

Abstract
The vascular endothelium is a critical regulator of vascular function. Diverse stimuli such as proinflammatory cytokines and hemodynamic forces modulate endothelial phenotype and thereby impact on the development of vascular disease states. Therefore, identification of the regulatory factors that mediate the effects of these stimuli on endothelial function is of considerable interest. Transcriptional profiling studies identified the Kruppel-like factor (KLF)2 as being inhibited by the inflammatory cytokine interleukin-1β and induced by laminar shear stress in cultured human umbilical vein endothelial cells. Overexpression of KLF2 in umbilical vein endothelial cells robustly induced endothelial nitric oxide synthase expression and total enzymatic activity. In addition, KLF2 overexpression potently inhibited the induction of vascular cell adhesion molecule-1 and endothelial adhesion molecule E-selectin in response to various proinflammatory cytokines. Consistent with these observations, in vitro flow assays demonstrate that T cell attachment and rolling are markedly attenuated in endothelial monolayers transduced with KLF2. Finally, our studies implicate recruitment by KLF2 of the transcriptional coactivator cyclic AMP response element–binding protein (CBP/p300) as a unifying mechanism for these various effects. These data implicate KLF2 as a novel regulator of endothelial activation in response to proinflammatory stimuli.
Keywords: cytokine, endothelium, adhesion, nitric oxide, transcription
Introduction
As the interface between blood and all tissues, the vascular endothelium is critically involved in the biological response to inflammation (1). Proinflammatory stimuli such as cytokines induce endothelial dysfunction and confer a proadhesive and prothrombotic phenotype (2, 3). Although these events are important in certain adaptive responses such as wound healing, sustained endothelial activation can lead to deleterious consequences as seen in several chronic inflammatory disease states such as rheumatoid arthritis, inflammatory bowel disease, and atherosclerosis (4).
One of the initial and key events in the endothelium's response to inflammatory stimuli is the expression of adhesion molecules such as vascular cell adhesion molecule (VCAM)-1 and E-selectin. These proteins mediate early leukocyte attachment and rolling on the endothelial surface (5). Subsequent events such as firm adhesion and transmigration across the endothelial lining then set the stage for a developing inflammatory response within tissues (5). Given the importance of these adhesion molecules in inflammation, the molecular mechanisms regulating their expression has been the subject of considerable investigation. Several studies have shown that the induction of VCAM-1 and E-selectin by inflammatory stimuli occurs principally at the level of transcription (6, 7). Furthermore, these studies highlight the central importance of nuclear factor κB (NF-κB) in mediating the induction of these adhesion molecules (6, 7, 8).
Accumulating evidence also suggests that in addition to biochemical stimuli such as inflammatory cytokines, fluid mechanical forces regulate key aspects of endothelial function (9). Unbranched segments of the vascular tree are exposed to laminar shear stress (LSS), whereas branch points experience disturbed/turbulent shear stress and flow reversal (9). Previous studies show that LSS induces specific gene products such as endothelial nitric oxide synthase (eNOS), which confers antiinflammatory and antithrombotic properties to the endothelium (10, 11). In addition, eNOS is an essential regulator of vascular reactivity and tone (12, 13). The ability of laminar flow to induce genetic factors that impart favorable properties to the endothelium is particularly relevant in certain inflammatory disease states such as atherosclerosis (14, 15). Indeed, atherosclerotic lesion development is accentuated at branch points that do not experience LSS (2, 14). Interestingly, these regions also exhibit increased levels of proinflammatory mediators such as NF-κB and adhesion molecules such as VCAM-1 (16–18). These observations suggest that hemodynamic forces such as LSS may be protective against atherosclerotic disease development (19).
Kruppel-like factors (KLFs) are a subclass of the zinc finger family of transcription factors which regulates cellular differentiation and tissue development (20). For example, KLF1/erythroid KLF is essential for red blood cell maturation (21, 22), whereas KLF4/gut KLF regulates the differentiation and maturation of dermal and gastrointestinal epithelial cells (23, 24). Targeted disruption of KLF2/lung KLF revealed an essential role in programming the quiescent phenotype of single-positive T cells and lung development (25, 26). Furthermore, within the blood vessel wall KLF2 expression is exclusively within endothelial cells, and KLF2-null mice exhibit abnormal blood vessel formation, resulting in embryonic hemorrhage and death (embryonic day 12.5 [27]). Recently, KLF2 expression was shown to be increased when cultured endothelial cells were exposed to sustained shear stress (28). However the targets and function of KLF2 in endothelial cell biology have not been elucidated.
Using a transcriptional profiling approach to assess global patterns of gene expression in cultured human umbilical vein endothelial cells (HUVECs) exposed to different humoral and biomechanical stimuli, we identified KLF2 as being inhibited by the inflammatory cytokine IL-1β and induced by LSS. Our studies demonstrate that KLF2 induces eNOS and inhibits the cytokine-mediated induction of adhesion molecules such as E-selectin and VCAM-1. Furthermore, we provide evidence that recruitment of the coactivator CBP/p300 may serve as a unifying mechanism underlying KLF2 function. Together, these observations support a critical role for KLF2 as a “molecular switch,” regulating critical aspects of endothelial cell function in inflammatory disease states.
Materials and Methods
Cell Culture and Reagents.
Human umbilical vein endothelial cells, human umbilical arterial endothelial cells, human aortic endothelial cells, and the tissue culture components were obtained from Cambrex Company. Bovine aortic endothelial cells (BAECs) were from Cell Systems and studied between passages 4 and 7. COS-7 cells were obtained from American Type Culture Collection. Human IL-1β (Calbiochem), human TNFα (R&D Systems), and LPS (Sigma-Aldrich) were used at final concentrations of 2.5 ng/ml, 10 ng/ml, and 1 μg/ml, respectively. Antibodies recognizing VCAM-1, intercellular cell adhesion molecule (ICAM)-1, p50, p65, IκB-α, phospho–IκB-α, and α-HA were purchased from Santa Cruz Biotechnology, Inc., p300 antibody (Labvision), eNOS antibody (BD Biosciences), anti–E-selectin mAb is clone H18/7 (developed in the laboratory of M.A. Gimbrone Jr.), and anti–α-tubulin antibody (Sigma-Aldrich). Adenoviral constructs were generated by the Harvard Gene Therapy Group. The VCAM-1 promoter was a gift from W.C. Aird (Beth Israel Deaconess Medical Center, Boston, MA). The eNOS promoter constructs were provided by C.J. Lowenstein (The Johns Hopkins University School of Medicine, Baltimore, MD) (29). The eNOS cDNA was provided by J.K. Liao (Brigham and Women's Hospital, Boston, MA).
RNA Isolation, Real-Time PCR, and Microarrays.
After exposure to the appropriate stimulus, endothelial cells were rinsed twice with ice cold PBS and scrapped in Trizol (GIBCO BRL). RNA was isolated by ethanol precipitation and DNase treated and purified on column (QIAGEN). The RNA integrity was assessed by a microfluidics RNA 6000 Nano-Assay using a 2100 Bioanalyzer (Agilent Technologies). For real-time PCR, RNA was transcribed using a MultiScribe-based RT reaction (PE Biosystems). PCR reactions were performed under conditions established previously in a GeneAmp 5700 sequence detection system (Applied Biosystems). For microarrays studies, 20 μg of total RNA were used for making fluorescently labeled (Cy3 and Cy5) cDNA probes. The probes, deposition control targets, and human Cot-1 DNA were added to the hybridization buffer (Agilent) and incubated for 2 min at 98°C before the solution was added to the surface of the microarrays. Hybridization to human cDNA microarrays (Agilent) was performed for 17 h at 65°C. Arrays were washed, dried, and scanned at 5 μm resolution using an Agilent G2505A microarray scanner. The scanned images were processed using Agilent feature extraction software. The resulting feature intensities, background intensities, and artificial flags were imported into Argus microarray analysis software for further data analysis as described previously (30, 19).
Northern and Western Blot Analysis.
HUVECs were infected with Ad-GFP (control) and Ad-KLF2 (K2) for 24 h, treated with IL-1β for 4 h, and then harvested for RNA or protein analysis. Northern blot studies were performed as described previously (31). Exogenous KLF2 mRNA expression was detected using a mouse KLF2 cDNA probe, whereas exogenous protein was detected using a polyclonal antibody (a gift from J. Leiden, Abbott Laboratories, Chicago, IL). The endogenous KLF2 mRNA expression was detected using a 3′ UTR fragment of the human KLF2 cDNA. For eNOS inhibitor experiments, L-NNA (100 μM–5 mM; Sigma-Aldrich) was added to the corresponding plates 4 h before the treatment with IL-1β. After experimental treatment of HUVECs, cellular protein extraction was performed as described, and Western blot analyses were performed (32).
Transient Transfection Assays.
BAECs or COS-7 cells were plated at a density of 5 × 104/well in 6-well plates 1 d before transfection. Transient transfection was performed using Fugene™6 reagent (Roche Molecular Biochemicals) according to instructions by the manufacturer. A total 1–2 μg of plasmid DNA was used in transfections, and total DNA was always kept constant. Cells were harvested 48 h after transfection, assayed for luciferase activity, and normalized to total protein as analyzed by BCA kit (Pierce Chemical Co.) in each sample. In some experiments, cells were treated with human TNFα (10 ng/ml; R&D Systems) for 6 h before harvest. All transfections were performed in triplicate for at least three independent experiments.
eNOS Activity Assays.
eNOS enzyme activity was measured by monitoring the conversion of L-[3H] arginine to L-[3H] citrulline in labeled cells as described previously (33). Briefly, HUVECs were incubated in Hepes buffer (25 mM Hepes, pH 7.3, 109 mM NaCl, 5.4 mM KCl, 0.9 mM CaCl2, 1 mM MgSO4, and 25 mM glucose) for 1 h at 37°C and then labeled with L-[3H] arginine (10 μCi/ml) and stimulated with 1 μM calcium ionophore A23187 for 10 min at 37°C. Immediately thereafter, cells were washed two times with ice cold PBS containing 5 mM EDTA and 5 mM l-arginine, scraped into 2 ml of stop buffer (20 mM sodium acetate, pH 5.5, 1 mM l-citrulline, 2 mM EDTA, and 2 mM EGTA) and sonicated. Aliquots of these lysates were withdrawn to determine total cellular protein abundance and 3H incorporation. L-[3H] citrulline was isolated from the remaining lysate by anion exchange chromatography with AG 50W-X8 resin (Bio-Rad Laboratories) and quantitated by liquid scintillation counting.
In Vitro Flow Assays.
HUVECs were infected for 24 h with KLF2 or control adenoviruses that both encoded green fluorescence protein (GFP). The GFP and KLF2 are expressed as separate proteins driven by a bidirectional ctyomegalovirus promoter. Cells were then washed twice with warm PBS, trypsinized, and resuspended in PBS containing 2% FBS. Cells in the two groups were individually sorted based on equal levels of GFP expression and plated on fibronectin-coated coverslips for adhesion and rolling experiments. The laminar flow chamber used has been described previously in detail (34, 35).
Glutathione S-Transferase Pull-Down and Coimmunoprecipitation Assays.
Glutathione S-transferase (GST) fusion proteins of KLF2, ZnF, and KLF2ΔZnF were generated using the pGEX-4T-1 vector according to manufacturer's recommendations (Amersham Biosciences). GST pull-down assays were performed as described previously by incubating whole cell extract from COS-7 cell expressing full-length P300 or FLAG-tagged KLF2 with GST fusion proteins containing different domains of KLF2 or P300 (a gift from Dr. Y. Shi, Harvard Medical School, Boston, MA) (36). For coimmunoprecipitation assays, COS-7 cells were transfected with Flag-tagged P300, HA-tagged KLF2, or both using Fugene. The presence of KLF2 and P300 complex was detected by immunoprecipitation with anti-FLAG antibody and immunoblotting with anti-HA antibody.
Gel Shift Studies.
For gel shift studies, ∼20 μg of the nuclear extract from HUVECs expressing Ad-GFP or Ad-KLF2 in the presence or absence of IL-1β was used for the gel shift assay. The experiment was performed as described previously using the NF-κB binding sites from the VCAM-1 promoter (31). Electrophoretic mobility shift assay using GST-KLF2 (2.5 μg) or adenovirally overexpressed KLF2 were performed using the following sequences: upper, 5′-GTCATGGGGGTGTGGGGGTTCCAGGAAAT-3′; lower: 5′-ATTTCCTGGAACCCCCACACCCCCATGAC-3′; upper (mutated) 5′-GTCATGGTATTTTAAGGGTTCCAGGAAAT-3′; and lower (mutated): 5′-ATT-TCCTGGAACCCTTAAAATACCATGAC-3′.
Results
KLF2 Expression in Endothelial Cells Is Regulated by IL-1β and LSS.
To better understand how biochemical and biomechanical stimuli modulate endothelial function, high throughput genomic transcriptional profiling studies were undertaken. In one set of experiments, cDNA microarray analysis was performed using mRNA from HUVECs exposed to vehicle or IL-1β for various time periods. In a second set of experiments, mRNA from HUVECs exposed to LSS (10 dynes/cm2 for 24 h) was compared with static (no flow) conditions. Results derived from the study of ∼15,000 genes identified several genes to be differentially regulated by these two stimuli. One of these factors, KLF2, was found to be inhibited by IL-1β treatment and induced by LSS. Using real-time PCR, we verified our transcriptional profiling results. As shown in Fig. 1 A, exposure of HUVECs to IL-1β for 3 h resulted in a 4.7-fold decrease in KLF2 expression. In contrast, KLF2 expression was induced 4.5-fold in endothelial cells exposed to LSS when compared with static (no flow) conditions (Fig. 1 B).
Figure 1.

KLF2 expression in endothelial cells in response to biochemcial and biomechanical stimuli. (A) Inhibition of KLF2 by IL-1β. HUVECs were treated with IL-1β (10 U/ml) for the indicated times, and expression was assessed by Taqman assay. *P < 0.05. (B) Induction of KLF2 by LSS. HUVEC monolayers were exposed to laminar flow (10 dynes/cm2) for 24 h, and the relative expression of KLF2 was assessed by Taqman assay. *P < 0.05.
KLF2 Induces eNOS Expression and Activity.
Because KLF2 was induced by LSS, we considered the possibility that this transcription factor may regulate the expression of certain LSS-inducible genes. Previous studies have identified several genes that are induced by LSS such as eNOS, a central regulator of endothelial cell function (10). As shown in Fig. 2 A, adenoviral overexpression of KLF2 resulted in a marked induction of eNOS mRNA and protein (Fig. 2 A). This effect was specific, since no induction was observed on the expression of another LSS-inducible gene COX-2 (not depicted). Furthermore, we consistently noted that exogenous KLF2 (Fig. 2 A, exo-KLF2) inhibited the expression of endogenous KLF2 (Fig. 2 A, endo-KLF2), perhaps via a negative feedback mechanism.
Figure 2.


KLF2 regulates eNOS expression and promoter activity. (A) KLF2 induces eNOS expression. HUVECs were infected with control (Ad-GFP; control) and KLF2 adenovirus (KLF2) at 10 MOI for 24 h and assessed for eNOS mRNA (Northern) and protein expression (Western). Exo-KLF2 refers to exogenously expressed mouse KLF2. Endo-KLF2 refers to endogenous human KLF2. (B) KLF2 induces eNOS enzymatic activity. HUVECs were infected with indicated adenoviral constructs, and eNOS activity was assessed by measuring the conversion of L-[3H] arginine to L-[3H] citrulline. *P < 0.0001. (C) Deletion and mutational analyses of eNOS promoter. Transient transfection and mutational studies identify the KLF site between −644 and −652 as critical for KLF2-mediated induction of the eNOS promoter activity. Top graph indicate studies in COS-7, whereas bottom graph is in BAECs. n = 6–12 per group. *P < 0.00005; **P < 0.00001; ‡P < 0.001. (D) KLF2 induction of the eNOS promoter is dependent on DNA binding. Transient transfection studies performed in BAECs and COS-7 cells demonstrate that KLF2 but not mutant constructs (DBD–DNA binding domain; ZnF–DNA binding domain alone; KLF2ΔZnF–non-DNA binding domain) can induce the eNOS promoter. n = 6–12 per group. *P < 0.0001. (E) KLF2 binds the eNOS promoter. Probe is derived from the sequence between −644 and −652 in the eNOS promoter. Gel shift studies were performed using GST–KLF2 fusion protein (left) and nuclear extracts from adenovirally overexpressed GFP (Ctrl) or KLF2 (right). Arrow denotes major retarded band, and arrowhead identifies the supershifted band using α-KLF2 antibody.
To determine if the increase in eNOS expression translates into an increase in catalytically active protein, we assayed for enzymatic activity by formation of L-[3H] citrulline from L-[3H] arginine (33, 37). As shown in Fig. 2 B, endothelial cells overexpressing KLF2 (Ad-KLF2) exhibited an ∼79-fold induction of eNOS activity relative to control overexpression (Ad-GFP). These data demonstrate that KLF2 overexpression leads to a dramatic increase in eNOS expression and activity.
KLF2 Regulates the eNOS Promoter.
KLFs are transcription factors that bind DNA and regulate target genes. To understand how KLF2 can induce eNOS expression, we assessed the effect on the eNOS promoter activity. For these studies we used the –1.6-kB fragment of the human eNOS promoter, which has been shown to confer expression in endothelial cells in vitro and in vivo (38–40). As shown in Fig. 2 C (top), KLF2 transactivated the –1.6-kB Luc eNOS promoter in COS-7 cells. This effect was specific, since two other family members (KLF6 and KLF15) were unable to transactivate the eNOS promoter (not depicted). Using a series of deletion constructs, we found that KLF2-mediated transactivation is partially reduced with deletion from –0.73-kB Luc to –0.66-kB Luc construct and essentially lost with further deletion with the –0.64-kB Luc promoter. A similar marked reduction in activity was seen in BAECs (Fig. 2 C, bottom). The ability to transactivate the –0.66-kB Luc construct requires full-length KLF2, since neither the DNA-binding domain alone (ZnF) nor the non-DNA binding region (KLF2ΔZnF) alone was able to induce the eNOS promoter in both BAECs and COS cells (Fig. 2 D).
The region between –0.66 and –0.64 kB of the eNOS promoter contains a consensus KLF binding site. To assess the importance of this site, the sequence was mutated in the context of the full-length and the –0.66-kB Luc promoter constructs. As shown in Fig. 2 C, mutation of this site in the full-length promoter (–1.6-kBmut-Luc) resulted in an ∼66% reduction in KLF2-mediated transactivation. Furthermore, mutation of this site within the –0.66-kB construct (–0.66-kBmut) resulted in nearly complete loss of KLF2-mediated transactivation. Finally, the ability of KLF2 to bind this site was verified by gel shift studies using purified GST-KLF2 (Fig. 2 E, left) and nuclear extracts from adenovirally infected cells (Fig. 2 E, right).
KLF2 Inhibits IL-1β–mediated Induction of Adhesion Molecules and T Cell Rolling and Adhesion to Endothelial Cells.
As shown in Fig. 1 A, KLF2 mRNA expression is inhibited by IL-1β. Inflammatory cytokines are known to induce several effects on endothelial cells, such as the expression of key adhesion molecules like VCAM-1, E-selectin, and ICAM-1 (41, 5, 14). To determine the effect of KLF2 on endothelial activation in response to inflammatory cytokines, HUVECs were infected with Ad-GFP (control; C) or Ad-KLF2 (K2) for 24 h, stimulated with IL-1β for an additional 4 h, and assessed for adhesion molecule mRNA and protein abundance. As shown in Fig. 3 A, treatment of HUVECs with IL-1β strongly induced VCAM-1, E-selectin, and ICAM-1 in both uninfected and control virus-infected cells. In contrast, adenoviral overexpression of KLF2 (10 MOI) strongly inhibited the induction of VCAM-1 and E-selectin but not ICAM-1 mRNA (Fig. 3 A). This effect was dose dependent, since no significant effect was observed at a dose of 1 MOI. Consistent with these observations, KLF2 inhibited VCAM-1 and E-selectin protein expression but did not affect ICAM-1 (Fig. 3 B). We also found that the ability of KLF2 to inhibit adhesion molecule expression was seen with other proinflammatory agents such as LPS and TNFα (Fig. 3 C).
Figure 3.


Effect of KLF2 on cytokine-mediated induction of adhesion molecules. (A) KLF2 inhibits VCAM-1 and E-selectin but not ICAM-1 mRNA. HUVECs were infected with the adenovirus (C, Ad-GFP; K2, Ad-KLF2) at the indicated dose, stimulated with IL-1β for 4 h, and total RNA was assessed for adhesion molecule expression. In contrast to VCAM-1 and E-selectin, no effect is observed on ICAM-1 expression. Exo-KLF2 refers to exogenously expressed mouse KLF2. Endo-KLF2 refers to endogenous human KLF2. (B) KLF2 inhibits IL-1β–mediated induction of VCAM-1 and E-selectin protein levels. Experiments were performed as described in A except cells were harvested for total protein and Western blot analysis was performed. (C) KLF2 inhibits VCAM-1 and E-selectin in response to multiple cytokines. HUVECs infected with the adenovirus at the indicated dose, stimulated with cytokine for 4 h, and total RNA was assessed for adhesion molecule expression. (D) KLF2 inhibits IL-1β–mediated T cell attachment and rolling to endothelial cells. HUVECs infected with the indicated adenovirus (Ctrl, Ad-GFP), stimulated with IL-1β, and then perfused with T cells under flow conditions (0.75 dynes/cm2). *P < 0.05.
Adhesion molecules such as VCAM-1 mediate lymphocyte and leukocyte attachment to endothelial cells (41, 5). To determine the functional consequence of KLF2's effect on adhesion molecule expression, in vitro flow assays were performed (42, 43). HUVECs infected with Ad-GFP (Ctrl) or Ad-KLF2 were FACS® sorted based on comparable levels of GFP expression and exposed to IL-1β and incubated with primary human T cells under flow conditions. As shown in Fig. 3 D, HUVECs infected with control Ad-GFP exhibited robust T cell attachment and rolling. In contrast, T cell attachment and rolling was markedly attenuated in KLF2-overexpressing cells. These data demonstrate that KLF2 can inhibit adhesion molecule expression and T cell attachment and rolling to activated endothelial cells.
KLF2 Inhibits NF-κB–mediated Activation.
Previous studies have demonstrated that NF-κB is critical for the activation of both the VCAM-1 and E-selectin promoters in response to inflammatory stimuli (44, 45, 6, 7, 8). This factor normally exists in the cytoplasm as a heterodimer of the p50 and p65 subunits. Activation is controlled by a family of inhibitors termed IκB that bind to NF-κB dimers, thereby retaining the entire complex in the cytoplasm. Phosphorylation and subsequent degradation of IκB in response to inflammatory cytokines liberates NF-κB heterodimers, which can then translocate to the nucleus, bind specific DNA sequences, and thereby affect target gene expression (45).
As a first step, we assessed the effect of adenovirally expressed KLF2 (versus control virus; Fig. 4 A, right, C) on p50, p65, IκB, and phosphorylated IκB levels at several time points after IL-1β treatment. As shown in Fig. 4 A (left), we did not observe any significant differences among the two groups. In addition, we did not observe any effect on nuclear accumulation of p65 after IL-1β treatment (Fig. 4 A, right). We next assessed if KLF2 can affect NF-κB DNA binding. Nuclear extracts were harvested from HUVECs overexpressing Ad-GFP and Ad-KLF2 in the presence or absence of IL-1β stimulation, and gel shift assays were performed using a NF-κB site derived from the VCAM-1 promoter (6). As expected, NF-κB binding was not detected in either Ad-GFP (Ctrl) or Ad-KLF2–infected cells in the absence of cytokine treatment (not depicted). However, as shown in Fig. 4 B (left) a DNA–protein complex is induced in Ad-GFP cells after treatment with IL-1β, and the presence of p50 and p65 in this complex was verified by competition and supershift studies. In the presence of KLF2 (right), a nearly identical binding pattern was observed. These data suggest that KLF2's inhibitory effect is not due to effects on NF-κB expression or DNA binding.
Figure 4.


KLF2 inhibits NF-κB function. (A) KLF2 does not affect expression or nuclear translocation of various components of the NF-κB pathway. HUVECs were infected with the adenovirus (C, Ad-GFP; K2, Ad-KLF2), stimulated with IL-1β for 30 or 60 min, and assessed for expression of the indicated factors by Western blot analysis. NE, nuclear extracts; Cyto, cytoplasmic extracts. (B) KLF2 does not affect NF-κB binding to DNA. Nuclear extracts were harvested from HUVECs overexpressing Ad-GFP (control) and Ad-KLF2 (KLF2) in the presence or absence of IL-1β. Induced NF-κB band is designated by the arrow. Specificity was verified by competition and supershift studies. (C) KLF2 inhibits TNFα-mediated induction of the VCAM-1 promoter and NF-κB concatemer. Transient transfection studies were performed in BAECs with the indicated constructs. A similar degree of inhibition is seen with KLF2 and KLFΔZnF. n = 6–12 per group; *P < 0.00005; **P < 0.00001. (D) KLF2 inhibits p65-mediated induction of the VCAM-1 and NF-κB concatemer. Transient transfection studies were performed in COS-7 cells with the indicated constructs. A similar degree of inhibition is seen with KLF2 and KLFΔZnF. n = 6–12 per group; *P < 0.00005; **P < 0.00001.
Previous studies have demonstrated that nitric oxide (NO) can partially inhibit VCAM-1 and E-selectin induction by inflammatory cytokines (46). To test this possibility, we pretreated cells with two eNOS inhibitors, L-NAME and L-NNA, over a broad concentration range (100 μM–5 mM). Consistent with previous reports in the literature, pretreatment with either inhibitor resulted in a small recovery of the KLF2-mediated inhibition of VCAM-1 and E-selectin protein expression (∼10–15%; not depicted) (46). Thus, although the production of NO may contribute in small part to the inhibitory effect of KLF2 on adhesion molecule expression, NO does not account for the large majority of KLF2's inhibitory effect.
We next explored the effects of KLF2 on cytokine and p65-mediated induction of the VCAM-1 promoter and an NF-κB reporter construct. As shown in Fig. 4 C, treatment of BAECs with TNFα induced both the VCAM-1 promoter and NF-κB concatemer. Cotransfection with KLF2 markedly attenuated this induction. Interestingly cotransfection of the non-DNA binding domain of KLF2 (KLF2ΔZnF) was equally effective in inhibiting promoter activity, whereas the DNA binding domain alone was not sufficient (Fig. 4 C). Similar results were seen in the heterologous cell line, COS 7. Transfection with p65 resulted in a robust induction of both the VCAM-1 and NF-κB promoter (∼130- and 160-fold, respectively; Fig. 4 D). Cotransfection with KLF2 or KLF2ΔZnF (but not ZnF alone) potently inhibited p65-mediated transactivation of the VCAM-1 promoter and NF-κB concatemer. These data suggest that KLF2-mediated inhibition of NF-κB does not require DNA-binding and is mediated via the non-DNA binding domain.
Recruitment of the Coactivator CBP/p300 by KLF2 as a Unifying Mechanism.
The data presented above demonstrate that KLF2 can induce eNOS and inhibit cytokine-mediated induction of endothelial adhesion molecules. The induction of eNOS requires DNA binding, whereas the inhibition of VCAM-1 does not. Furthermore, the inhibitory effect occurs in the absence of any effect on p50/p65 expression or DNA binding. To reconcile these observations, we considered the possibility that KLF2 may recruit away from NF-κB, a critical cofactor that is required for its function. Previous studies have demonstrated that NF-κB activity is critically modulated by several cofactors such as p300/CBP, PCAF-1, and SRC-1, (47, 48, 8). We chose to focus on p300/CBP, since it is essential for optimal NF-κB–mediated transcriptional activity in endothelial cells (8). In addition, previous studies demonstrate that the interaction of other KLF family members with p300 is important for the ability of these factors to transactivate reporter genes (49–51). Consistent with this hypothesis, cotransfection of CBP/p300 rescued both the KLF2 and KLF2ΔZnF-mediated repression of the NF-κB concatemer (Fig. 5 A). Furthermore, cotransfection of KLF2 (but not ZnF) and CBP/p300 augmented eNOS promoter activity to a greater degree than either factor alone (Fig. 5 B). These data suggest that KLF2 and p300 can work in a cooperative fashion.
Figure 5.
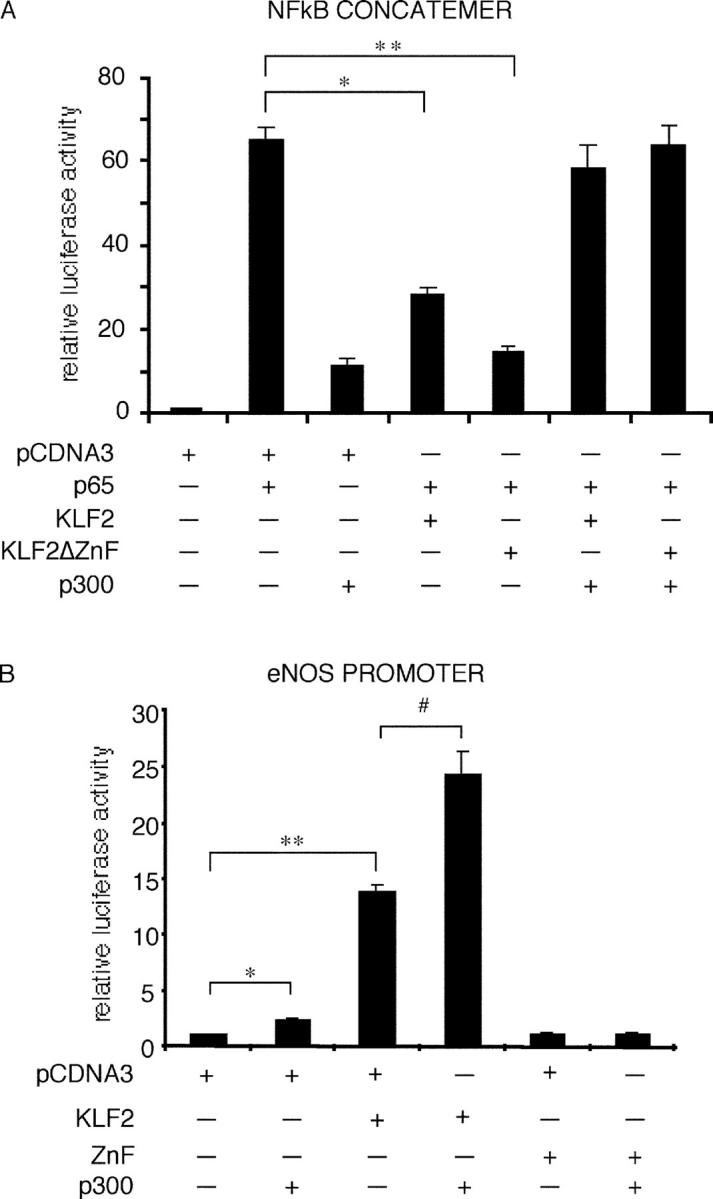

KLF2 interacts directly with p300. (A) p300 rescues KLF2-mediated inhibition. Transient transfection studies with the indicated plasmids were performed in COS-7 cells. Cotransfection studies demonstrate that p300 can rescue KLF2 and KLFΔZnF-mediated inhibition of the NF-κB concatemer. n = 6–12 per group. *P < 0.00001; **P < 0.00002. (B) KLF2 and p300 cooperate to induce the eNOS promoter. Cotransfection studies were performed in COS-7 cells. Cotransfection of KLF2 and p300 induces eNOS promoter activity greater than either factor alone. n = 6–12 per group; *P < 0.001; **P < 0.0001; #P < 0.05. (C) KLF2 and p300 interact. GST fusion proteins were generated for KLF2 and p300. Both KLF2 and KLFΔZnF can interact with p300 (top). Conversely, KLF2 interacts specifically with the NH2 terminus of p300. (D) KLF2 and p300 interact in cells. COS-7 cells were transfected with HA-KLF2 and Flag-p300. Immunoprecipitation was performed using the α-Flag antibody (or isotype control) followed by Western blot for KLF2 with α-HA antibody.
To determine if KLF2 and CBP/p300 interact directly, we next performed GST pull-down and coimmunoprecipitation studies. As shown in Fig. 5 C (top), full-length KLF2 (GST-KLF2) and the non-DNA binding region (GST-KLF2ΔZnF) can interact with p300. This effect is specific, since no interaction was seen with GST alone or the DNA-binding domain alone (GST-ZnF). Reverse binding studies showed that KLF2 interacted with the NH2 terminus of p300 (Fig. 5 C, bottom). This interaction is specific, since no interaction was noted with GST alone or other regions of p300 (GST-p300M and GST-p300C). To verify that this interaction occurs in cells, coimmunoprecipitation experiments were performed. As shown in Fig. 5 D, KLF2 could be immunoprecipitated from cellular extracts using an anti-Flag antibody to p300 but not with an IgG1 isotype control antibody. Together, these data suggest that KLF2 and p300 can physically interact in cells.
Discussion
The appreciation that inflammatory cytokines and fluid mechanical forces can modulate endothelial gene phenotype served here as the basis for the use of transcriptional profiling approaches to assess for global patterns of gene expression. From these studies we identified KLF2 as being differentially regulated by IL-1β and defined LSS. We provide evidence that KLF2 overexpression regulates the expression of key endothelial factors implicated in inflammatory states such as eNOS and VCAM-1/E-selectin. Furthermore, we suggest that competition for rate-limiting amounts of the coactivator p300 is the underlying mechanism for these distinct effects of KLF2. As such, these studies identify KLF2 as a novel regulator of proinflammatory endothelial activation.
Recruitment of immune cells is a critical feature common to most inflammatory disease states. This occurs through endothelial expression of certain adhesion molecules such as VCAM-1 and E-selectin (41, 5, 14, 44). These molecules allow for the attachment of leukocytes and lymphocytes to the blood vessel wall. Subsequently, these cells migrate across the endothelial barrier, invade adjacent tissues, and thereby sustain the inflammatory response. In this context, our observation that KLF2 can potently inhibit VCAM-1 and E-selectin expression is of considerable interest (Fig. 3, A–C). Importantly, KLF2-mediated inhibition is observed for several major physiologic proinflammatory stimuli such as IL-1β, TNFα, and LPS (Fig. 3 C). As a functional consequence, T cell adhesion to an endothelial cell monolayer is impaired (Fig. 3 D). The effect of KLF2 is specific because, in contrast to VCAM-1 and E-selectin, no significant inhibition is seen on ICAM-1 expression. This is an intriguing observation, although the basis for this differential inhibition has not been fully elucidated by our studies. Finally, our observations may be particularly relevant in the context of atherosclerosis. Recent studies by Dekker et al. demonstrated using normal human aortic tissue that KLF2 expression is robust in the endothelium except at branch points where expression is nearly absent (28). These regions are known to be particularly susceptible to atherosclerotic lesion formation and exhibit increased levels of NF-κB and VCAM-1 expression in animal models of atherosclerosis (16, 17). Our observations that KLF2 overexpression inhibits NF-κB–mediated activation of VCAM-1 coupled with the distinct pattern of KLF2 expression within the vascular tree support the possibility that KLF2 may contribute to the focal nature of early atherosclerotic disease.
Previous studies from several laboratories demonstrate that LSS can confer antiinflammatory, antithrombotic, and antiproliferative properties (10, 11). As such, our observations that laminar flow induces KLF2 led us to consider potential targets of KLF2. Although KLF2 did not induce some LSS-inducible factors such as COX-2 (unpublished data), we did observe a robust induction of eNOS (Fig. 2 A). Indeed, to our knowledge the magnitude of eNOS induction by KLF2 exceeds that observed by any other factor or stimulus. This enzyme is a well-established central regulator of vascular function (52–54). Mice deficient in eNOS are hypertensive, lack endothelium-dependent vasodilation, respond poorly to inflammatory challenge, and exhibit enhanced atherosclerotic lesion formation in susceptible mouse strains (55–59, 12, 13). Therefore, the KLF2-mediated induction of eNOS expression may have important functional consequences in vascular health and disease. However, despite its central importance our understanding of eNOS gene regulation remains incomplete. Transgenic studies suggest that the proximal 1.6 kB of the human eNOS promoter is sufficient to confer endothelial expression in small and large blood vessels in some tissues such as heart, brain, skeletal muscle, and aorta (40). Previous studies examining the human –1.6-kB promoter highlight the importance of GATA and Sp1 factors for basal eNOS expression (39). More recently, members of the Smad family have been shown to induce eNOS expression and promoter activity (29). Our dissection of the –1.6-kB human eNOS promoter highlights the importance of a KLF binding site (−644 to −652) as critical for KLF2-mediated induction of the eNOS promoter (Fig. 2, C and E). Whether some of these previously identified factors may cooperate with KLF2 has yet to be determined. However, a cooperative interaction between KLFs and members of the GATA family and Sp1 has been reported in other cellular systems (60–63). It is also noteworthy that this KLF2 binding site lies next to an AP-1 element, which has been implicated recently as mediating the LSS induction of the eNOS promoter, raising the possibility that these factors may cooperate to regulate eNOS gene expression (64). Finally, our studies do not exclude the possibility that KLF2 may also bind to regulatory regions outside of the –1.6-kB promoter. For example, Laumonnier et al. reported the identification of an enhancer element in the eNOS promoter located ∼4.9 kB upstream of the transcription start site (65). Indeed, a potential KLF binding site is present within this enhancer, raising the potential that KLF2 may bind additional regions in regulating endogenous eNOS expression.
To reconcile the ability of KLF2 to simultaneously inhibit VCAM-1 and induce eNOS (Fig. 3 B) we considered several unifying hypotheses. For example, the induction of eNOS by KLF2 may result in higher levels of NO, which itself may confer antiinflammatory properties. Previous studies demonstrate that NO can partially inhibit inflammatory cytokine-mediated induction of endothelial adhesion molecules such as VCAM-1 and E-selectin (46). Using two eNOS inhibitors, we observed a mild rescue of KLF2-mediated inhibition of VCAM-1 and E-selectin. These results are consistent with previous studies (46) but do suggest that mechanism(s) independent of NO production account for the vast majority of KLF2's inhibitory effects. Importantly, we observed that despite potent inhibition of the TNFα or p65-mediated induction of VCAM-1 and NF-κB promoter activity (Fig. 4, C and D), no effect was observed on various components of the NF-κB pathway (Fig. 4 A) or NF-κB DNA binding (Fig. 4 B). In light of the fact that NF-κB activity is critically dependent on transcriptional coactivator CBP/p300 (8) coupled with the observation that interaction of KLFs with CBP/p300 augments their transcriptional activity (50) led us to consider competition for this coactivator as the basis for the observed effects. This mechanism has been implicated in other systems such as the mutual antagonism observed between nuclear hormone receptors and AP-1 pathways (66). Indeed, overexpression of p300 was able to rescue KLF2-mediated inhibition of the NF-κB concatemer and augmented KLF2 induction of the eNOS promoter (Fig. 5, A and B). Conversely, NF-κB is able to potently inhibit KLF2 promoter activity (unpublished data). These results suggest that KLF2 recruits p300 away from p65 to a target gene such as eNOS and thereby promotes opposing effects on endothelial gene expression.
In summary, we have identified KLF2 as a cytokine-inhibited and LSS-induced transcription factor that regulates the expression of key genes involved in the endothelial response to inflammation. These studies support the possibility that KLF2 may serve as an important “molecular switch” regulating important aspects of vascular physiology and pathology.
Acknowledgments
This work was supported by the National Institutes of Health grants HL-03747 (to M.K. Jain), HL46957 (to T.M. Michel), and HL53993 (to F.W. Luscinskas), American Heart Association grant 0250030N (to M.K. Jain), American Diabetes Association grant 1-02-JF-40 (to M.K. Jain), National Institutes of Health-National Heart, Lung, and Blood Institute grants P50-HL56985 and R37-HL511509 (to M.A. Gimbrone), and the Dual-Mentored grant from the Brigham and Women's Hospital Research Council (to G. Garcia-Cardena and M.K. Jain).
S. SenBanerjee and Z. Lin contributed equally to this work.
Abbreviations used in this paper: BAEC, bovine aortic endothelial cell; eNOS, endothelial nitric oxide synthase; GFP, green fluorescence protein; GST, glutathione S-transferase; HUVEC, umbilical vein endothelial cell; ICAM, intercullular cell adhesion molecule; KLF, Kruppel-like factor; LSS, laminar shear stress; NF-κB, nuclear factor κB; NO, nitric oxide; VCAM, vascular cell adhesion molecule.
References
- 1.DiChiara, M.R., J.M. Kiely, M.A. Gimbrone, Jr., M.E. Lee, M.A. Perrella, and J.N. Topper. 2000. Inhibition of E-selectin gene expression by transforming growth factor beta in endothelial cells involves coactivator integration of Smad and nuclear factor kappaB-mediated signals. J. Exp. Med. 192:695–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gimbrone, M.A., Jr., J.N. Topper, T. Nagel, K.R. Anderson, and G. Garcia-Cardena. 2000. Endothelial dysfunction, hemodynamic forces, and atherogenesis. Ann. NY Acad. Sci. 902:230–239; discussion 239–240. [DOI] [PubMed] [Google Scholar]
- 3.Berk, B.C., J.J. Abe, W. Min, J. Suprapisitchat, and C. Yan. 2001. Endothelial atheroprotective and anti-inflammatory mechanisms. Ann. NY Acad. Sci. 947:93–111. [DOI] [PubMed] [Google Scholar]
- 4.Ross, R. 1999. Atherosclerosis—an inflammatory disease. N. Engl. J. Med. 340:115–126. [DOI] [PubMed] [Google Scholar]
- 5.Springer, T.A. 1994. Traffic signals for lymphocyte recirculation and leukocyte emigration: the multistep paradigm. Cell. 76:301–314. [DOI] [PubMed] [Google Scholar]
- 6.Neish, A.S., A.J. Williams, H.J. Palmer, M.Z. Whitley, and T. Collins. 1992. Functional analysis of the human vascular cell adhesion molecule 1 promoter. J. Exp. Med. 176:1583–1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Read, M.A., M.Z. Whitley, A. Williams, and T. Collins. 1994. NF-kappa B and I kappa B alpha: an inducible regulatory system in endothelial activation. J. Exp. Med. 179:503–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sheppard, K.A., D.W. Rose, Z.K. Haque, R. Kurokawa, E. McInerney, S. Westin, D. Thanos, M.G. Rosenfeld, C.K. Glass, and T. Collins. 1999. Transcriptional activation by NF-kappaB requires multiple coactivators. Mol. Cell. Biol. 19:6367–6378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gimbrone, M.A., Jr., T. Nagel, and J.N. Topper. 1997. Biomechanical activation: an emerging paradigm in endothelial adhesion biology. J. Clin. Invest. 100:S61–S65. [PubMed] [Google Scholar]
- 10.Topper, J.N., J. Cai, D. Falb, and M.A. Gimbrone, Jr. 1996. Identification of vascular endothelial genes differentially responsive to fluid mechanical stimuli: cyclooxygenase-2, manganese superoxide dismutase, and endothelial cell nitric oxide synthase are selectively up-regulated by steady laminar shear stress. Proc. Natl. Acad. Sci. USA. 93:10417–10422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lin, K., P.P. Hsu, B.P. Chen, S. Yuan, S. Usami, Y.J. Shyy, Y.S. Li, and S. Chien. 2000. Molecular mechanism of endothelial growth arrest by laminar shear stress. Proc. Natl. Acad. Sci. USA. 97:9385–9389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang, P.L., Z. Huang, H. Mashimo, K. Bloch, M.A. Moskowitz, J.A. Bevan, and M.C. Fishman. 1995. Hypertension in mice lacking the gene for endothelial nitric oxide synthase. Nature. 377:239–242. [DOI] [PubMed] [Google Scholar]
- 13.Shesely, E.G., N. Maeda, H.S. Kim, K.M. Desai, J.H. Krege, V.E. Laubach, P.A. Sherman, W.C. Sessa, and O. Smithies. 1996. Elevated blood pressures in mice lacking endothelial nitric oxide synthase. Proc. Natl. Acad. Sci. USA. 93:13176–13181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Libby, P. 2002. Inflammation in atherosclerosis. Nature. 420:868–874. [DOI] [PubMed] [Google Scholar]
- 15.Traub, O., and B.C. Berk. 1998. Laminar shear stress: mechanisms by which endothelial cells transduce an atheroprotective force. Arterioscler. Thromb. Vasc. Biol. 18:677–685. [DOI] [PubMed] [Google Scholar]
- 16.Hajra, L., A.I. Evans, M. Chen, S.J. Hyduk, T. Collins, and M.I. Cybulsky. 2000. The NF-kappa B signal transduction pathway in aortic endothelial cells is primed for activation in regions predisposed to atherosclerotic lesion formation. Proc. Natl. Acad. Sci. USA. 97:9052–9057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Iiyama, K., L. Hajra, M. Iiyama, H. Li, M. DiChiara, B.D. Medoff, and M.I. Cybulsky. 1999. Patterns of vascular cell adhesion molecule-1 and intercellular adhesion molecule-1 expression in rabbit and mouse atherosclerotic lesions and at sites predisposed to lesion formation. Circ. Res. 85:199–207. [DOI] [PubMed] [Google Scholar]
- 18.Li, H., M.I. Cybulsky, M.A.J. Gimbrone, Jr., and P. Libby. 1993. An atherogenic diet rapidly induces VCAM-1, a cytokine-regulatable mononuclear leukocyte adhesion molecule, in rabbit aortic endothelium. Arterioscler. Thromb. Vasc. Biol. 13:197–204. [DOI] [PubMed] [Google Scholar]
- 19.Garcia-Cardena, G., J.I. Comander, K.R. Anderson, B.R. Blackman, and M.A.J. Gimbrone, Jr. 2001. Biomechanical activation of vascular endothelium as a determinant of its functional phenotype. Proc. Natl. Acad. Sci. USA. 98:4478–4485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bieker, J.J. 2001. Kruppel-like factors: three fingers in many pies. J. Biol. Chem. 276:34355–34358. [DOI] [PubMed] [Google Scholar]
- 21.Nuez, B., D. Michalovich, A. Bygrave, R. Ploemacher, and F. Grosveld. 1995. Defective haematopoiesis in fetal liver resulting from inactivation of the EKLF gene. Nature. 375:316–318. [DOI] [PubMed] [Google Scholar]
- 22.Perkins, A.C., A.H. Sharpe, and S.H. Orkin. 1995. Lethal beta-thalassaemia in mice lacking the erythroid CACCC-transcription factor EKLF. Nature. 375:318–322. [DOI] [PubMed] [Google Scholar]
- 23.Segre, J.A., C. Bauer, and E. Fuchs. 1999. Klf4 is a transcription factor required for establishing the barrier function of the skin. Nat. Genet. 22:356–360. [DOI] [PubMed] [Google Scholar]
- 24.Katz, J.P., N. Perreault, B.G. Goldstein, C.S. Lee, P.A. Labosky, V.W. Yang, and K.H. Kaestner. 2002. The zinc-finger transcription factor Klf4 is required for terminal differentiation of goblet cells in the colon. Development. 129:2619–2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kuo, C.T., M.L. Veselits, and J.M. Leiden. 1997. LKLF: a transcriptional regulator of single-positive T cell quiescence and survival. Science. 277:1986–1990. [DOI] [PubMed] [Google Scholar]
- 26.Wani, M.A., S.E. Wert, and J.B. Lingrel. 1999. Lung Kruppel-like factor, a zinc finger transcription factor, is essential for normal lung development. J. Biol. Chem. 274:21180–21185. [DOI] [PubMed] [Google Scholar]
- 27.Kuo, C.T., M.L. Veselits, K.P. Barton, M.M. Lu, C. Clendenin, and J.M. Leiden. 1997. The LKLF transcription factor is required for normal tunica media formation and blood vessel stabilization during murine embryogenesis. Genes Dev. 11:2996–3006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dekker, R.J., S. Von Soest, R.D. Fontijn, S. Salamanca, P.G. de Groot, E. VanBavel, H. Pannekoek, and A.J.G. Horrevoets. 2002. Prolonged fluid shear stress induces a distinct set of endothelial cell genes, most specifically lung Kruppel-like factor (KLF2). Blood. 100:1689–1698. [DOI] [PubMed] [Google Scholar]
- 29.Saura, M., C. Zaragoza, W. Cao, C. Bao, M. Rodriguez-Puyol, D. Rodriguez-Puyol, and C.J. Lowenstein. 2002. Smad2 mediates transforming growth factor-beta induction of endothelial nitric oxide synthase expression. Circ. Res. 91:806–813. [DOI] [PubMed] [Google Scholar]
- 30.Comander, J., G.M. Weber, M.A. Gimbrone, Jr., and G. Garcia-Cardena. 2001. Argus—a new database system for Web-based analysis of multiple microarray data sets. Genome Res. 11:1603–1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Banerjee, S.S., M.W. Feinberg, M. Watanabe, S. Gray, R.L. Haspel, D.J. Denkinger, R. Kawahara, H. Hauner, and M.K. Jain. 2003. The Kruppel-like factor KLF2 inhibits peroxisome proliferator-activated receptor-gamma expression and adipogenesis. J. Biol. Chem. 278:2581–2584. [DOI] [PubMed] [Google Scholar]
- 32.Yoshida, M., W.F. Westlin, N. Wang, D.E. Ingber, A. Rosenzweig, N. Resnick, and M.A. Gimbrone, Jr. 1996. Leukocyte adhesion to vascular endothelium induces E-selectin linkage to the actin cytoskeleton. J. Cell Biol. 133:445–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Greif, D.M., R. Kou, and T. Michel. 2002. Site-specific dephosphorylation of endothelial nitric oxide synthase by protein phosphatase 2A: evidence for crosstalk between phosphorylation sites. Biochemistry. 41:15845–15853. [DOI] [PubMed] [Google Scholar]
- 34.Goetz, D.J., D.M. Greif, H. Ding, R.T. Camphausen, S. Howes, K.M. Comess, K.R. Snapp, G.S. Kansas, and F.W. Luscinskas. 1997. Isolated P-selectin glycoprotein ligand-1 dynamic adhesion to P- and E-selectin. J. Cell Biol. 137:509–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Luscinskas, F.W., G.S. Kansas, H. Ding, P. Pizcueta, B.E. Schleiffenbaum, T.F. Tedder, and M.A. Gimbrone, Jr. 1994. Monocyte rolling, arrest and spreading on IL-4-activated vascular endothelium under flow is mediated via sequential action of L-selectin, beta 1-integrins, and beta 2-integrins. J. Cell Biol. 125:1417–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Atkins, G.B., X. Hu, M.G. Guenther, C. Rachez, L.P. Freedman, and M.A. Lazar. 1999. Coactivators for the orphan nuclear receptor RORalpha. Mol. Endocrinol. 13:1550–1557. [DOI] [PubMed] [Google Scholar]
- 37.Igarashi, J., H.S. Thatte, P. Prabhakar, D.E. Golan, and T. Michel. 1999. Calcium-independent activation of endothelial nitric oxide synthase by ceramide. Proc. Natl. Acad. Sci. USA. 96:12583–12588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Robinson, L.J., S. Weremowicz, C.C. Morton, and T. Michel. 1994. Isolation and chromosomal localization of the human endothelial nitric oxide synthase (NOS3) gene. Genomics. 19:350–357. [DOI] [PubMed] [Google Scholar]
- 39.Zhang, R., W. Min, and W.C. Sessa. 1995. Functional analysis of the human endothelial nitric oxide synthase promoter. Sp1 and GATA factors are necessary for basal transcription in endothelial cells. J. Biol. Chem. 270:15320–15326. [DOI] [PubMed] [Google Scholar]
- 40.Guillot, P.V., L. Liu, J.A. Kuivenhoven, J. Guan, R.D. Rosenberg, W.C. Sessa, and W.C. Aird. 2000. A vascular bed-specific pathway. J. Clin. Invest. 103:799–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gimbrone, M.A., Jr., M.P. Bevilacqua, and M.I. Cybulsky. 1990. Endothelial-dependent mechanisms of leukocyte adhesion in inflammation and atherosclerosis. Ann. NY Acad. Sci. 598:77–85. [DOI] [PubMed] [Google Scholar]
- 42.Shaw, S.K., P.S. Bamba, B.N. Perkins, and F.W. Luscinskas. 2001. Real-time imaging of vascular endothelial-cadherin during leukocyte transmigration across endothelium. J. Immunol. 167:2323–2330. [DOI] [PubMed] [Google Scholar]
- 43.Allport, J.R., W.A. Muller, and F.W. Luscinskas. 2000. Monocytes induce reversible focal changes in vascular endothelial cadherin complex during transendothelial migration under flow. J. Cell Biol. 148:203–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Collins, T., and M.I. Cybulsky. 2001. NF-kB: pivotal mediator or innocent bystander in atherogenesis? J. Clin. Invest. 107:255–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tak, P.P., and G.S. Firestein. 2001. NF-kappaB: a key role in inflammatory diseases. J. Clin. Invest. 107:7–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.De Caterina, R., P. Libby, H.B. Peng, V.J. Thannickal, T.B. Rajavashisth, M.A. Gimbrone, Jr., W.S. Shin, and J.K. Liao. 1995. Nitric oxide decreases cytokine-induced endothelial activation. Nitric oxide selectively reduces endothelial expression of adhesion molecules and proinflammatory cytokines. J. Clin. Invest. 96:60–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Glass, C.K., D.W. Rose, and M.G. Rosenfeld. 1997. Nuclear receptor coactivators. Curr. Opin. Cell Biol. 9:222–232. [DOI] [PubMed] [Google Scholar]
- 48.Torchia, J., C.K. Glass, and M.G. Rosenfeld. 1998. Co-activators and co-repressors in the integration of transcriptional responses. Curr. Opin. Cell Biol. 10:373–383. [DOI] [PubMed] [Google Scholar]
- 49.Zhang, W., S. Kadam, B.M. Emerson, and J.J. Bieker. 2001. Site-specific acetylation by p300 or CREB binding protein regulates erythroid Kruppel-like factor transcriptional activity via its interaction with the SWI-SNF complex. Mol. Cell. Biol. 21:2413–2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Song, C.Z., K. Keller, K. Murata, H. Asano, and G. Stamatoyannopoulos. 2002. Functional interaction between coactivators CBP/p300, PCAF, and transcription factor FKLF2. J. Biol. Chem. 277:7029–7036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Geiman, D.E., H. Ton-That, A.W. Johnson, and X. Yang. 2000. Transactivation and growth suppression by the gut-enriched Kruppel-like factor (Kruppel-like factor 4) are dependent on acidic amino acid residues and protein-protein interaction. Nucleic Acids Res. 28:1106–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Michel, T. 1999. Targeting and translocation of endothelial nitric oxide synthase. Braz. J. Med. Biol. Res. 32:1361–1366. [DOI] [PubMed] [Google Scholar]
- 53.Cirino, G., S. Fiorucci, and W.C. Sessa. 2003. Endothelial nitric oxide synthase: the Cinderella of inflammation? Trends Pharmacol. Sci. 24:91–95. [DOI] [PubMed] [Google Scholar]
- 54.Albrecht, E.W.J.A., C.A. Stegeman, P. Heeringa, R.H. Henning, and H. van Goor. 2003. Protective role of endothelial nitric oxide synthase. J. Pathol. 199:8–17. [DOI] [PubMed] [Google Scholar]
- 55.Sasaki, M., S. Bharwani, P. Jordan, T. Joh, K. Manas, A. Warren, H. Harada, P. Carter, J.W. Elrod, M. Wolcott, et al. 2003. The 3-hydroxy-3-methylglutaryl-CoA reductase inhibitor pravastatin reduces disease activity and inflammation in dextran-sulfate induced colitis. J. Pharmacol. Exp. Ther. 305:78–85. [DOI] [PubMed] [Google Scholar]
- 56.Koedel, U., R. Paul, F. Winkler, S. Kastenbauer, P.L. Huang, and H.W. Pfister. 2001. Lack of endothelial nitric oxide synthase aggravates murine pneumococcal meningitis. J. Neuropathol. Exp. Neurol. 60:1041–1050. [DOI] [PubMed] [Google Scholar]
- 57.Heeringa, P., H. van Goor, Y. Itoh-Lindstrom, N. Maeda, R.J. Falk, K.J. Assmann, C.G. Kallenberg, and J.C. Jennette. 2000. Lack of endothelial nitric oxide synthase aggravates murine accelerated anti-glomerular basement membrane glomerulonephritis. Am. J. Pathol. 156:879–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Knowles, J.W., R.L. Reddick, J.C. Jennette, E.G. Shesely, O. Smithies, and N. Maeda. 2000. Enhanced atherosclerosis and kidney dysfunction in eNOS(−/−)Apoe(−/−) mice are ameliorated by enalapril treatment. J. Clin. Invest. 105:451–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kuhlencordt, P.J., R. Gyurko, F. Han, M. Scherrer-Crosbie, T.H. Aretz, R. Hajjar, M.H. Picard, and P.L. Huang. 2001. Accelerated atherosclerosis, aortic aneurysm formation, and ischemic heart disease in apolipoprotein E/endothelial nitric oxide synthase double-knockout mice. Circulation. 104:448–454. [DOI] [PubMed] [Google Scholar]
- 60.Goodwin, A.J., J.M. McInerney, M.A. Glander, O. Pomerantz, and C.H. Lowrey. 2001. In vivo formation of a human beta-globin locus control region core element requires binding sites for multiple factors including GATA-1, NF-E2, erythroid Kruppel-like factor, and Sp1. J. Biol. Chem. 276:26883–26892. [DOI] [PubMed] [Google Scholar]
- 61.Gregory, R.C., D.J. Taxman, D. Seshasayee, M.H. Kensinger, J.J. Bieker, and D.M. Wojchowski. 1996. Functional interaction of GATA1 with erythroid Kruppel-like factor and Sp1 at defined erythroid promoters. Blood. 87:1793–1801. [PubMed] [Google Scholar]
- 62.Yasuda, K., K. Hirayoshi, H. Hirata, H. Kubota, N. Hosokawa, and K. Nagata. 2002. The Kruppel-like factor Zf9 and proteins in the Sp1 family regulate the expression of HSP47, a collagen-specific molecular chaperone. J. Biol. Chem. 277:44613–44622. [DOI] [PubMed] [Google Scholar]
- 63.Higaki, Y., D. Schullery, Y. Kawata, M. Shnyreva, C. Abrass, and K. Bomsztyk. 2002. Synergistic activation of the rat laminin gamma1 chain promoter by the gut-enriched Kruppel-like factor (GKLF/KLF4) and Sp1. Nucl. Acid Res. 30:2270–2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wedgwood, S., C.J. Mitchell, J.R. Fineman, and S.M. Black. 2003. Developmental differences in the shear strees-induced expression of endothelial NO synthase: changing role of AP-1. Am. J. Physiol. Lung Cell. Mol. Physiol. 284:L650–L662. [DOI] [PubMed] [Google Scholar]
- 65.Laumonnier, Y., S. Nadaud, M. Agrapart, and F. Soubrier. 1999. Characterization of an upstream enhancer region in the promoter of the human endothelial nitric-oxide synthase gene. J. Biol. Chem. 275:40732–40741. [DOI] [PubMed] [Google Scholar]
- 66.Kamei, Y., L. Xu, T. Heinzel, J. Torchia, R. Kurokawa, B. Gloss, S.C. Lin, R.A. Heyman, D.W. Rose, C.K. Glass, and M.G. Rosenfeld. 1996. A CBP integrator complex mediates transcriptional activation and AP-1 inhibition by nuclear receptors. Cell. 85:403–414. [DOI] [PubMed] [Google Scholar]