

Abstract
Helicobacter pylori infection is associated with gastric epithelial damage, including apoptosis, ulceration, and cancer. Although bacterial factors and the host response are believed to contribute to gastric disease, no receptor has been identified that explains how the bacteria attach and signal the host cell to undergo apoptosis. Using H. pylori as “bait” to capture receptor proteins in solubilized membranes of gastric epithelial cells, class II major histocompatibility complex (MHC) molecules were identified as a possible receptor. Signaling through class II MHC molecules leading to the induction of apoptosis was confirmed using cross-linking IgM antibodies to surface class II MHC molecules. Moreover, binding of H. pylori and the induction of apoptosis were inhibited by antibodies recognizing class II MHC. Since type 1 T helper cells are present during infection and produce interferon (IFN)-γ, which increases class II MHC expression, gastric epithelial cell lines were exposed to H. pylori in the presence or absence of IFN-γ. IFN-γ increased the attachment of the bacteria as well as the induction of apoptosis in gastric epithelial cells. In contrast to MHC II–negative cell lines, H. pylori induced apoptosis in cells expressing class II MHC molecules constitutively or after gene transfection. These data describe a novel receptor for H. pylori and provide a mechanism by which bacteria and the host response interact in the pathogenesis of gastric epithelial cell damage.
Keywords: gastric, epithelium, apoptosis, Helicobacter pylori, T helper cell type 1
H elicobacter pylori is a gram-negative bacterium that colonizes human gastric epithelium and is strongly associated with peptic ulceration, gastric lymphoma, and gastric carcinoma (1–3). Several studies have suggested that specific virulence factors expressed by certain strains are more likely to cause disease. Strains of H. pylori expressing the cagA and vacA genes have been reported to have an association with gastric disease (4–6), including peptic ulcers (4, 7) and gastric cancer (3). More recently, cagA has been shown to mark a “pathogenicity island” of genes (8), which may encode virulence factors that contribute to the more severe gastric diseases associated with H. pylori infection. However, the association between CagA and disease or gastric inflammation has not been observed in all studies (9), indicating that other environmental or host factors, including the gastric immune and inflammatory responses, also contribute to the pathogenesis of gastroduodenal disease associated with H. pylori infection.
Since infection with H. pylori persists for life, the host immune and inflammatory responses to natural infection appear to provide little protection. Moreover, the host response has been implicated even in the pathogenesis of gastric disease associated with H. pylori. This notion was supported by studies reporting that patients infected with strains expressing cagA, which may increase the risk of disease, produce increased levels of inflammatory cytokines (5, 10). In addition, autoantibodies are induced after infection and contribute to gastritis (11, 12). Furthermore, increased numbers of IFN-γ–producing Th1 cells have been shown to be present during infection (13, 14). As H. pylori bacteria are noninvasive and remain in the gastric lumen, a persistent local Th1 response may inflict considerable tissue damage via cell-mediated immune responses. Several changes that have been observed in gastric epithelial cells, including an increase in class II MHC expression (15–17) and IL-8 production (18), could be mediated in part by the cytokines produced by Th1 cells. The increase in apoptosis reported in gastric epithelial cells during infection with H. pylori (19–22) could similarly be attributed to cellular immune responses mediated by Th1 cells (23).
Class II MHC molecules are best known for their ability to bind and present antigens to CD4+ T cells. However, engagement of class II MHC by T cells also has functional consequences for the antigen-presenting cell. For example, cross-linking class II MHC molecules with molecules such as microbial superantigens has been reported to induce apoptosis (24, 25). Thus, the attachment of H. pylori to class II MHC molecules could explain the induction of apoptosis in gastric epithelial cells.
Although infection with H. pylori is associated with significant epithelial cell damage, including increased levels of apoptosis, it is not clear how the bacteria bind to the host cells and trigger the deleterious responses. Several receptors for H. pylori have been described (26, 27), but no specific molecule has yet been shown to bind the organism or signal any changes in gastric epithelial cells. In this report, we used a strategy whereby H. pylori was used as “bait” to capture epithelial cell proteins that bind the bacteria. Class II MHC molecules were shown to be capable of binding H. pylori and necessary for the induction of apoptosis in gastric epithelial cell lines. The effect of IFN-γ on the attachment and induction of apoptosis provides additional insight into the interactions between the bacteria and host response that have implications for bacterial colonization and the pathogenesis of epithelial cell damage.
Materials and Methods
Bacterial Culture
H. pylori, strain LC-11 (28), was grown at 37°C as described previously (28). A nongastric pathogen, Campylobacter jejuni, originally obtained from the American Type Culture Collection (33291; Rockville, MD), was provided by the Microbiology Laboratories in the Department of Pathology at University of Texas Medical Branch. Both species of bacteria were grown on blood agar base (Becton Dickinson, San Jose, CA) at 37°C under microaerobic conditions and harvested on day 3 into PBS. After centrifugation at 2,500 g for 15 min, bacteria were resuspended in sterile PBS (pH 7.4) to a concentration of 2 × 108 bacteria/ml. The estimation of bacterial number was determined by measuring the absorbance (at 530 nm) using a spectrophotometer (DU-65; Beckman Instruments, Inc., Fullerton, CA), and comparing the value to a standard curve generated by quantifying viable organisms from aliquots of bacteria at varying concentrations that were also assessed for absorbance. The motility of both organisms was confirmed by phase–contrast microscopy before experimental use. In some experiments, H. pylori or C. jejuni were killed by heat (5 min at 100°C) before use.
Cell Lines and Hybridomas
The Kato III, NCI N87, and AGS gastric epithelial cell lines were obtained from the American Type Culture Collection and maintained in RPMI 1640 medium supplemented with 10% FCS. Additional cell lines differing in their expression of class II MHC molecules were also used: COS-1 cells (provided by Dr. M. Falzon, Department of Pharmacology and Toxicology, University of Texas Medical Branch), and class II–expressing cells, including Jesthom B cells (29) and COS-1 cells transfected with the gene encoding DR1 (ID12; provided by Dr. M. Xu, Antigen Express, Inc., Worcester, MA) (29). To compare the effect of different isoforms encoded by genes within the subregions of the class II MHC region, the binding of H. pylori and the induction of apoptosis were compared in bare lymphocyte syndrome–derived cells (BLS-1) before or after transfection (30). These cells included BLS-DR4 (BLS-1 transfected with the gene encoding DR4) and BLS-DQ2 (transfected with the gene encoding DQ2). The BLS cell lines were provided by Dr. Gerald Nepom (Virginia Mason Research Center, Seattle, WA) and maintained in RPMI 1640–Hepes (20 mM) medium supplemented with 10% FCS with 1 mM sodium pyruvate. The hybridomas HB55, HB145, and HB180 (American Type Culture Collection) were used as the source of anti–human class II MHC antibodies (L243, IVA12, and 9.3F10, respectively). Antibodies were purified from mouse ascites fluid over a protein G–Sepharose column.
Stimulation of Gastric Epithelial Cell Lines
To study the induction of apoptosis, we determined an optimal bacteria to epithelial cell ratio and concentration of IFN-γ for the stimulation of epithelial cells. Subsequently, cells were exposed to H. pylori (∼300 bacteria/epithelial cell) or IFN-γ (100 U/ml; R&D Systems, Minneapolis, MN) either alone or in combination. After incubation for varying lengths of time, the cells were harvested, and cell recovery was assessed by counting the cells and evaluating their viability by trypan blue exclusion. Cells were evaluated for the presence of DNA fragmentation as described below. In some experiments, gastric epithelial cells were exposed to IgM anti-class II MHC (RFD1; Serotec, Inc., Raleigh, NC) at 10 μg/ml for 72 h or an IgM isotype control. To block the induction of apoptosis by H. pylori without cross-linking, Fab fragments were prepared from the IVA12 antibody using standard techniques.
Detection of DNA Fragmentation
ELISA.
DNA fragmentation was measured using a commercially available ELISA assay (Boehringer Mannheim Biochemicals, Indianapolis, IN). This assay detects small molecular weight nucleosome fragments in the cytoplasmic fractions of affected cells that arise during apoptosis but not as a result of necrosis. The absorbance was measured at 405 nm by Multiskan® (model MCC/ 340; Titertek Instruments, Inc., Irvine, CA) and compared with substrate solution as a blank. The apoptotic index was calculated according to the manufacturer's instructions by dividing the absorbance of stimulated cells by the absorbance for control cells.
Gel Electrophoresis.
Agarose gel electrophoresis to detect DNA fragmentation was conducted according to a standard procedure for assaying fragmentation in total genomic DNA (31). In brief, cells were harvested from different culture conditions and pelleted by centrifugation at 400 g for 5 min. Subsequently, 106 epithelial cells were washed with PBS, pelleted, and resuspended in sample buffer comprised of Tris-buffered glycerol with 1.4 mg/ ml ZnSO4, bromphenol blue, and RNase (Sigma Chemical Co., St. Louis, MO) at 10 mg/ml. Nucleosomal ladders were visualized in 2% agarose gels in TBE buffer (0.09 M Tris, 0.09 mM boric acid, 0.24 M EDTA, pH 8.0). Cells were lysed by 2% SDS and 53 μg/ml of proteinase K (Sigma Chemical Co.) contained within a 1% agarose gel slice above the wells. The gel was run for 10–14 h at 30 V, stained with 2 μg/ml ethidium bromide for 1 h, and washed overnight with a large volume of distilled water to remove excess stain. Gels were illuminated with UV light to visualize DNA fragments that were photographed and their migration compared with the fX174 HaeIII standard (Promega Corp., Madison, WI).
Binding of H. pylori to Gastric Epithelial Cells
Binding to Solubilized Epithelial Membrane Proteins.
Solubilized membrane proteins were prepared as described elsewhere (29). In brief, Kato III cells were incubated in methionine and cysteine– free RPMI 1640 medium and then metabolically radiolabeled with [35S]methionine and cysteine (0.5 mCi/108 cells). Cells were then washed and lysed in ice-cold 10 mM Tris-HCl buffer, pH 8.1, containing protease inhibitors (2 mM PMSF and 1.0 mM N-ethyl maleimide) for 30 min on ice. Nuclei were removed by centrifugation at 500 g for 10 min. This process was repeated once to ensure all cells were lysed. Microsomal membranes were obtained from the supernatants by ultracentrifugation at 100,000 g for 45 min. The membrane pellet was solubilized in the lysis buffer containing 0.5% Triton X-100, followed by a second centrifugation at 100,000 g for 45 min to remove the unsolubilized material. Membranes were precleared by incubation with normal rabbit serum, followed by three passages over Staphylococcus aureus beads and one passage over protein A–Sepharose beads (Sigma Chemical Co.). H. pylori was then incubated with Kato III cell membrane proteins or affinity-purified class II MHC (32) for 2 h at 4°C in the presence of the protease inhibitor PMSF (2 mM). After incubation, the bacteria were washed with PBS five times at 17,000 rpm for 10 min until the cpm of the wash reached background levels. The protein bound to the bacteria was eluted with either 0.1 M citrate (Sigma Chemical Co.) buffer, pH 3.0, or 0.1 M glycine (Sigma Chemical Co.) buffer, pH 2.2. The eluted proteins were run on 12% SDS-PAGE and analyzed by autoradiography.
Binding to Epithelial Cell Surface.
Binding of H. pylori to the surface of epithelial cells was evaluated by flow cytometry using a modification of a technique described elsewhere (33). In brief, gastric epithelial cell lines were cultured in the presence or absence of IFN-γ (100 U/ml) for 48 h and then washed twice with PBS containing sodium azide (0.02%). The cells were then incubated for 30 min on ice with a cocktail of the anti–MHC II antibodies IVA12, L243, and 9.3F10 or an isotype control. Cells were subsequently incubated with H. pylori for an additional 30 min in ice. After washing with PBS, bound bacteria were stained with human anti–H. pylori serum or isotype controls followed by FITC-conjugated rabbit anti–human IgG. In some experiments, class II MHC expression was measured simultaneously with binding of H. pylori, bacteria were harvested from broth culture, washed with PBS, and labeled with the PKH26 Red Fluorescent Cell Linker kit according to the manufacturer's instructions (Sigma Chemical Co.). Subsequently, both the bacterial and eucaryotic cells were incubated separately with isotype control IgG to prevent nonspecific binding. Cells were then incubated with anti–class II MHC antibody (IVA12) or an isotype control for 45 min on ice, followed by FITC-conjugated goat anti–mouse IgG, then were incubated with fluorochrome-labeled bacteria for 10 min at 37°C, washed with PBS three times, and fixed with 2% paraformaldehyde. Finally, the cells were resuspended and analyzed by flow cytometry (FACScan®; Becton Dickinson) (34).
Evaluation of Class II MHC Antigen Expression by Flow Cytometry
To evaluate the expression of class II MHC, cells treated with 100 U/ml of IFN-γ or media alone were grown as described above, and 106 cells were harvested, washed, and stained with an optimal amount of mouse anti–human class II MHC (1 μg, clone L243) or an appropriate isotype control. Subsequently, cells were washed with PBS with 0.1% BSA and incubated for 30 min at 4°C with 10 μl FITC-conjugated anti–mouse IgG1 (clone ZAP1; Chemicon International Inc., Temecula, CA). After incubation, cells were washed and fixed in 1% paraformaldehyde in PBS, and specific fluorescence was measured using a FACScan® after correcting for nonspecific fluorescence using the appropriate isotype controls.
Studies on Gastric Epithelium from Freshly Isolated Tissue
Class II MHC expression was also assessed in gastric tissue by immunohistochemistry. Biopsy specimens of the gastric antrum were obtained from consenting patients undergoing esophagogastroduodenoscopy for various clinical indications using a protocol approved by the Institutional Review Board at the University of Texas Medical Branch. Subjects were considered infected if H. pylori was detected in gastric biopsy samples by a rapid urease test or by histopathology. At least two biopsies were collected, fixed in buffered formalin, cut, and processed by the immunohistochemistry service in the Department of Pathology at University of Texas Medical Branch. Subsequently, slides were put through an automated staining process in which they were labeled with mAbs recognizing human class II MHC molecules or an appropriate isotype control (Ventana Medical Systems, Tucson, AZ) as described elsewhere (34). Slides were then counterstained with hematoxylin and eosin.
In other studies, gastric epithelial cells were partially enriched using a modification of previously described techniques (34, 35). In brief, biopsy specimens from subjects infected with H. pylori were collected into sterile collection medium (calcium and magnesium–free HBSS with 5% FCS and penicillin plus streptomycin). Biopsy specimens were rinsed with HBSS medium containing 1 mM dithiothreitol and 1 mM ethylenediamine tetraacetic acid (Sigma Chemical Co.). The specimens were agitated for 1 h at 37°C to obtain epithelial cells. The resulting cell suspensions were washed, and the viability of the gastric epithelial cells was determined by trypan blue exclusion. Cells were not used if viability did not exceed 80%. The yield from a single subject varied from 0.5–1 × 106, which precluded further purification by density gradients. To determine the purity, freshly isolated cells were stained for epithelial specific antigen using an FITC-conjugated mAb (clone Ber-EP4; Dakopatts A/S, Glostrup, Denmark) as described previously (34). Purity ranged from 50 to 80%, epithelial lymphocytes being the major contaminating cell (35). Cells stained by the epithelial cell antigen were selected by electronic gating, and this population was examined by flow cytometry for the expression of class II MHC and the binding of H. pylori as described above.
Statistical Analyses
Results are expressed as the mean ± SEM. The binding of H. pylori as well as the induction of apoptosis were compared using a two-tailed Student's t test and considered significant if P values were <0.05. Correlation of the binding of H. pylori and class II MHC expression was evaluated by multiple regression analysis.
Results
Induction of Apoptosis in Gastric Epithelial Cells Is Mediated through Class II MHC.
To determine whether MHC II on the surface of human gastric epithelial cells could trigger apoptotic signals, Kato III cells were exposed to anti–human class II MHC (IgM, RFD1) antibody. As shown in a representative experiment (Fig. 1), antibodies to class II MHC induced DNA degradation, whereas the isotype control did not. Cross-linking with anti–MHC II mAb also resulted in decreased cell viability (data not shown).
Figure 1.
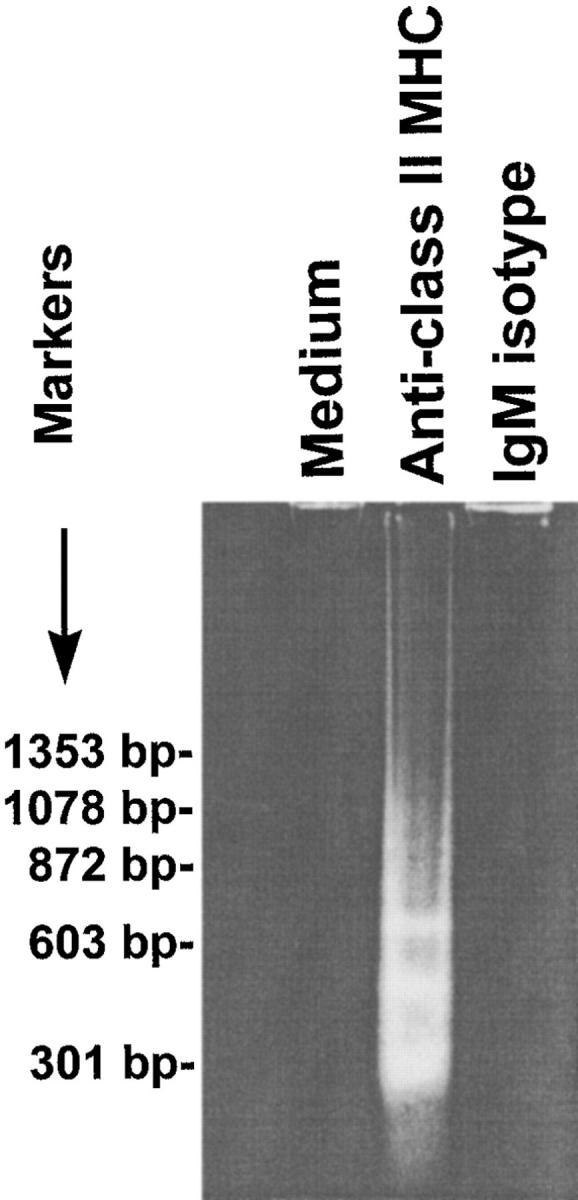

Induction of gastric epithelial cell apoptosis by cross-linking with anti–class II MHC. DNA degradation in Kato III cells grown in RPMI 1640 media alone or with anti–class II MHC (RFD1) at 10 μg/ml for 72 h was confirmed by low molecular weight DNA ladders resolved on a 2% agarose gel (left). The treatment conditions were as indicated above the figure. Data presented are representative of one of four separate experiments. In another series of experiments, the level of apoptosis in Kato III cells exposed to H. pylori (Hp) was measured by ELISA and compared with cells stimulated with H. pylori in the presence of varying concentrations of Fab fragments recognizing class II MHC. The Fab fragments decreased the induction of apoptosis relative to the positive control in a concentration-dependent manner (right).
To determine if the interaction between H. pylori and class II MHC was directly responsible for apoptosis, Kato III cells were stimulated with H. pylori either in the presence or absence of noncross-linking Fab fragments generated from antibodies (IVA12) recognizing various class II MHC isoforms (DR, DP, and DQ). Whereas H. pylori stimulated significant apoptosis of Kato III cells, the induction of cell death was blocked by noncross-linking Fab fragments recognizing class II MHC (Fig. 1).
Binding of Purified Class II MHC Molecules by H. pylori.
To confirm that class II MHC proteins may be candidates for a receptor for H. pylori, freshly grown bacteria were used as bait to bind solubilized membrane proteins from a gastric epithelial cell line (Kato III). H. pylori bacteria were incubated with metabolically labeled Kato III cell membrane proteins and washed, and bacteria-associated Kato III proteins were eluted and separated by gel electrophoresis. Solubilized membrane proteins from gastric epithelial cell lines that bound to H. pylori were separated by SDS-PAGE and yielded bands that coincided in size with MHC II α and β chains (data not shown). To examine whether H. pylori binds selectively to MHC II molecules from gastric epithelial cells, we incubated H. pylori with metabolically labeled, affinity-purified MHC II complexes from a gastric epithelial cell line (Kato III) and a B cell line. Gel electrophoresis analysis of the eluted material confirmed the presence of MHC II α and β chains among the proteins bound to H. pylori and not to a related bacteria, C. jejuni (Fig. 2).
Figure 2.

H. pylori binds to MHC II. H. pylori or C. jejuni were incubated with affinity-purified [35S]methionine and cysteine–labeled class II MHC (32) purified from Kato III gastric epithelial cells or Jesthom B cells as described in Materials and Methods. The bound MHC II molecules were eluted before bacteria-associated proteins were run on a 12% SDS-PAGE gel and detected by autoradiography. MWS, Molecular weight standards. α and β, α and β chains of class II. Class II, Immunoprecipitated class II MHC. Hp, Purified class II MHC eluted from H. pylori. Cj, Purified class II MHC adherent to C. jejuni.
Class II MHC Expression Is Associated with Binding of H. pylori to Gastric Epithelial Cells.
Since H. pylori binds purified class II MHC, we postulated that the regulation of surface class II MHC expression would affect binding of the bacteria to gastric epithelial cells. To pursue this possibility, we used three gastric epithelial cell lines, two of which express class II MHC (Kato III and N87) and one, a variant of the AGS line, that does not. Class II MHC expression was increased significantly in response to treatment of the Kato III and N87 cells with IFN-γ, whereas AGS cells continued to express very little (Fig. 3 A). As shown in Fig. 3 B, there was a significant increase in binding of H. pylori by the IFN-γ–treated Kato III and N87 cells relative to the same cells treated with media alone. In contrast, IFN-γ had little effect on bacterial binding to the class II MHC-deficient AGS cells. In the absence of bacteria, control or IFN-γ–treated cells did not bind significant amounts of the anti– H. pylori antisera or secondary antibody, indicating that the assay was specific for the detection of bound H. pylori. The role of class II MHC molecules as a receptor for H. pylori was further supported by the fact that mAbs recognizing class II MHC blocked the binding of H. pylori to Kato III cells (Fig. 3 C).
Figure 3.



Binding of H. pylori to the surface of gastric epithelial cells. To determine if the binding of H. pylori was associated with the increased expression of class II MHC molecules, Kato III, N87, and AGS cells were assayed for class II MHC expression before (filled curves) or after (open curves) stimulation with IFN-γ (A). Subsequently, Kato III, N87, and AGS cells were cultured with or without IFN-γ (100 U/ml) for 48 h, washed, and incubated with H. pylori for 2 h at 4°C. Cells were then labeled with anti–H. pylori antisera as described in Materials and Methods and evaluated by flow cytometry. Data (B) are presented as the percent mean fluorescence intensity of cells treated with IFN-γ compared with those treated with media alone. There was significantly increased fluorescence, indicating more H. pylori binding to Kato III and N87 (*P <0.05 compared with control cells) but not to AGS cells. Inset, Fluorescence histogram that was typical for the binding of bacteria to Kato III or N87 cells. The results are representative of three separate experiments. (C) Kato III cells were cultured with IFN-γ (100 U/ml) for 48 h, pretreated with a cocktail of anti–MHC II antibodies (Blocked) or an irrelevant antibody (Unblocked), and evaluated for H. pylori. Isotype, The background fluorescence of Kato III cells with H. pylori and the isotype control for the binding assay. Representative of two separate experiments.
Correlation of Class II MHC Expression and Binding of H. pylori on Freshly Isolated Gastric Epithelial Cells.
To confirm that class II MHC molecules had the potential to serve as receptors in vivo, sections obtained from biopsy specimens of gastric antrum isolated from an infected subject were examined for class II MHC expression by immunohistochemistry. Fig. 4 A shows that class II MHC molecules are not only expressed by the epithelial cells in situ, but can also be observed on the apical surface, where they could be in contact with luminal bacteria. To confirm that the gastric epithelium could bind H. pylori, partially purified preparations of epithelial cells were isolated from biopsy specimens obtained from an infected subject. The purity of the epithelium was determined using an antibody to an epithelial specific antigen as described in Materials and Methods. Purity ranged from 50 to 80%. Simultaneous two-color flow cytometry detecting class II MHC expression and binding of H. pylori on the gated gastric epithelial cells showed that the cells were usually positive for both parameters (77% ± 4.0, n = 4). Moreover, regression analysis comparing the mean fluorescence intensity for class II MHC and binding of H. pylori showed a strong correlation for both variables (Fig. 4 B). Using forward scatter as an estimate of size, it was shown that the correlation between class II MHC expression and binding was independent of the size of the cells. These data suggest that binding of H. pylori to gastric epithelial cells is associated with class II MHC expression.
Figure 4.


Binding of H. pylori to gastric epithelium correlates with class II MHC expression. Sections obtained from gastric antral biopsy from H. pylori–infected subjects were examined for the expression of class II MHC molecules by immunohistochemistry or the appropriate isotype control as described in Materials and Methods. Although no staining was detected with the isotype control, the photomicrograph in A demonstrates that specific antibodies detect class II MHC on gastric epithelial cells and that staining can be detected on the apical surface (arrows in lumen). Examples of the lumen are indicated (L); ×400. To document the correlation between class II MHC expression and the binding of H. pylori, gastric epithelial cells were isolated from biopsy specimens collected from an infected subject. Subsequently, class II MHC expression and H. pylori binding were measured simultaneously by flow cytometry. The data in B show the correlation of the fluorescence intensity for the two parameters (r 2 = 0.93, P <0.05). Representative of four separate experiments.
Induction of Apoptosis Is Mediated by a Specific Interaction between H. pylori and Class II MHC.
In light of the data that class II MHC molecules bind H. pylori but not C. jejuni and that cross-linking class II MHC molecules leads to apoptosis, we postulated that the induction of apoptosis would be restricted to H. pylori. As IFN-γ can enhance the expression of class II MHC molecules, gastric epithelial cells were stimulated with H. pylori or C. jejuni, either alone or in combination with IFN-γ before being assayed for DNA fragmentation. Fig. 5 A shows that both live and killed H. pylori stimulated apoptosis in Kato III cells relative to cells treated with media alone (P <0.05), whereas C. jejuni alone had no effect. Apoptosis was enhanced approximately fourfold when H. pylori and IFN-γ were applied simultaneously. This increase in apoptotic index (mean 6.7) was significantly greater (P <0.05) than the index (mean 1.5) observed with either stimulus alone. Moreover, IFN-γ was capable of synergistically increasing the level of apoptosis that was induced with different strains of H. pylori (unpublished observations). The level of apoptosis induction observed in the cells treated with C. jejuni and IFN-γ was no greater than that observed with IFN-γ alone (see below).
Figure 5.


IFN-γ promotes the induction of apoptosis by H. pylori. (A) Kato III cells were exposed to live or heat-killed H. pylori (Hp) or live C. jejuni (Cj), either alone or in the presence of 100 U/ml IFN-γ. After 48 h, apoptosis was evaluated by ELISA. Both live and killed H. pylori induced apoptosis (*P <0.05 compared with cells in media alone), whereas IFN-γ enhanced the induction of apoptosis relative to the bacteria alone (**P <0.05). Treatment of Kato III cells with live C. jejuni did not induce apoptosis; however, when combined with IFN-γ, C. jejuni induced apoptosis to the same degree as IFN-γ alone (***P <0.05 compared with media controls). (B) To compare the ability of H. pylori to induce apoptosis in gastric cells lines that differed in relative expression of class II MHC molecules, Kato III, N87, and AGS cells were treated as described for A. Both H. pylori and IFN-γ alone induced significant levels of apoptosis in Kato III and N87 cells compared with cells treated with media alone (*P <0.05). The effects of H. pylori were enhanced by IFN-γ in Kato III and N87 compared with control cells (**P <0.05). Data presented are the mean apoptotic index ± SEM of three (C. jejuni) or four (H. pylori) experiments. Absolute values for nucleosomal DNA detection in all three cell types stimulated with media alone were comparable (OD = 0.100–0.250 for Kato III, N87, and AGS).
One possible explanation for the synergy in the induction of apoptosis between IFN-γ and H. pylori could be the enhanced bacterial binding attributed to the increase in expression of class II MHC. To evaluate this, the effect of H. pylori on apoptosis was studied using the two gastric epithelial cell lines that express class II MHC molecules (Kato III and N87) and the one that does not (AGS). As shown in Fig. 5 B, apoptosis was induced only in the lines expressing class II MHC molecules. To evaluate the interaction between H. pylori and IFN-γ in the induction of apoptosis in gastric epithelial cells, the recovery of viable cells and DNA fragmentation were examined by ELISA assay in Kato III cells at different times after exposure. Compared with cells maintained in media alone, exposure to H. pylori decreased viability significantly as measured by trypan blue exclusion (61.5% of control, P <0.05) by 48 h, and this was associated with a progressive increase in the apoptotic index assayed by ELISA (1.47 by 48 h; Fig. 6 A). IFN-γ also decreased the recovery of viable cells (70% of controls) and induced DNA fragmentation (1.9 by 48 h; Fig. 6 B). However, when epithelial cells were exposed to both H. pylori and IFN-γ, as occurs in vivo, there were more marked changes in cell viability (<40% of controls) and apoptotic index (2.8) by 48 h (Fig. 6 C). To confirm that the detection of nucleosomes by ELISA was associated with DNA fragmentation, the effects of H. pylori and IFN-γ alone or in combination were detected by gel electrophoresis. Low molecular weight DNA was formed in cells stimulated with H. pylori and IFN-γ either alone or in combination (Fig. 6, bottom), as occurred when cells were exposed to cross-linking antibodies to class II MHC (Fig. 1).
Figure 6.


The kinetics for the induction of apoptosis by H. pylori and IFN-γ. Viability was measured by trypan blue exclusion (circles, solid lines), and DNA fragmentation was examined by ELISA (triangles, dashed lines) in Kato III cells at different times (from 6 to 48 h) after exposure. (A) Kato III cells were exposed to H. pylori (∼300 bacteria/epithelial cell) for 6–48 h. (B) The results when Kato III cells were exposed to IFN-γ (100 U/ml) for 6–48 h alone. In both A and B, there was a significant decrease in cell viability by 48 h associated with an increase in the apoptotic index as evaluated by ELISA. (C) By 24 h, there was an enhanced induction of cell death when H. pylori was combined with 100 U/ml of IFN-γ compared with treatment with either stimulus alone. Data presented are from one representative experiment. To confirm that the detection of nucleosomes by ELISA was associated with DNA fragmentation characteristic of apoptosis, low molecular weight DNA from treated Kato III cells was separated on agarose gels (bottom). Lane 1, Molecular weight standards; lane 2, the degradation of DNA in Kato III cells grown in media alone; lane 3, in the presence of H. pylori plus IFN-γ (100 U/ml) for 48 h; lane 4, in the presence of H. pylori plus IFN-γ for 24 h; lane 5, H. pylori alone; lane 6, in the presence of IFN-γ alone. The gel is representative of five separate experiments.
Using a complementary approach to implicate the ability of H. pylori to bind class II MHC molecules and stimulate the induction of apoptosis, cells lacking class II MHC were compared with the same lines transfected with genes encoding different class II MHC molecules. Using flow cytometry, H. pylori was shown to bind modestly to class II– deficient COS-1 or BLS-1 cells (Fig. 7 A). However, BLS-1 cells transfected with the gene encoding HLA-DR4 or -DQ2 as well as COS-1 cells transfected with the gene encoding HLA-DR1 (ID12) bound H. pylori at higher levels. Similarly, transfected cells expressing class II MHC became susceptible to apoptosis (Fig. 7 B). Thus, consistent with the results shown in Fig. 3, apoptosis occurred only in the class II MHC–bearing cell lines. Moreover, apoptosis was associated with a decrease in viability, whereas class II MHC− cell lines did not undergo any change in viability. This response was not restricted by the different alleles or isoforms tested.
Figure 7.


Different alleles and isoforms of HLA bind H. pylori and signal the induction of apoptosis. As human class II MHC is represented by different isoforms as well as distinct allelic forms, the interaction between H. pylori and different class II MHC was compared. (A) H. pylori only bound modestly to class II–deficient cells (BLS-1 and COS-1). However, BLS-1 cells, a B cell line, transfected with the gene encoding HLA-DR4 or -DQ2 as well as COS-1 cells transfected with the gene encoding HLA-DR1 (ID12) bound H. pylori at significantly higher levels. (B) Similarly, transfected cells expressing class II MHC became susceptible to apoptosis. Thus, class II MHC enhanced binding, while apoptosis was dependent on the presence of these molecules. This response was not restricted by the different alleles or isoforms tested. *P <0.05 compared with class II–deficient cells.
Discussion
H. pylori induces functional changes in gastric epithelial cells, although a specific receptor that binds and induces these changes has not yet been identified. The data reported here suggest that H. pylori can bind either purified or cell surface–associated class II MHC molecules. Moreover, class II MHC molecules are expressed in vivo, where they can act as functional receptors. While H. pylori may use other receptors to bind to gastric epithelial cells that lack class II MHC molecules, the data in this report suggest that class MHC molecules must also be considered as participants in the attachment of H. pylori. Importantly, the results presented suggest that the binding of H. pylori to class II MHC molecules is necessary for the transduction of signals leading to apoptosis.
Bacteria and viruses are known to produce proteins that can bind to class II MHC molecules directly. Some of these proteins are referred to as “superantigens” due to their affinity for class II MHC and their effect on T cell proliferation. Based on the observation that H. pylori can bind class II MHC, it is possible that these bacteria express superantigen-like proteins. Indeed, preliminary experiments indicate that outer membrane proteins from H. pylori are capable of binding directly to class II MHC and may be responsible for the attachment described in this report. Moreover, H. pylori can bind to all of the isoforms and alleles of class II MHC tested, thereby leading to apoptosis. However, H. pylori binding to human class II MHC molecules and the subsequent signaling may differ depending on the haplotype of the host, as has been observed with other microbial superantigens. This could lead to variations in the host response to H. pylori that affect the manifestation of gastric disease in an infected individual (36). Furthermore, as microbial superantigens have been implicated in the pathogenesis of chronic inflammatory diseases, adhesins of H. pylori that bind class II MHC molecules may represent virulence factors which contribute to both colonization and the pathogenesis of epithelial damage. Further studies into the interactions of the various domains of class II MHC and the H. pylori proteins that bind to class II will help elucidate the genetic events that determine colonization, persistence of the infection, and the diseases associated with this prevalent human pathogen.
Apoptosis is believed to be essential in the regulation of epithelial cell number in the mammalian gastrointestinal tract (37–39). It has also been shown that bacteria can induce apoptosis in intestinal epithelial cells (40) or monocytes (41). Previous studies suggest that apoptosis is increased in the gastric epithelium during infection with H. pylori (19–21), indicating that this process may play an important role in the regulation of gastric epithelial cell function. Apoptosis is also widely recognized as one of the major mechanisms by which immune and inflammatory responses can damage or eliminate host cells. For example, apoptosis of epithelial cells can be triggered through surface receptors for Fas ligand (42) or by cytokines (38). In fact, data presented in this report and elsewhere (43) show that IFN-γ alone is capable of stimulating apoptosis. Evidence is also provided showing that H. pylori is capable of inducing apoptosis in gastric epithelial cells and that this response is augmented by IFN-γ. The role of IFN-γ is significant, since it is one of the major cytokines produced by the T cells recruited to the gastric mucosa during infectious and noninfectious gastritis (14, 44). Recently, Jones and colleagues have reported that apoptosis of gastric epithelial cells is also increased in graft-versus-host disease as well as H. pylori– induced gastritis (22). These findings support the notion that apoptosis may be regulated locally by inflammatory mediators as well as by H. pylori.
H. pylori does not invade gastric epithelial cells, including Kato III cells (28, 45, 46), suggesting that invasion is not involved in the induction of the changes observed in the epithelium. This notion is supported by the fact that killed H. pylori, as well as the secreted vacuolating toxin encoded by the vacA gene, are capable of damaging epithelial cells through the induction of vacuolization (47, 48). As indicated in this report, killed bacteria also induce apoptosis of gastric epithelial cells, while other results suggest that sonicates of H. pylori, as well as both vacA − strains and an isogenic mutant of H. pylori that lacked a functional vacA gene, were still capable of triggering apoptosis in different gastric epithelial cells lines (reference 43, and unpublished observations). Thus, the mechanism whereby H. pylori binds to gastric epithelial cells and signals them to undergo apoptosis does not require invasion and appears to differ from that associated with vacuolization.
Other studies have suggested that the binding of H. pylori to host cells stimulates tyrosine phosphorylation of host proteins (49) and that IL-8 is induced via a tyrosine kinase– dependent pathway that leads to the activation of activator protein-1 and nuclear factor-κB transcription factors (50). As the induction of apoptosis and IL-8 in epithelial cells are often linked (40), one might predict that the phosphorylation of host proteins regulating IL-8 expression plays a role in the induction of apoptosis. However, previous data suggest that viable H. pylori bacteria were necessary for the induction of IL-8 in gastric epithelial cells (46), whereas this report demonstrated that both live and killed bacteria induced apoptosis. Moreover, the induction of IL-8 production by epithelial cells appears to be limited to strains of H. pylori bearing the Cag pathogenicity island (8, 51), but the level of apoptosis in vivo is not elevated in proportion to the presence of Cag-bearing strains (21). The fact that the viability of H. pylori and the reported strain variations may differentially affect apoptosis and IL-8 induction would agree with other reports that the mechanisms responsible for the induction of these responses in epithelial cells can differ (38). Thus, H. pylori may interact with different surface molecules in order to stimulate the various responses in gastric epithelial cells.
Previous studies have reported the presence of H. pylori– specific T cells in the gastric mucosa during infection with H. pylori (52) and an increase in T cell numbers in response to infection (16, 17, 53). Several reports suggest IFN-γ is induced by H. pylori (44, 53, 54), and our own results show that Th1-like cells producing IFN-γ and IL-2 but not IL-4 or IL-5 predominate in the gastric mucosa during infection (14, 35). Since the H. pylori infection resides in the lumen, a Th1 cell–mediated immune response is counterintuitive and may in fact account for the life-long persistence of the infection. In addition, local cell-mediated immune response could contribute to epithelial cell death through apoptosis. It is possible that numerous interactions between the host and the organism, including those governed by hormones, inflammatory mediators, and the expression of bcl-2 or related proteins, may be important in the eventual outcome in vivo. This would be consistent with other reports on the differential expression of bcl-2 in intestinal epithelium and the lack of apoptosis in cells expressing this molecule (55). Thus, in addition to an interaction between H. pylori and class II MHC, our ability to detect apoptosis in vitro may have been similarly affected by differences in the susceptibility of the different cell lines due to factors such as the expression of bcl-2.
The expression of class II MHC molecules is largely restricted to the epithelial cells of the neck region of the gastric glands in the antrum of the noninflamed stomach, and spreads throughout the gastric glands and into the body during infection with H. pylori (15–17). Infection with H. pylori is believed to commence in the antrum and then spread throughout the gastric body. T cells are also more numerous in the neck of the antral gastric pits, where one might predict that the release of IFN-γ would stimulate the increased levels of class II MHC molecules. These observations suggest a model by which IFN-γ–producing T cells play a role in increasing the expression of a cellular receptor for H. pylori and in so doing, contribute to the induction of apoptosis. Given that the kinetics of apoptosis induction in vitro was maximal at 24–48 h, one might predict that the apoptosis in vivo would be detected in the superficial epithelium after the cells have migrated from the proliferative zone. This prediction fits with the reported data showing that the preponderance of cells stained by the TUNEL (terminal deoxynucleotidyl transferase–mediated dUTP nick end labeling) method were, in fact, located in the superficial epithelium of the gastric pit (19, 20).
Although apoptosis may play a constructive role in balancing increased cellular proliferation and the prevention of gastric cancers, excessive disruptions that increase apoptosis could overwhelm mechanisms of epithelial cell restitution. This could predispose the host to peptic ulceration or, as a consequence of aberrant cellular repair mechanisms or mutations that lead to apoptosis resistance, result in the development of atrophy and intestinal metaplasia. This association between Th1 cells, H. pylori attachment, and epithelial damage provide a novel opportunity to control the infection through immunization. Vaccines that induce Th2-like cytokine responses antagonizing these effects could decrease colonization and epithelial damage, thereby creating a unique opportunity for therapeutic vaccination. Future studies will determine if receptor expression, bacterial binding, and signaling via class II MHC molecules can be attenuated by cytokines, thereby providing a novel mechanism for antimicrobial immunity.
Acknowledgments
The authors wish to thank Kim Palkowetz for her technical assistance with flow cytometry.
This work was supported by National Institutes of Health grants DK-50669, DK-51577, and CHD-35741 and a John Sealy Memorial Endowment Recruitment grant. X. Fan is a recipient of a McLaughlin Fellowship. S.E. Crowe is a recipient of the American Gastroenterological Association/Industry Research Scholar Award. N. Van Houten is a recipient of the Claribel and Saul Simkin Research Scholar Award in Crohn's Disease of the American Digestive Health Foundation and the Crohn's and Colitis Foundation of America.
References
- 1.Marshall BJ, Warren JR. Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration. Lancet. 1984;8390:1311–1315. doi: 10.1016/s0140-6736(84)91816-6. [DOI] [PubMed] [Google Scholar]
- 2.Cover TL, Blaser MJ. Helicobacter pyloriand gastroduodenal disease. Annu Rev Med. 1992;43:135–145. doi: 10.1146/annurev.me.43.020192.001031. [DOI] [PubMed] [Google Scholar]
- 3.Blaser MJ, Perez-Perez GI, Kleanthous H, Cover TL, Peek RM, Chyou PH, Stemmermann GN, Nomura A. Infection with Helicobacter pyloristrains possessing cagA is associated with an increased risk of developing adenocarcinoma of the stomach. Cancer Res. 1995;55:2111–2115. [PubMed] [Google Scholar]
- 4.Covacci A, Censini S, Bugnoli M, Petracca R, Buroni D, Macchia G, Massone A, Papini E, Xiang Z, Figura N. Molecular characterization of the 128-kDa immunodominant antigen of Helicobacter pyloriassociated with cytotoxicity and duodenal ulcer. Proc Natl Acad Sci USA. 1993;90:5791–5795. doi: 10.1073/pnas.90.12.5791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Peek RM, Jr, Miller GG, Tham KT, Perez-Perez GI, Zhao X, Atherton JC, Blaser MJ. Heightened inflammatory response and cytokine expression in vivo to cagA+ Helicobacter pyloristrains. Lab Invest. 1995;73:760–770. [PubMed] [Google Scholar]
- 6.Weel JF, van der Hulst RW, Gerrits Y, Roorda P, Feller M, Dankert J, Tytgat GN, van der Ende A. The interrelationship between cytotoxin-associated gene A, vacuolating cytotoxin, and Helicobacter pylori-related diseases. J Infect Dis. 1996;173:1171–1175. doi: 10.1093/infdis/173.5.1171. [DOI] [PubMed] [Google Scholar]
- 7.Crabtree JE, Taylor JD, Wyatt JI, Heatley RV, Shallcross TM, Tompkins DS, Rathbone BJ. Mucosal IgA recognition of Helicobacter pylori120 kDa protein, peptic ulceration, and gastric pathology. Lancet. 1991;338:332–335. doi: 10.1016/0140-6736(91)90477-7. [DOI] [PubMed] [Google Scholar]
- 8.Censini S, Lange C, Xiang Z, Crabtree JE, Ghiara P, Borodovsky M, Rappuoli R, Covacci A. Cag, a pathogenicity island of Helicobacter pylori, encodes type I-specific and disease-associated virulence factors. Proc Natl Acad Sci USA. 1996;93:14648–14653. doi: 10.1073/pnas.93.25.14648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Graham DY, Genta RM, Graham DP, Crabtree JE. Serum CagA antibodies in asymptomatic subjects and patients with peptic ulcer: lack of correlation of IgG antibody in patients with peptic ulcer or asymptomatic Helicobacter pylorigastritis. J Clin Pathol. 1996;49:829–832. doi: 10.1136/jcp.49.10.829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yamaoka Y, Kita M, Sawai N, Imanishi J. Helicobacter pylori cagAgene and expression of cytokine messenger RNA in gastric mucosa. Gastroenterology. 1996;110:1744–1752. doi: 10.1053/gast.1996.v110.pm8964399. [DOI] [PubMed] [Google Scholar]
- 11.Negrini R, Lisato L, Zanella I, Cavazzini L, Gullini S, Villanacci V, Poiesi C, Albertini A, Ghielmi S. Helicobacter pyloriinfection induces antibodies cross-reacting with human gastric mucosa. Gastroenterology. 1991;101:437–445. doi: 10.1016/0016-5085(91)90023-e. [DOI] [PubMed] [Google Scholar]
- 12.Appelmelk BJ, Simoons-Smit I, Negrini R, Moran AP, Aspinall GO, Forte JG, De Vries T, Quan H, Verboom T, Maaskant JJ, et al. Potential role of molecular mimicry between Helicobacter pylorilipopolysaccharide and host Lewis blood group antigens in autoimmunity. Infect Immun. 1996;64:2031–2040. doi: 10.1128/iai.64.6.2031-2040.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.D'Elios MM, Manghetti M, De Carli M, Costa F, Baldari CT, Burroni D, Telford J, Romagnani S, Del Prete G. T helper 1 effector cells specific for Helicobacter pyloriin gastric antrum of patients with peptic ulcer disease. J Immunol. 1997;158:962–967. [PubMed] [Google Scholar]
- 14.Haeberle H, Crowe SE, Luthra R, Gourley WK, Garofalo R, Karttunen R, El-Zaateri F, Graham DY, Kubin M, Trinchieri G, et al. Induction of IL-12 and selection of Th1 cells in the gastric mucosa in response to H. pylori. . Infect Immun. 1997;65:4229–4235. doi: 10.1128/iai.65.10.4229-4235.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brandtzaeg, P., K. Valnes, H. Scott, T.O. Rognum, K. Bjerke, and K. Baklien. 1986. The human gastrointestinal secretory immune system in health and disease. In Basic Science in Gastroenterology: Diseases of the Gut. J.M. Polak, S.R. Bloom, N.A. Wright, and A.G. Butler, editors. Page Bros., Norwich. 179–200.
- 16.Engstrand L, Scheynius A, Pahlson C, Grimelius L, Schwan A, Gustavsson S. Association of Campylobacter pyloriwith induced expression of class II transplantation antigens on gastric epithelial cells. Infect Immun. 1989;57:827–832. doi: 10.1128/iai.57.3.827-832.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Valnes K, Huitfeldt HS, Brandtzaeg P. Relation between T cell number and epithelial HLA class II expression quantified by image analysis in normal and inflamed human gastric mucosa. Gut. 1990;31:647–652. doi: 10.1136/gut.31.6.647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yasumoto K, Okamoto S, Mukaida N, Murakami S, Mai M, Matsushima K. Tumor necrosis factor α and interferon τ synergistically induce interleukin 8 production in a human gastric cancer cell line through acting concurrently on AP-1 and NF-κB-like binding sites of the interleukin 8 gene. J Biol Chem. 1992;267:22506–22511. [PubMed] [Google Scholar]
- 19.Moss SF, Calam J, Agarwal B, Wang S, Holt PG. Induction of gastric epithelial apoptosis by Helicobacter pylori. . Gut. 1996;38:498–501. doi: 10.1136/gut.38.4.498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mannick EE, Bravo LE, Zarama G, Realpe JL, Zhang X-J, Ruiz B, Fontham ETH, Mera R, Miller MJS, Correa P. Inducible nitric oxide synthase, nitrotyrosine and apoptosis in Helicobacter pylorigastritis: effect of antibiotics and antioxidants. Cancer Res. 1996;56:3238–3243. [PubMed] [Google Scholar]
- 21.Peek RM, Jr, Moss SF, Tham KT, Perez-Perez GI, Wang S, Miller GG, Atherton JC, Holt PR, Blaser MJ. Helicobacter pylori cagA+strains and dissociation of gastrointestinal epithelial cell proliferation from apoptosis. J Natl Cancer Inst. 1997;89:863–868. doi: 10.1093/jnci/89.12.863. [DOI] [PubMed] [Google Scholar]
- 22.Jones NL, Shannon PT, Cutz E, Yeger H, Sherman PM. Increase in proliferation and apoptosis of gastric epithelial cells early in the natural history of Helicobacter pyloriinfection. Am J Pathol. 1997;151:1695–1703. [PMC free article] [PubMed] [Google Scholar]
- 23.Ernst PB, Crowe SE, Reyes VE. The immunopathogenesis of gastroduodenal disease associated with Helicobacter pyloriinfection. Curr Opin Gastroenterol. 1995;11:512–518. [Google Scholar]
- 24.Scholl PR, Geha RS. MHC class II signaling in B-cell activation. Immunol Today. 1994;15:418–423. doi: 10.1016/0167-5699(94)90271-2. [DOI] [PubMed] [Google Scholar]
- 25.Newell MK, Vanderwall J, Beard KS, Freed JH. Ligation of major histocompatibility complex class II molecules mediates apoptotic cell death in resting B lymphocytes. Proc Natl Acad Sci USA. 1993;90:10459–10463. doi: 10.1073/pnas.90.22.10459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sherman, P.M. 1994. Adherence and internalization of H. pylori by epithelial cells. In H. pylori: Basic Mechanisms to Clinical Cure. R.H. Hunt and G.N.J. Tytgat, editors. Kluwer Academic Publishers, Boston. 148–162.
- 27.Falk P, Roth KA, Boren T, Westblom TU, Gordon JI, Normark S. An in vitro adherence assay reveals that Helicobacter pyloriexhibits cell lineage-specific tropism in the human gastric epithelium. Proc Natl Acad Sci USA. 1993;90:2035–2039. doi: 10.1073/pnas.90.5.2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dytoc M, Gold B, Louie M, Heusca M, Fedorko L, Crowe SE, Lingwood C, Brunton J, Sherman PM. Comparison of Helicobacter pylori and attaching-effacing Escherichia coliadhesion to eukaryotic cells. Infect Immun. 1992;61:448–456. doi: 10.1128/iai.61.2.448-456.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xu M, Capraro GA, Daibata B, Reyes VE, Humphreys RE. Cathepsin B cleavage and release of invariant chain from MHC Class II molecules follow a staged pattern. Mol Immunol. 1994;31:723–731. doi: 10.1016/0161-5890(94)90146-5. [DOI] [PubMed] [Google Scholar]
- 30.Kwok WK, Kovats S, Thurtle P, Nepom GT. HLA-DQ allelic polymorphisms constrain patterns of class II heterodimer formation. J Immunol. 1993;150:2263–2272. [PubMed] [Google Scholar]
- 31.Van Houten N, Budd RC. Accelerated programmed cell death of MRL-lpr/lpr T lymphocytes. J Immunol. 1992;149:2513–2517. [PubMed] [Google Scholar]
- 32.Reyes VE, Lu S, Humphreys RE. Cathepsin B cleavage of Ii form class II MHC α- and β-chains. J Immunol. 1991;146:3877–3880. [PubMed] [Google Scholar]
- 33.Dunn, B.E., M. Altmann, and G.P. Campbell. 1991. Adherence of Helicobacter pylori to gastric carcinoma cells: analysis by flow cytometry. Rev. Infect. Dis. 13(Suppl. 8):S657–S664. [DOI] [PubMed]
- 34.Ye G, Barrera C, Fan XJ, Crowe SE, Ernst PB, Reyes VE. Gastric epithelial cells express the costimulatory molecules B7.1 and B7.2 during infection with Helicobacter pyloriand activate CD4 T cells. J Clin Invest. 1997;99:1628–1636. doi: 10.1172/JCI119325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bamford KB, Fan XJ, Crowe SE, Leary JF, Gourley WK, Luthra GK, Brooks EG, Graham DY, Reyes VE, Ernst PB. Lymphocytes in the human gastric mucosa during Helicobacter pylorihave a T helper cell 1 phenotype. Gastroenterology. 1998;114:482–492. doi: 10.1016/s0016-5085(98)70531-1. [DOI] [PubMed] [Google Scholar]
- 36.Azuma T, Konishi J, Tanaka Y, Hirai M, Ito S, Kato T, Kohli Y. Contribution of HLA-DQA gene to host's response against Helicobacter pylori. . Lancet. 1994;343:542–543. doi: 10.1016/s0140-6736(94)91496-6. [DOI] [PubMed] [Google Scholar]
- 37.Hall PA, Coates PJ, Ansari B, Hopwood D. Regulation of cell number in the mammalian gastrointestinal tract: the importance of apoptosis. J Cell Sci. 1994;107:3569–3577. doi: 10.1242/jcs.107.12.3569. [DOI] [PubMed] [Google Scholar]
- 38.Abreu-Martin MT, Vidrich A, Lynch DH, Targan SR. Divergent induction of apoptosis and IL-8 secretion in HT-29 cells in response to TNF-α and ligation of Fas antigen. J Immunol. 1995;155:4147–4154. [PubMed] [Google Scholar]
- 39.O'Connell J, O'Sullivan GC, Collins JK, Shanahan F. The Fas counter-attack: Fas-mediated T cell killing by colon cancer cells expressing Fas ligand. J Exp Med. 1996;184:1075–1082. doi: 10.1084/jem.184.3.1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mahida YR, Makh S, Hyde S, Gray T, Boriello SP. Effect of Clostridium difficiletoxin A on human intestinal epithelial cells: induction of interleukin 8 production and apoptosis after cell detachment. Gut. 1996;38:337–347. doi: 10.1136/gut.38.3.337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Baran J, Guzik K, Hryniewicz W, Ernst M, Flad HD, Pryjma J. Apoptosis of monocytes and prolonged survival of granulocytes as a result of phagocytosis of bacteria. Infect Immun. 1996;64:4242–4248. doi: 10.1128/iai.64.10.4242-4248.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Itoh N, Yonehara S, Ishii A, Yonehara M, Mizushima S, Sameshima M, Hase A, Seto Y, Nagata S. The polypeptide encoded by the cDNA for human cell surface antigen Fas can mediate apoptosis. Cell. 1991;66:233–243. doi: 10.1016/0092-8674(91)90614-5. [DOI] [PubMed] [Google Scholar]
- 43.Wagner S, Beil W, Westermann J, Logan RPH, Bock CT, Trautwein C, Bleck JS, Manns MP. Regulation of epithelial cell growth by Helicobacter pylori: evidence for a major role of apoptosis. Gastroenterology. 1997;113:1836–1847. doi: 10.1016/s0016-5085(97)70003-9. [DOI] [PubMed] [Google Scholar]
- 44.Karttunen R, Karttunen T, Ekre HP, MacDonald TT. Interferon gamma and interleukin 4 secreting cells in the gastric antrum in Helicobacter pyloripositive and negative gastritis. Gut. 1995;36:341–345. doi: 10.1136/gut.36.3.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hemalatha SG, Drumm B, Sherman PM. Adherence of Helicobacter pylori to human gastric epithelial cells in vitro. . J Med Microbiol. 1991;35:197–202. doi: 10.1099/00222615-35-4-197. [DOI] [PubMed] [Google Scholar]
- 46.Crowe SE, Alvarez L, Sherman PM, Jin Y, Dytoc M, Hunt RH, Patel J, Muller MJ, Ernst PB. Expression of interleukin-8 and CD54 by human gastric epithelium after H. pyloriinfection in vitro. Gastroenterology. 1995;108:65–74. doi: 10.1016/0016-5085(95)90009-8. [DOI] [PubMed] [Google Scholar]
- 47.Leunk RD, Johnson PT, David BC, Kraft WG, Morgan DR. Cytotoxic activity in broth-culture filtrates of Campylobacter pylori. . J Med Microbiol. 1988;36:93–99. doi: 10.1099/00222615-26-2-93. [DOI] [PubMed] [Google Scholar]
- 48.Cover TL, Halter SA, Blaser MJ. Characterization of HeLa cell vacuoles induced by Helicobacter pyloribroth culture supernatant. Hum Pathol. 1992;23:1004–1010. doi: 10.1016/0046-8177(92)90261-z. [DOI] [PubMed] [Google Scholar]
- 49.Segal ED, Falkow S, Tompkins LS. Helicobacter pyloriattachment to gastric cells induces cytoskeletal rearrangements and tyrosine phosphorylation of host cell proteins. Proc Natl Acad Sci USA. 1996;93:1259–1264. doi: 10.1073/pnas.93.3.1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Aihara M, Tsuchimoto D, Takizawa H, Azuma A, Wakebe H, Ohmoto Y, Imagawa K, Kikuchi M, Mukaida N, Matsushima K. Mechanisms involved in Helicobacter pylori-induced interleukin-8 production by a gastric cancer cell line, MKN45. Infect Immun. 1997;65:3218–3224. doi: 10.1128/iai.65.8.3218-3224.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Crabtree JE, Covacci A, Farmery SM, Xiang Z, Tompkins DS, Perry S, Lindley IJD, Rappuoli R. Helicobacter pyloriinduced interleukin-8 expression in gastric epithelial cells is associated with CagA positive phenotype. J Clin Pathol. 1994;48:41–45. doi: 10.1136/jcp.48.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Di Tommaso A, Xiang Z, Bugnoli M, Pileri P, Figura N, Bayeli PF, Rappuoli R, Abrignani S, De Magistris MT. Helicobacter pylori-specific CD4+T-cell clones from peripheral blood and gastric biopsies. Infect Immun. 1995;63:1102–1106. doi: 10.1128/iai.63.3.1102-1106.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fan XJ, Chua A, Shahi CN, McDevitt J, Keeling PWN, Kelleher D. Gastric T lymphocyte response to Helicobacter pylori in patients with H. pyloricolonisation. Gut. 1994;35:1379–1384. doi: 10.1136/gut.35.10.1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tarkkanen J, Kosunen TU, Saksela E. Contact of lymphocytes with Helicobacter pyloriaugments natural killer cell activity and induces production of gamma interferon. Infect Immun. 1993;61:3012–3016. doi: 10.1128/iai.61.7.3012-3016.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Merritt AJ, Potten CS, Watson AJM, Loh DY, Nakayama K-I, Nakayama K, Hickman JA. Differential expression of bcl-2 in intestinal epithelia. Correlation with attenuation of apoptosis in colonic crypts and incidence of colonic neoplasia. J Cell Sci. 1995;108:2261–2271. doi: 10.1242/jcs.108.6.2261. [DOI] [PubMed] [Google Scholar]