low-temperature unfolding of a mutant of phage T4 lysozyme. 2. Kinetic investigations. Biochemistry 28: 691–699. [DOI] [PubMed] [Google Scholar]
- Chung E.W., Nettleton, E.J., Morgan, C.J., Gross, M., Miranker, A., Radford, S.E., Dobson, C.M., and Robinson, C.V. 1997. Hydrogen exchange properties of proteins in native and denatured states monitored by mass spectrometry and NMR. Protein Sci. 6: 1316–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark P.L. 2004. Protein folding in the cell: Reshaping the folding funnel. Trends Biochem. Sci. 29: 527–534. [DOI] [PubMed] [Google Scholar]
- Clarke J. and Fersht, A.R. 1996. An evaluation of the use of hydrogen exchange at equilibrium to probe intermediates on the protein folding pathway. Fold. Des. 1: 243–254. [DOI] [PubMed] [Google Scholar]
- Cliff M.J., Higgins, L.D., Sessions, R.B., Waltho, J.P., and Clarke, A.R. 2004. Beyond the EX1 limit: Probing the structure of high-energy states in protein unfolding. J. Mol. Biol. 336: 497–508. [DOI] [PubMed] [Google Scholar]
- Dai S.Y. and Fitzgerald, M.C. 2006. Accuracy of SUPREX (stability of unpurified proteins from rates of H/D exchange) and MALDI mass spectrometry-derived protein unfolding free energies determined under non-EX2 exchange conditions. J. Am. Soc. Mass Spectrom. 17: 1535–1542. [DOI] [PubMed] [Google Scholar]
- Deng Y. and Smith, D.L. 1998. Identification of unfolding domains in large proteins by their unfolding rates. Biochemistry 37: 6256–6262. [DOI] [PubMed] [Google Scholar]
- Dill K.A. and Chan, H.S. 1997. From Levinthal to pathways to funnels. Nat. Struct. Biol. 4: 10–19. [DOI] [PubMed] [Google Scholar]
- Dobson C.M. 1999. Protein misfolding, evolution and disease. Trends Biochem. Sci. 24: 329–332. [DOI] [PubMed] [Google Scholar]
- Eigen M. 1964. Proton transfer, acid-base catalysis, and enyzmatic hydrolysis. Angew. Chem. Int. Ed. Engl. 3: 1–19. [Google Scholar]
- Engen J.R., Bradbury, E.M., and Chen, X. 2002. Using stable isotope-labeled proteins for hydrogen exchange studies in complex mixtures. Anal. Chem. 74: 1680–1686. [DOI] [PubMed] [Google Scholar]
- Englander S.W. 2000. Protein folding intermediates and pathways studied by hydrogen exchange. Annu. Rev. Biophys. Biomol. Struct. 29: 213–238. [DOI] [PubMed] [Google Scholar]
- Englander S.W., Downer, N.W., and Teitelbaum, H. 1972. Hydrogen exchange. Annu. Rev. Biochem. 41: 903–924. [DOI] [PubMed] [Google Scholar]
- Englander S.W., Mayne, L., Bai, Y., and Sosnick, T.R. 1997. Hydrogen exchange: The modern legacy of Linderstrom-Lang. Protein Sci. 6: 1101–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyles S.J. and Kaltashov, I.A. 2004. Methods to study protein dynamics and folding by mass spectrometry. Methods 34: 88–99. [DOI] [PubMed] [Google Scholar]
- Ferraro D.M. and Robertson, A.D. 2004. EX1 hydrogen exchange and protein folding. Biochemistry 43: 587–594. [DOI] [PubMed] [Google Scholar]
- Ghaemmaghami S. and Oas, T.G. 2001. Quantitative protein stability measurement in vivo. Nat. Struct. Biol. 8: 879–882. [DOI] [PubMed] [Google Scholar]
- Gross M., Robinson, C.V., Mayhew, M., Hartl, F.U., and Radford, S.E. 1996. Significant hydrogen exchange protection in GroEL-bound DHFR is maintained during iterative rounds of substrate cycling. Protein Sci. 5: 2506–2513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidary D.K., Gross, L.A., Roy, M., and Jennings, P.A. 1997. Evidence for an obligatory intermediate in the folding of interleukin-1 β. Nat. Struct. Biol. 4: 725–731. [DOI] [PubMed] [Google Scholar]
- Hoang L., Bedard, S., Krishna, M.M., Lin, Y., and Englander, S.W. 2002. Cytochrome c folding pathway: Kinetic native-state hydrogen exchange. Proc. Natl. Acad. Sci. 99: 12173–12178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoofnagle A.N., Resing, K.A., and Ahn, N.G. 2003. Protein analysis by hydrogen exchange mass spectrometry. Annu. Rev. Biophys. Biomol. Struct. 32: 1–25. [DOI] [PubMed] [Google Scholar]
- Huyghues-Despointes B.M., Scholtz, J.M., and Pace, C.N. 1999. Protein conformational stabilities can be determined from hydrogen exchange rates. Nat. Struct. Biol. 6: 910–912. [DOI] [PubMed] [Google Scholar]
- Hvidt A. 1964. A discussion of the pH dependence of the hydrogen-deuterium exchange of proteins. C. R. Trav. Lab. Carlsberg 34: 299–317. [PubMed] [Google Scholar]
- Hvidt A. and Nielsen, S.O. 1966. Hydrogen exchange in proteins. Adv. Protein Chem. 21: 287–386. [DOI] [PubMed] [Google Scholar]
- Jackson S.E. 1998. How do small single-domain proteins fold? Fold. Des. http://biomednet.com/elecref/13590278003R0081. [DOI] [PubMed]
- Karplus M. and McCammon, J.A. 1983. Dynamics of proteins: Elements and function. Annu. Rev. Biochem. 52: 263–300. [DOI] [PubMed] [Google Scholar]
- Katta V. and Chait, B.T. 1991. Conformational changes in proteins probed by hydrogen-exchange electrospray-ionization mass spectrometry. Rapid Commun. Mass Spectrom. 5: 214–217. [DOI] [PubMed] [Google Scholar]
- Kelly J.W. 1996. Alternative conformations of amyloidogenic proteins govern their behavior. Curr. Opin. Struct. Biol. 6: 11–17. [DOI] [PubMed] [Google Scholar]
- Kim M.Y., Maier, C.S., Reed, D.J., and Deinzer, M.L. 2002. Conformational changes in chemically modified Escherichia coli thioredoxin monitored by H/D exchange and electrospray ionization mass spectrometry. Protein Sci. 11: 1320–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanman J. and Prevelige Jr, P.E. 2004. High-sensitivity mass spectrometry for imaging subunit interactions: Hydrogen/deuterium exchange. Curr. Opin. Struct. Biol. 14: 181–188. [DOI] [PubMed] [Google Scholar]
- Lee T., Hoofnagle, A.N., Kabuyama, Y., Stroud, J., Min, X., Goldsmith, E.J., Chen, L., Resing, K.A., and Ahn, N.G. 2004. Docking motif interactions in MAP kinases revealed by hydrogen exchange mass spectrometry. Mol. Cell 14: 43–55. [DOI] [PubMed] [Google Scholar]
- Li R. and Woodward, C. 1999. The hydrogen exchange core and protein folding. Protein Sci. 8: 1571–1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh S.N., Rohl, C.A., Kiefhaber, T., and Baldwin, R.L. 1996. A general two-process model describes the hydrogen exchange behavior of RNase A in unfolding conditions. Proc. Natl. Acad. Sci. 93: 1982–1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandell J.G., Falick, A.M., and Komives, E.A. 1998. Identification of protein-protein interfaces by decreased amide proton solvent accessibility. Proc. Natl. Acad. Sci. 95: 14705–14710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez J.C., Viguera, A.R., Berisio, R., Wilmanns, M., Mateo, P.L., Filimonov, V.V., and Serrano, L. 1999. Thermodynamic analysis of α-spectrin SH3 and two of its circular permutants with different loop lengths: Discerning the reasons for rapid folding in proteins. Biochemistry 38: 549–559. [DOI] [PubMed] [Google Scholar]
- Matthews C.R. and Hurle, M.R. 1987. Mutant sequences as probes of protein folding mechanisms. Bioessays 6: 254–257. [DOI] [PubMed] [Google Scholar]
- Maxwell K.L., Wildes, D., Zarrine-Afsar, A., De Los Rios, M.A., Brown, A.G., Friel, C.T., Hedberg, L., Horng, J.C., Bona, D., Miller, E.J., et al. 2005. Protein folding: Defining a “standard” set of experimental conditions and a preliminary kinetic data set of two-state proteins. Protein Sci. 14: 602–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranker A., Robinson, C.V., Radford, S.E., Aplin, R.T., and Dobson, C.M. 1993. Detection of transient protein folding populations by mass spectrometry. Science 262: 896–900. [DOI] [PubMed] [Google Scholar]
- Miranker A., Robinson, C.V., Radford, S.E., and Dobson, C.M. 1996. Investigation of protein folding by mass spectrometry. FASEB J. 10: 93–101. [DOI] [PubMed] [Google Scholar]
- Myers J.K., Pace, C.N., and Scholtz, J.M. 1995. Denaturant m values and heat capacity changes: Relation to changes in accessible surface areas of protein unfolding. Protein Sci. 4: 2138–2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nettleton E.J., Sunde, M., Lai, Z., Kelly, J.W., Dobson, C.M., and Robinson, C.V. 1998. Protein subunit interactions and structural integrity of amyloidogenic transthyretins: Evidence from electrospray mass spectrometry. J. Mol. Biol. 281: 553–564. [DOI] [PubMed] [Google Scholar]
- Rist W., Graf, C., Bukau, B., and Mayer, M.P. 2006. Amide hydrogen exchange reveals conformational changes in Hsp70 chaperones important for allosteric regulation. J. Biol. Chem. 281: 16493–16501. [DOI] [PubMed] [Google Scholar]
- Robinson C.V., Gross, M., Eyles, S.J., Ewbank, J.J., Mayhew, M., Hartl, F.U., Dobson, C.M., and Radford, S.E. 1994. Conformation of GroEL-bound α-lactalbumin probed by mass spectrometry. Nature 372: 646–651. [DOI] [PubMed] [Google Scholar]
- Rodriguez H.M., Robertson, A.D., and Gregoret, L.M. 2002. Native state EX2 and EX1 hydrogen exchange of Escherichia coli CspA, a small β-sheet protein. Biochemistry 41: 2140–2148. [DOI] [PubMed] [Google Scholar]
- Scalley M.L. and Baker, D. 1997. Protein folding kinetics exhibit an Arrhenius temperature dependence when corrected for the temperature dependence of protein stability. Proc. Natl. Acad. Sci. 94: 10636–10640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons D.A., Dunn, S.D., and Konermann, L. 2003. Conformational dynamics of partially denatured myoglobin studied by time-resolved electrospray mass spectrometry with online hydrogen-deuterium exchange. Biochemistry 42: 5896–5905. [DOI] [PubMed] [Google Scholar]
- Sivaraman T., Arrington, C.B., and Robertson, A.D. 2001. Kinetics of unfolding and folding from amide hydrogen exchange in native ubiquitin. Nat. Struct. Biol. 8: 331–333. [DOI] [PubMed] [Google Scholar]
- Swint-Kruse L. and Robertson, A.D. 1996. Temperature and pH dependences of hydrogen exchange and global stability for ovomucoid third domain. Biochemistry 35: 171–180. [DOI] [PubMed] [Google Scholar]
- Tsui V., Garcia, C., Cavagnero, S., Siuzdak, G., Dyson, H.J., and Wright, P.E. 1999. Quench-flow experiments combined with mass spectrometry show apomyoglobin folds through an obligatory intermediate. Protein Sci. 8: 45–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wales T.E. and Engen, J.R. 2006. Hydrogen exchange mass spectrometry for the analysis of protein dynamics. Mass Spectrom. Rev. 25: 158–170. [DOI] [PubMed] [Google Scholar]
- Weber-Ban E.U., Reid, B.G., Miranker, A.D., and Horwich, A.L. 1999. Global unfolding of a substrate protein by the Hsp100 chaperone ClpA. Nature 401: 90–93. [DOI] [PubMed] [Google Scholar]
- Weis D.D., Wales, T.E., Engen, J.R., Hotchko, M., and Ten Eyck, L.F. 2006. Identification and characterization of EX1 kinetics in H/D exchange mass spectrometry by peak width analysis. J. Am. Soc. Mass Spectrom. 17: 1498–1509. [DOI] [PubMed] [Google Scholar]
- Woods V.L. and Hamuro, Y. 2001. High resolution, high-throughput amide deuterium exchange-mass spectrometry (DXMS) determination of protein binding site structure and dynamics: Utility in pharmaceutical design. J. Cell. Biochem. Suppl. Suppl. 37: 89–98. [DOI] [PubMed] [Google Scholar]
- Woodward C., Carulla, N., and Barany, G. 2004. Native state hydrogen-exchange analysis of protein folding and protein motional domains. Methods Enzymol. 380: 379–400. [DOI] [PubMed] [Google Scholar]
- Xiao H., Hoerner, J.K., Eyles, S.J., Dobo, A., Voigtman, E., Mel'cuk, A.I., and Kaltashov, I.A. 2005. Mapping protein energy landscapes with amide hydrogen exchange and mass spectrometry. I. A generalized model for a two-state protein and comparison with experiment. Protein Sci. 14: 543–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan S., Kennedy, S.D., and Koide, S. 2002. Thermodynamic and kinetic exploration of the energy landscape of Borrelia burgdorferi OspA by native-state hydrogen exchange. J. Mol. Biol. 323: 363–375. [DOI] [PubMed] [Google Scholar]
- Yao Z.P., Tito, P., and Robinson, C.V. 2005. Site-specific hydrogen exchange of proteins: Insights into the structures of amyloidogenic intermediates. Methods Enzymol. 402: 389–402. [DOI] [PubMed] [Google Scholar]
- Yi Q. and Baker, D. 1996. Direct evidence for a two-state protein unfolding transition from hydrogen-deuterium exchange, mass spectrometry, and NMR. Protein Sci. 5: 1060–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]