

Abstract
Gangliosides are a family of sialic acid-containing glycosphingolipids highly enriched in the mammalian nervous system. Although they are the major sialoglycoconjugates in the brain, their neurobiological functions remain poorly defined. By disrupting the gene for a key enzyme in complex ganglioside biosynthesis (GM2/GD2 synthase; EC 2.4.1.92) we generated mice that express only simple gangliosides (GM3/GD3) and examined their central and peripheral nervous systems. The complex ganglioside knockout mice display decreased central myelination, axonal degeneration in both the central and peripheral nervous systems, and demyelination in peripheral nerves. The pathological features of their nervous system closely resemble those reported in mice with a disrupted gene for myelin-associated glycoprotein (MAG), a myelin receptor that binds to complex brain gangliosides in vitro. Furthermore, GM2/GD2 synthase knockout mice have reduced MAG expression in the central nervous system. These results indicate that complex gangliosides function in central myelination and maintaining the integrity of axons and myelin. They also support the theory that complex gangliosides are endogenous ligands for MAG. The data extend and clarify prior observations on a similar mouse model, which reported only subtle conduction defects in their nervous system [Takamiya, K., Yamamoto, A., Furukawa, K., Yamashiro, S., Shin, M., Okada, M., Fukumoto, S., Haraguchi, M., Takeda, N., Fujimura, K., et al. (1996) Proc. Natl. Acad. Sci. USA 93, 10662–10667].
Gangliosides are major cell surface determinants and the predominant sialoglycoconjugates in the mammalian nervous system (1–3). The most abundant gangliosides in the adult mammalian nervous system, GM1, GD1a, GD1b, and GT1b, are closely related and contain a ceramide lipid, a neutral tetrasaccharide core (Gal β3 GalNAc β4 Gal β4 Glc), and one or more sialic acids (2).‖ They are biosynthesized by the sequential action of a series of specific glycosyltransferases (4). Although a clear neurobiological role for the major nervous system gangliosides has not been defined, recent studies have proposed that GD1a and GT1b serve as complementary ligands for myelin-associated glycoprotein (MAG) (5, 6). MAG, a minor constituent of both peripheral and central myelinating glia (Schwann cells and oligodendrocytes), is localized predominantly to the adaxonal (periaxonal) glial plasmalemma (7). Because of its periaxonal location, it is postulated that MAG may mediate axon–glial interactions (8). This hypothesis is supported by the observations that MAG-deficient mice develop age-dependent axonal atrophy, axonal degeneration, and demyelination in sciatic nerves (9, 10) and delayed myelination of optic nerves (11). The axonal atrophy seen in the sciatic nerves of these animals is associated with decreased neurofilament phosphorylation and spacing (9). Based on these results it has been proposed that MAG plays an important role in maintaining the integrity of myelin and axons and is one of the glial signals that regulates axonal caliber and, in part, myelination.
If complex gangliosides such as GD1a and GT1b are axonal ligands for MAG, animals deficient in these complex gangliosides might be expected to have phenotypic features similar to those of MAG-deficient animals. However, mice engineered to lack GM2/GD2 synthase (UDP-N-acetyl-d-galactosamine:GM3/GD3 N-acetyl-d-galactosaminyltransferase, EC 2.4.1.92), which express no complex gangliosides but, rather, high concentrations of the simpler gangliosides GM3 and GD3, have been reported to develop only subtle neurological defects, restricted largely to decreased central conduction velocity (12). Because conduction velocity depends on both myelination and axonal diameter (13–16), myelination defects, axonal atrophy, or both may underlie the observed decrease. In a prior study animals were examined at the young age of 10 weeks (12) whereas the morphologic abnormalities observed in MAG-deficient animals became more prominent with increasing age (9, 10). To determine whether mice lacking complex gangliosides have morphologic changes similar to MAG deficient animals, we generated an independent strain of GM2/GD2 synthase knockout mice (GalNAcT–/–) and examined their central and peripheral nervous systems (CNS and PNS, respectively). Morphological examination of these animals revealed axonal degeneration in both optic and sciatic nerves. Further, central myelination was decreased (apparently delayed), and there was an increase in the myelination threshold in optic nerves. These data support the hypothesis that complex gangliosides are functionally important to nervous system maintenance and are consistent with the theory that gangliosides are functional ligands for MAG.
MATERIALS AND METHODS
Mice.
Gene targeting and generation of GalNAcT−/− mice (17) was accomplished as shown in Fig. 1A. Clones from a 129/sv mouse genomic library (Stratagene) were isolated by using a mouse GalNAcT cDNA probe (18). A clone containing the 7.8-kb genomic DNA sequence was used to construct the targeting vector. The GalNAcT gene was disrupted by a replacement-type vector in which exon 6, exon 7, and part of exon 8 were deleted and replaced with the MC1NeoPolyA selection cassette (19). The targeting vector was transfected into embryonic stem cells, antibiotic-resistant cells were selected, and chimeric mice were established by using standard techniques (20). Chimeric animals transmitted the GalNAcT mutation to progeny. Mice heterozygous for the disrupted GalNAcT gene were mated, and homozygous mutant progeny were identified by Southern blot and/or PCR analysis of DNA isolated from mouse tails. Except as noted, experiments used 12- to 16-week-old GM2/GD2 synthase knockout (GalNAcT−/−) mice, with age-matched heterozygote (GalNAcT+/−) and wild-type littermates as controls.
Figure 1.

Targeted disruption of the GalNAcT locus and brain glycosphingolipid analysis of offspring. (A) GalNAcT targeting vector (Top), GalNAcT locus (Middle), and predicted homologously recombined locus (Bottom). (B) Biosynthetic pathways for major brain gangliosides. The biosynthetic relationships between major brain gangliosides and their precursors is shown schematically, along with the ganglioside nomenclature of Svennerholm (1). The block in ganglioside biosynthesis because of disruption at the GalNAcT locus is indicated by a double line. GalNAcT–/– mice lack all major brain gangliosides, expressing instead a corresponding increase in GM3 and GD3 (17).
Morphological Analysis and Morphometrics.
Three animals from each group were perfused with 4% paraformaldehyde or 4% paraformaldehyde and 3% glutaraldehyde in Sorenson’s buffer. Lumbosacral spinal roots, dorsal root ganglia and spinal cord, sciatic nerves, and optic nerves were harvested from all animals. Eyes and 3-mm skin punches from foot pads were obtained from paraformaldehyde-only perfused animals to examine retinal ganglion cells and unmyelinated nerve fibers, respectively. Paraformaldehyde-fixed tissues other than skin were embedded in paraffin, stained with hematoxylin and eosin, and examined by light microscopy. Paraformaldehyde–glutaraldehyde-fixed nerves were postfixed in osmium and embedded in Epon. Sections (1-μm thick) were stained with toluidine blue for light microscopy, and 80-nm (thin) sections were examined by electron microscopy. Skin was prepared and stained with PGP 9.5 as an axonal marker as described previously (21) and examined by light microscopy.
Axonal Caliber and Myelination.
The sciatic and optic nerves from three different animals in each group were analyzed at light and ultrastructural levels, respectively. One-micrometer Epon cross-sections of sciatic nerve were sampled and nerve fiber counts were obtained at light level by standard stereological methods as described previously (22–25). The mean axonal caliber of myelinated fibers in the sciatic nerves was calculated from the diameter of a circle with an area equivalent to that of each axon. The groups were compared by the two-tailed Student’s t test. A systematic random-sampling method was used (23, 24) to determine the number and spectrum of mean axonal calibers of myelinated and unmyelinated fibers in the optic nerves from digitized electron microscopic (EM) images of cross-sectionally oriented, nonoverlapping regions at a final magnification of ×18,000. Axonal area and diameter of myelinated and unmyelinated fibers were determined by using a computerized morphometric program (bioquant megx) as described previously (25, 26). Greater than 1,200 myelinated axons were measured in the optic nerves of each group. The number of unmyelinated axons per optic nerve was calculated and compared between the two groups. Statistical analysis was performed by the two-tailed Student’s t test. Myelination threshold was defined as the axonal diameter at which 50% of the total fibers were myelinated. Curves for myelination were generated by plotting the percentage of total unmyelinated axon against axonal diameter for GalNAcT−/− and control animals.
Neurofilament Spacing.
EM images of myelinated axons at a final magnification of ×100,000 were used to determine neurofilament spacing. Nearest-neighbor distance was used as a measure of neurofilament spacing in myelinated axons in optic and sciatic nerves as described previously (9, 25). For statistical analysis, the mean neurofilament nearest-neighbor distance for 15 or more axons from three animals in each group were compared by the two-tailed Student’s t test.
Quantification of Wallerian Degeneration.
For sciatic nerves, the number of degenerating myelinated nerve fibers per total nerve cross section was determined on Epon cross-sections (1 μm). For optic nerves, the number of degenerating fibers per total nerve cross-section was determined on thin sections by electron microscopy. Three animals from each group were analyzed and compared by the two-tailed Student’s t test.
MAG.
Purified myelin was prepared from fresh mouse brains as described (27). The protein content of each preparation was quantified (BCA assay, Pierce), and equal protein amounts from GalNAcT−/− and control animals were subjected to SDS/PAGE (28). Resolved proteins were either stained with Coomassie brilliant blue or transferred to poly(vinylidene difluoride) membranes for immunoblotting with GenS3, a mAb that binds denatured MAG (29, 30). Gels and blots were digitally scanned and band densities were quantified by using scion image software. Some immunoblots were developed by using the enhanced chemiluminescence system (DuPont), radiographic films were exposed for various times, and band density was quantified by laser densitometry.
RESULTS
Targeted Mutation of the GalNAcT Gene.
Inactivation of the GalNAcT gene was demonstrated by the absence of GalNAcT-related transcripts in the GalNAcT−/− mice. As expected from the pathways for ganglioside biosynthesis (Fig. 1B), TLC analysis confirmed a lack of GM2 or more complex gangliosides in brains from these animals and a corresponding increase in the presence of GD3 and GM3 (data not shown, see ref. 17). Although these mice were created by multiexon gene deletion, these mutant animals were grossly normal as has been reported for similar animals generated through neo gene insertion into exon 4 of the same gene (12).
Morphological Analysis of the Nervous System.
Wallerian degeneration of myelinated axons was the predominant pathologic feature in the sciatic nerves of GalNAcT−/− mice (Fig. 2). Features of Wallerian degeneration observed included myelin figures and ovoids representing various stages of myelin degeneration (Fig. 2 A and B), macrophages containing myelin debris (Fig. 2C), bands of Bungner, and regenerating sprouts (Fig. 2D). The extent of axonal degeneration increased in a proximodistal direction with minimal changes in the spinal roots and increasing numbers of degenerating axons in successively more distal sections of the sciatic nerves. Sciatic nerves from control GalNAcT+/− mice infrequently revealed a degenerating fiber, but were otherwise normal. Quantitative analysis (Table 1) revealed a 43-fold increase in axon degeneration in GalNAcT−/− mice. The sciatic nerves of these mice had occasional demyelinated fibers with surrounding Schwann cell processes forming minor onion bulbs (Fig. 2 A–C). Supernumerary Schwann cell processes also were seen surrounding thinly and, occasionally, normally myelinated fibers (Fig. 2 A–C). These features in GalNAc−/− mice are consistent with ongoing demyelination and remyelination. Similar changes were not evident in control animals. The lumbosacral anterior horn cells and dorsal root ganglia, which are the neuronal cell bodies of motor and sensory fibers in sciatic nerves, respectively, were normal in GalNAcT−/− mice (data not shown), a finding that suggests that a primary neuropathic process underlies the Wallerian degeneration seen in the sciatic nerves. The unmyelinated axons were normal in sciatic nerves and epidermis of foot pad skin in both mutant and control animals. Examination of internodal compact myelin showed normal formation of major dense lines, intraperiod lines, and myelin periodicity (i.e., myelin ultrastructure) in both mutant and control animals.
Figure 2.

Pathological features in sciatic nerves of GalNAcT−/− mice. (A) Toluidine blue-stained 1-μm epon section showing several myelinated fibers undergoing axonal degeneration (arrowheads) and a myelinated fiber surrounded by supernumerary Schwann cell processes (arrow). (Bar = 5 μm.) (B) Low-power EM image showing myelin figures and collapsed myelin at different stages of degeneration (arrowheads) and a thinly myelinated fiber surrounded by supernumerary Schwann cell processes (arrow) (Bar = 2.5 μm.) (C) EM image showing a macrophage containing myelin debris (arrow) and a minor onion bulb around a myelinated fiber (arrowheads point to Schwann cell processes). An endoneurial fibroblast also is apparent (∗) (Bar = 1 μm.) (D) EM image showing a thinly myelinated fiber and an axonal sprout (∗) invested by a Schwann cell (Bar = 200 nm). Mice were 12–16 weeks of age at the time nerves were harvested.
Table 1.
Quantitative morphometric analysis of Wallerian degeneration in sciatic and optic nerves of control (GalNAcT+/−) and GM2/GD2 synthase knockout (GalNAcT−/−) mice
| GalNAcT+/− | GalNAcT−/− | Ratio | P178 | |
|---|---|---|---|---|
| Sciatic nerves | ||||
| Axonal caliber of myelinated fibers, μm | 3.07 ± 0.09 | 2.92 ± 0.06 | 0.95 | 0.18 |
| Neurofilament spacing, nm | 42.7 ± 1.7 | 36.8 ± 1.3 | 0.86 | 0.008 |
| Degenerating fibers† | 0.66 ± 1.15 | 28.7 ± 4.6 | 43.5 | 0.013 |
| Optic nerves | ||||
| Axonal caliber of myelinated fibers, μm | 0.76 ± 0.10 | 0.70 ± 0.03 | 0.92 | 0.38 |
| Axonal caliber of unmyelinated fibers, μm | 0.36 ± 0.09 | 0.43 ± 0.17 | 1.19 | 0.02 |
| Unmyelinated axons† | 53 ± 7 | 123 ± 5.3 | 2.32 | 0.0001 |
| Neurofilament spacing, nm | 38.0 ± 0.36 | 37.9 ± 0.41 | 1.00 | 0.98 |
| Degenerating fibers† | 1.7 ± 0.6 | 10.3 ± 1.52 | 6.06 | 0.04 |
Mice were 12–16 weeks of age at the time nerves were harvested. Morphometric values for control and knockout mice are shown as mean ± SD.
Means were compared statistically on a per-animal basis.
Per animal.
A highly significant increase in the number of unmyelinated axons was the most striking abnormality noted in the optic nerves of GalNAcT−/− mice compared with controls (Fig. 3 A and B; Table 1). Some of these unmyelinated axons achieved a caliber comparable to that of large myelinated axons (see quantitative analyses, below). These large-caliber unmyelinated axons (Fig. 3B) were virtually absent in controls. The examination of longitudinal sections confirmed that the unmyelinated profiles seen on cross-sections represented unmyelinated axons. On occasion, segmental myelination defects were observed in a single fiber in longitudinal section. Wallerian degeneration of myelinated axons (retinal ganglion cell axons) also was seen in the optic nerves of GalNAcT−/− mice. Features of Wallerian degeneration observed included empty myelin profiles with axonal loss and collapsed and degenerating myelin (Fig. 3C). The retinal ganglion cells from which these axons originate were normal (data not shown), suggesting a primary neuropathic process underlying the axonal degeneration seen in the optic nerves. Control optic nerves had few degenerating fibers. A quantitative comparison (Table 1) revealed a 6-fold increase in axon degeneration in the knockout mice. Axons surrounded by two concentrically arranged myelin sheaths with cytoplasmic pockets between the myelin sheaths also were observed in the optic nerves of GalNAcT−/− mice, but not in controls (Fig. 3D). Redundant myelin loops, i.e., compact myelin sheaths that coursed away from the axon, were seen more frequently in the knockout animals than in controls. As in the PNS, the ultrastructure of CNS myelin was normal in both groups of animals. There was no significant difference in the width of the periaxonal space in optic nerves of GalNAT−/− mice (14.4 ± 1.2 nm) compared with control (heterozygote) mice (15.4 ± 0.3 nm).
Figure 3.

Pathological features in optic nerves of GalNAcT−/− mice. EM image demonstrating numerous unmyelinated axons (*) of varying caliber (A) (Bar = 1 μm), a pocket of relatively large-caliber unmyelinated axons (B) (∗) (Bar = 1 μm), axonal degeneration and resultant myelin figures (C) (a number of unmyelinated axons also can be seen; Bar = 200 nm), and a doubly myelinated axon (arrow) with cytoplasm between the two compact myelin sheaths (D) (Bar = 200 nm). Mice were 12–16 weeks of age at the time nerves were harvested.
Neurofilament Spacing, Axon Caliber, and Myelination.
Although the mean axonal caliber was not reduced significantly in the sciatic nerves of knockout animals (Fig. 4A), their neurofilament spacing, as determined by nearest neurofilament neighbor distance, was reduced significantly in the myelinated internodal axons of GalNAcT−/− animals compared with controls (Fig. 4B; Table 1). The number of myelinated fibers was similar in both groups of animals. Unmyelinated fibers were not quantified in the sciatic nerves.
Figure 4.

Morphometric analyses of sciatic and optic nerves. (A) Axonal diameters of myelinated fibers in sciatic nerves from GalNAcT+/− (control, gray line) and GalNAcT−/− (black line) mice. (B) Neurofilament nearest-neighbor distances in sciatic nerve axons from GalNAcT−/− (black line) and control (gray line) mice. (C) Histogram of the diameters of unmyelinated axons in optic nerves from GalNAcT−/− (black line) and control (gray line) mice. (D) Myelination threshold (percentage of total fibers remaining unmyelinated for each 0.1-μm increment in axonal diameter) in optic nerves from GalNAcT−/− (black line) and control (gray line) mice. Mice were 12–16 weeks of age at the time nerves were harvested.
In optic nerves, the number of unmyelinated fibers in GalNAcT−/− mice was more than twice that in controls (Fig. 4C). The mean diameter of unmyelinated fibers and the diameter at which 50% of the total fibers were myelinated were increased significantly in knockout animals compared with controls (Fig. 4 C and D; Table 1). The total number of myelinated fibers, axonal caliber of myelinated fibers, and neurofilament spacing in the CNS were not significantly different between the two groups.
MAG Expression.
Immunoblotting revealed a decrease in MAG expression in the brains of GalNAcT−/− mice compared with controls (Fig. 5A), whereas expression of other myelin proteins, detected by Coomassie staining, was not reduced (Fig. 5B). When quantified densitometrically and expressed relative to major myelin proteins, MAG expression was reduced 43% in GalNAcT−/− mice compared with wild-type littermates, whereas MAG expression was increased 10% in GalNAcT+/− mice. In three other sets of mice similarly evaluated, MAG expression was reduced compared with control in every case, with a maximum reduction of 75%. Preliminary Northern blot analyses revealed only modest decreases of MAG mRNA in GalNAcT−/− (−18%) and GalNAcT+/− (−6%) compared with wild-type mice (data not shown).
Figure 5.
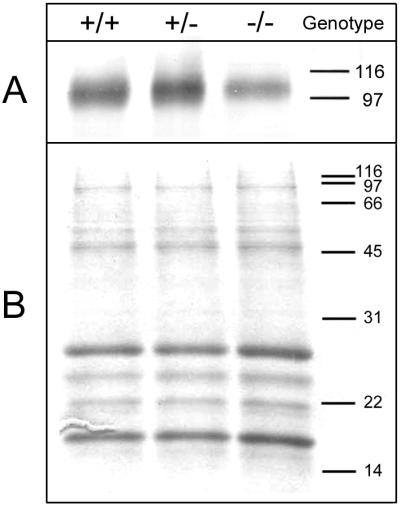
Expression of MAG in GalNAcT−/− and control mice. Equivalent amounts of myelin from wild-type (+/+), GalNAcT+/−, and GalNAcT−/− mice (1.3 μg protein for immunoblotting or 5 μg protein for Coomassie staining) were subjected to SDS/PAGE (28). The positions of molecular mass standards are indicated in kDa. (A) Immunodetection of MAG. After resolution on a 10% polyacrylamide gel, proteins were transferred to a poly(vinylidene difluoride) membrane by using a semidry transfer apparatus. MAG, which migrates at 100 kDa, was detected by incubation of the blot with GenS3 mAb followed by an alkaline phosphatase-conjugated secondary antibody. Antibody binding was detected by development with nitro blue tetrazolium and 5-bromo-4-chloro-3-indolyl phosphate. (B) Staining of major myelin proteins. After resolution on an 8–16% polyacrylamide gradient gel, proteins were detected by using Coomassie brilliant blue stain. The major myelin proteins myelin basic protein (MBP) and proteolipid protein (PLP) are indicated. Mice were 15–23 weeks of age when brain tissue was collected for analysis.
DISCUSSION
Gangliosides, a complex family of sialylated glycosphingolipids, are highly enriched in the mammalian nervous system, where their expression is developmentally regulated and cell type-specific (reviewed in ref. 31). As major determinants at the nerve cell surface (32, 33), they have been proposed to play important roles in neural cell–cell recognition, development, and function. These proposals were challenged by a recent study in which mice were engineered (via GalNAcT knockout) to lack complex gangliosides (including all of the major adult brain gangliosides) and, instead, express a comparable amount of the simpler gangliosides, GD3 and GM3 (12). Despite this dramatic biochemical change, the knockout mice were reported to undergo normal brain development, differentiation, morphogenesis, histogenesis (including myelin), and synaptogenesis. The noted reduced central conduction velocity, as determined by somatosensory evoked responses, led the authors to postulate that complex gangliosides have subtle effects on normal neuronal function but no role in normal brain morphogenesis or organogenesis (12).
The present study shows that mice engineered to lack complex gangliosides have morphological abnormalities in both the CNS and PNS. Whereas complex gangliosides may not affect the gross development of the nervous system, their absence results in axonal degeneration in the CNS and PNS, decreased myelination in the CNS, and demyelination in peripheral nerves. The mice examined in the current study were relatively young (12–16 weeks old). We do not know whether the neurodegenerative processes we noted are ongoing, accelerating, or diminishing as the mice age. Longitudinal studies are underway to address this important question. The presence of CNS dysmyelination in this study provides a potential explanation for the reduced central conduction velocity reported in similar animals in a prior study (12). Limited examination of sciatic nerves from the previously published GalNAcT−/− mouse model (kindly provided by Koichi Furukawa, Nagoya University, Nagoya, Japan) indicated the same neuropathologies as those reported here (data not shown), precluding strain differences or artifacts of genetic engineering as the basis for the documented changes.
Complex gangliosides play a role in the initiation of myelination in the CNS, as indicated by a >2-fold increase in the number of unmyelinated fibers, a significant increase in the mean diameter of unmyelinated axons, and an increase in myelination threshold in the optic nerves of GalNAcT−/− mice compared with heterozygote controls. Further evidence that myelination of the CNS is dysregulated in these animals is provided by the presence of doubly myelinated axons and redundant myelin loops, features that also were observed in the CNS of mice lacking galactocerebrosides and sulfatides, the major glycosphingolipids of oligodendrocytes (34), and in mice lacking MAG (see below).
In contrast to the CNS, GalNAcT−/− mice have no apparent hallmarks of delayed PNS myelination at this developmental stage, but display ongoing demyelination and remyelination, as suggested by demyelinated and thinly myelinated fibers and the formation of onion bulbs. These observations suggest that in the PNS, complex gangliosides are necessary to maintain the integrity of myelin sheaths. Although the reason for the appearance of different forms of dysmyelination in the CNS vs. the PNS is unclear, our observations reflect similar dysmyelination phenotypes in MAG−/− mice (10, 11).
The ongoing Wallerian degeneration evident in both the central and peripheral nerves of GalNAcT−/− mice indicates that complex gangliosides are important in maintaining the integrity of axons (neuronal processes). An alternate explanation is that storage of excess, overexpressed simple gangliosides (GD3 and GM3) in GalNAcT−/− mice causes neuronal degeneration and, hence, axonal degeneration. This is not the case, because the neuronal cell bodies of optic and sciatic nerve axons (retinal ganglion cells, lumbosacral anterior horn cells, and dorsal root ganglia) have normal morphology, and no inclusions were seen. Therefore, a primary neuropathic (axonopathic) process underlies the observed nerve degeneration. Whether this axonopathic process is ongoing and whether it will result, eventually, in a significant depletion of the axon pool remain to be determined.
The extent of axonal degeneration in peripheral nerves increased in a proximodistal direction with minimal changes in spinal roots and maximal axonal degeneration in distal sciatic nerves. Although the basis for the observed differential susceptibility of distal axons (neuronal processes) compared with proximal axons and perikarya is not known, similar observations were made in animals genetically engineered to lack P0 expression (R. Martini, Wurzburg, Germany; personal communication). In both models, although the targeted mutation results in lack of expression of gene product at all levels of the neuraxis, the distal axons are more susceptible to the deleterious effects of the mutation.
It has been proposed that MAG modulates the axonal caliber of peripheral myelinated fibers by affecting neurofilament spacing via neurofilament phosphorylation: mice with a disrupted MAG gene have decreased axonal calibers that correlate with reduced neurofilament spacing and phosphorylation (9). As with MAG−/− mice, GalNAcT−/− mice also had decreased neurofilament spacing in the sciatic nerves compared with controls. The lack of a clear relationship between reduced neurofilament spacing and the axon caliber in sciatic nerves of GalNAcT−/− mice may reflect the younger age at the time of tissue collection compared with MAG−/− mice. A longitudinal study will be required to clarify this issue.
This study supports the hypothesis that complex gangliosides are functional ligands for MAG. Prior in vitro studies reported that, among the common brain gangliosides, GD1a and GT1b can serve as complementary ligands for MAG (5, 6). Our recent immunocytochemical studies using tetanus toxin C binding suggest that GT1b is expressed on the axolemma of peripheral myelinated fibers (ref. 35; similar studies on GD1a have not been done). It is attractive to postulate that GT1b and GD1a serve as axolemmal ligands for MAG, which is present on the apposing adaxonal glial plasmalemma. This hypothesis is supported by the striking similarity of the pathological features seen in the optic and sciatic nerves of GalNAcT−/− mice compared with those in MAG−/− mice. As with GalNAcT−/− mice, in the optic nerves of MAG−/− mice there is a significant increase in unmyelinated fibers, doubly myelinated fibers, and redundant myelin loops (11, 36–39) and the peripheral nerves of these animals show axonal degeneration, demyelination and remyelination, axonal sprouting, decreased neurofilament spacing, and axonal atrophy (9, 10). It is also notable that MAG expression in GalNAcT−/− myelin is selectively reduced. The neuropathologies documented in GalNAcT−/− mice are not likely to be secondary to this reduction in MAG expression, however, because the reduction is modest (≈50%) and mice heterozygous for the MAG allele were reported to have similarly reduced MAG expression, yet lack any of the neuropathological features of MAG−/− or GalNAcT−/− mice (39, 40). It is feasible that MAG is dysregulated downstream of depletion of its complex ganglioside ligands and that the neuropathological features in GalNAcT−/− mice are related to the absence of MAG’s response rather than the reduction of MAG expression. Because only a modest reduction in MAG mRNA expression was observed in preliminary experiments, the decrease in MAG expression in GalNAcT−/− mice may be a result of accelerated degradation, a hypothesis that has yet to be tested. Further evaluation of the relationship between the physiological roles of MAG and complex gangliosides is warranted in light of these observations.
Acknowledgments
We thank Ms. Susan Fromholt for assistance in breeding and genotyping mutant mice, Dr. Norman Latov for providing mAb Gen-S3, and Ms. Jenn Reed for assistance in figure design. This work was supported, in part, by grants from the Paralyzed Veterans of America Spinal Cord Research Foundation, the National Multiple Sclerosis Society, the National Science Foundation (IBN-9631745), and the National Institutes of Health (NS37096).
ABBREVIATIONS
- MAG
myelin-associated glycoprotein
- CNS
central nervous system
- PNS
peripheral nervous system
- EM
electron microscopic
Note Added in Proof
In collaboration with S. Chiavegatto (Departments of Neurology and Psychology) and R. J. Nelson (Departments of Psychology and Neuroscience), The Johns Hopkins University, we have determined that, compared with control littermates, GalNAcT−/− mice display significant deficits in motor behavior at the age of 7–9 months, including (i) marked loss of hindlimb reflex extension (visual scoring of hindlimb position upon suspension by the tail, P < 0.05); (ii) reduced latency to fall from a balance beam (50% reduction, P < 0.001); and (iii) reduced latency to fall from a rotarod apparatus (52% reduction, P < 0.001). Compared with controls, knockout animals tend to walk in small labored movements. These deficits are consistent with axon degeneration and/or dysmyelination.
Footnotes
Ganglioside nomenclature is that of Svennerholm (1).
References
- 1.Svennerholm L. Prog Brain Res. 1994;101:xi–xiv. doi: 10.1016/S0079-6123(08)61935-4. [DOI] [PubMed] [Google Scholar]
- 2.Yu R K, Saito M. In: Neurobiology of Glycoconjugates. Margolis R U, Margolis R K, editors. New York: Plenum; 1989. pp. 1–42. [Google Scholar]
- 3.Tettamanti G, Bonali F, Marchesini S, Zambotti V. Biochim Biophys Acta. 1973;296:160–170. doi: 10.1016/0005-2760(73)90055-6. [DOI] [PubMed] [Google Scholar]
- 4.van Echten G, Sandhoff K. J Biol Chem. 1993;268:5341–5344. [PubMed] [Google Scholar]
- 5.Yang L J S, Zeller C B, Shaper N L, Kiso M, Hasegawa A, Shapiro R E, Schnaar R L. Proc Natl Acad Sci USA. 1996;93:814–818. doi: 10.1073/pnas.93.2.814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Collins B E, Yang L J S, Mukhopadhyay G, Filbin M T, Kiso M, Hasegawa A, Schnaar R L. J Biol Chem. 1997;272:1248–1255. doi: 10.1074/jbc.272.2.1248. [DOI] [PubMed] [Google Scholar]
- 7.Trapp B D, Andrews S B, Cootauco C, Quarles R. J Cell Biol. 1989;109:2417–2426. doi: 10.1083/jcb.109.5.2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Trapp B D. Ann N Y Acad Sci. 1990;605:29–43. doi: 10.1111/j.1749-6632.1990.tb42378.x. [DOI] [PubMed] [Google Scholar]
- 9.Yin X, Crawford T O, Griffin J W, Tu P, Lee V Y, Li C, Roder J, Trapp B D. J Neurosci. 1998;18:1953–1962. doi: 10.1523/JNEUROSCI.18-06-01953.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fruttiger M, Montag D, Schachner M, Martini R. Eur J Neurosci. 1995;7:511–515. doi: 10.1111/j.1460-9568.1995.tb00347.x. [DOI] [PubMed] [Google Scholar]
- 11.Bartsch S, Montag D, Schachner M, Bartsch U. Brain Res. 1997;762:231–234. doi: 10.1016/s0006-8993(97)00484-8. [DOI] [PubMed] [Google Scholar]
- 12.Takamiya K, Yamamoto A, Furukawa K, Yamashiro S, Shin M, Okada M, Fukumoto S, Haraguchi M, Takeda N, Fujimura K, et al. Proc Natl Acad Sci USA. 1996;93:10662–10667. doi: 10.1073/pnas.93.20.10662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rushton W A H. J Physiol. 1951;115:101–122. doi: 10.1113/jphysiol.1951.sp004655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Waxman S G, Bennett M V. Nat New Biol. 1972;238:217–219. doi: 10.1038/newbio238217a0. [DOI] [PubMed] [Google Scholar]
- 15.Ritchie J M. Proc R Soc London B. 1982;217:29–35. doi: 10.1098/rspb.1982.0092. [DOI] [PubMed] [Google Scholar]
- 16.Hildebrand C, Remahl S, Persson H, Bjartmar C. Prog Neurobiol. 1993;40:319–384. doi: 10.1016/0301-0082(93)90015-k. [DOI] [PubMed] [Google Scholar]
- 17.Liu Y, Wada R, Kawai H, Sango K, Deng C, Tai T, McDonald M P, Araujo K, Crawley J N, Bierfreund U, et al. J Clin Invest. 1999;103:497–505. doi: 10.1172/JCI5542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sango K, Johnson O N, Kozak C A, Proia R L. Genomics. 1995;27:362–365. doi: 10.1006/geno.1995.1058. [DOI] [PubMed] [Google Scholar]
- 19.Capecchi M R. Science. 1989;244:1288–1292. doi: 10.1126/science.2660260. [DOI] [PubMed] [Google Scholar]
- 20.Deng C, Wynshaw-Boris A, Zhou F, Kuo A, Leder P. Cell. 1996;84:911–921. doi: 10.1016/s0092-8674(00)81069-7. [DOI] [PubMed] [Google Scholar]
- 21.Hsieh S T, Choi S, Lin W M, Chang Y C, McArthur J C, Griffin J W. J Neurocytol. 1996;25:513–524. doi: 10.1007/BF02284819. [DOI] [PubMed] [Google Scholar]
- 22.Mayhew T M, Sharma A K. J Anat. 1984;139:59–66. [PMC free article] [PubMed] [Google Scholar]
- 23.Mayhew T M, Sharma A K. J Anat. 1984;139:45–58. [PMC free article] [PubMed] [Google Scholar]
- 24.Mayhew T M. J Anat. 1988;157:127–134. [PMC free article] [PubMed] [Google Scholar]
- 25.Xu Z, Marszalek J R, Lee M K, Wong P C, Folmer J, Crawford T O, Hsieh S T, Griffin J W, Cleveland D W. J Cell Biol. 1996;133:1061–1069. doi: 10.1083/jcb.133.5.1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sanchez I, Hassinger L, Paskevich P A, Shine H D, Nixon R A. J Neurosci. 1996;16:5095–5105. doi: 10.1523/JNEUROSCI.16-16-05095.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Norton W T, Poduslo S E. J Neurochem. 1973;21:749–757. doi: 10.1111/j.1471-4159.1973.tb07519.x. [DOI] [PubMed] [Google Scholar]
- 28.Laemmli U K. Nature (London) 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 29.Nobile-Orazio E, Hays A P, Latov N, Perman G, Golier J, Shy M E, Freddo L. Neurology. 1984;34:1336–1342. doi: 10.1212/wnl.34.10.1336. [DOI] [PubMed] [Google Scholar]
- 30.Tropak M B, Roder J C. J Neurochem. 1994;62:854–862. doi: 10.1046/j.1471-4159.1994.62030854.x. [DOI] [PubMed] [Google Scholar]
- 31.Schnaar R L. Glycobiology. 1991;1:477–485. doi: 10.1093/glycob/1.5.477. [DOI] [PubMed] [Google Scholar]
- 32.Hansson H A, Holmgren J, Svennerholm L. Proc Natl Acad Sci USA. 1977;74:3782–3786. doi: 10.1073/pnas.74.9.3782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Seybold V, Rosner H, Greis C, Beck E, Rahmann H. J Neurochem. 1989;52:1958–1961. doi: 10.1111/j.1471-4159.1989.tb07286.x. [DOI] [PubMed] [Google Scholar]
- 34.Bosio A, Bussow H, Adam J, Stoffel W. Cell Tissue Res. 1998;292:199–210. doi: 10.1007/s004410051051. [DOI] [PubMed] [Google Scholar]
- 35.Sheikh K A, Deerinck T J, Ellisman M H, Griffin J W. Brain. 1999;122:449–460. doi: 10.1093/brain/122.3.449. [DOI] [PubMed] [Google Scholar]
- 36.Li C, Trapp B, Ludwin S, Peterson A, Roder J. J Neurosci Res. 1998;51:210–217. doi: 10.1002/(SICI)1097-4547(19980115)51:2<210::AID-JNR9>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 37.Bartsch U, Montag D, Bartsch S, Schachner M. Glia. 1995;14:115–122. doi: 10.1002/glia.440140206. [DOI] [PubMed] [Google Scholar]
- 38.Bartsch U. J Neurocytol. 1996;25:303–313. doi: 10.1007/BF02284804. [DOI] [PubMed] [Google Scholar]
- 39.Fujita N, Kemper A, Dupree J, Nakayasu H, Bartsch U, Schachner M, Maeda N, Suzuki K, Popko B. J Neurosci. 1998;18:1970–1978. doi: 10.1523/JNEUROSCI.18-06-01970.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Montag D, Giese K P, Bartsch U, Martini R, Lang Y, Bluthmann H, Karthingasan J, Kirschner D A, Wintergerst E S, Nave K-A, et al. Neuron. 1994;13:229–246. doi: 10.1016/0896-6273(94)90472-3. [DOI] [PubMed] [Google Scholar]