

Abstract
To investigate the possible involvement of DNA repair in the process of somatic hypermutation of rearranged immunoglobulin variable (V) region genes, we have analyzed the occurrence, frequency, distribution, and pattern of mutations in rearranged Vλ1 light chain genes from naive and memory B cells in DNA repair–deficient mutant mouse strains. Hypermutation was found unaffected in mice carrying mutations in either of the following DNA repair genes: xeroderma pigmentosum complementation group (XP)A and XPD, Cockayne syndrome complementation group B (CSB), mutS homologue 2 (MSH2), radiation sensitivity 54 (RAD54), poly (ADP-ribose) polymerase (PARP), and 3-alkyladenine DNA-glycosylase (AAG). These results indicate that both subpathways of nucleotide excision repair, global genome repair, and transcription-coupled repair are not required for somatic hypermutation. This appears also to be true for mismatch repair, RAD54-dependent double-strand–break repair, and AAG-mediated base excision repair.
Keywords: DNA repair, DNA repair–deficient mice, memory B cells, naive B cells, somatic mutations
In humans and mice, the primary Ig repertoire is generated by V(D)J recombination in B cell progenitors of the embryonic liver and adult bone marrow. Rearranged Ig genes of antigen-specific B cells can be further diversified by hypermutation in germinal center B cells, which develop in secondary lymphatic tissues during immune responses (for review see reference 1). The mutations are mainly point mutations, with rare deletions and insertions (2). The mutation rate has been estimated to be as high as 10−3 base pairs/generation, which is approximately six orders of magnitude higher than the spontaneous mutation rate (3–5).
Notwithstanding the extensive knowledge of the immunobiology (1), the molecular mechanism underlying somatic hypermutation remains speculative (6–9). Several models have been proposed: (a) “gratuitous” transcription-coupled repair (TCR1; 10, 11), (b) site-specific nicking and repair of DNA via an error prone DNA polymerase (12, 13), (c) gene conversion (14), (d) local DNA hyperreplication (15), (e) cDNA retrotransposition of reverse transcribed Ig mRNA (16), (f) altered DNA replication (17), and (g) inhibition of mismatch repair (8).
Recent advances in cloning, characterizing, and targeting eukaryotic DNA repair genes (18) has provided mouse models that allow us to test present concepts on the mechanism of hypermutation. The molecular mechanism underlying hypermutation is unlikely to be controlled by a single gene product and most likely has evolved from preexisting components involved in DNA modification. We speculate that mutations are introduced by a “site-specific mutator” that depends on other molecules involved in DNA repair and/or DNA synthesis, a scenario that is reminiscent of V(D)J-recombination, which uses the site-specific recombination activating gene (RAG) recombinase as well as components of DNA repair such as DNA-dependent protein kinase and x ray cross-complementing (XRCC)4 (19).
Therefore, seven mouse strains with predefined mutations in either one of the DNA repair genes xeroderma pigmentosum complementation group (XP)A (20), XPD (20a), Cockayne syndrome (CS)B (21), mutS homologue 2 (MSH2; 22), radiation sensitivity 54 (RAD54; 23), poly (ADP-ribose) polymerase (PARP; reference 24), and 3-alkyladenine DNA-glycosylase or 3-methyladenine DNA-glycosylase (AAG; 24a) were tested for the occurrence of somatic mutations in naive and memory B cells. The different repair pathways where XPA, XPD, CSB, MSH2, RAD54, PARP, and AAG are involved are briefly outlined below. For further details the reader is referred to Friedberg et al. (25).
The XPA protein is an essential component of the nucleotide-excision repair (NER) pathway. XPA protein functions in a preincision step of NER, i.e., the recognition of DNA damage and controls both NER pathways, global genome repair (GGR) and TCR. It recruits the basal transcription factor (TF)IIH, which among other downstream components is common to GGR and TCR (26). XP-deficient mice are highly susceptible to UV-B– or chemical-carcinogen–induced skin and eye tumors (20, 27).
XPD and XPB are components of the basal transcription factor TFIIH and both display helicase activity. Specific mutations in the XPD gene and in the XPB gene can cause the multisystemic disorder trichothiodystrophy (TTD) (28). Clinically, the defining feature of TTD is sulfur-deficient brittle hair, which is due to a reduced cysteine content. TTD is also associated with reduced size, mental retardation, unusual facial appearance, and ichthyosis. These and other findings led to the hypothesis that the clinical features underlying TTD relate to defective transcription, i.e., a repair/transcription syndrome concept (29).
Unlike XPD and XPB, the CSB gene is not essential for transcription but appears to play a key role in coupling transcription and NER. CSB is thought to control the functional transition of a RNA–polymerase II complex into a “repairosome” as soon as the polymerase becomes stalled at a DNA lesion. Alternatively, repairosomes might continuously be active in scanning-transcribed DNA for the occurrence of DNA damage. CS is a multisystemic disorder. CS patients are deficient in TCR. TCR is a subpathway of NER that accomplishes the fast and efficient repair of certain types of lesions from the transcribed strand of active genes (30). Mutations in either the CSA or CSB gene give rise to the classical form of CS. Specific mutations in the XPD, XPB, or XPG genes can also cause CS symptoms in combination with XP (28, 29). Consistent with a lack in TCR, CSB mutant mice are UV sensitive, show a selective inactivation of TCR but unaffected GGR, and are unable to resume RNA synthesis after UV exposure (21).
MSH2 is the eukaryotic homologue of the bacterial mutS gene. Like mutS, the MSH2 protein plays a central role in mismatch repair (MMR) and blocks recombination between nonhomologous DNA strands, i.e., antirecombination activity of MMR (22). MSH2 binds together with MSH6 to base mispairs and to extrahelical loops that occur during homologous recombination and replication. In contrast, the MSH3/MSH6 heterodimer (MSH3 is another mutS homologue) recognizes preferentially extrahelical loops (31–33). Upon recognition of the mismatch, the MSH2-containing heterodimer recruits other components of the mismatch repair system, i.e., the MLH1–PMS2 heterodimer (MLH, mutL homologue; PMS, postmeiotic segregation). This complex triggers the excision of the mismatched nucleotide by removal of a tract of single-stranded DNA that contains the mismatched nucleotide. Resynthesis of the excised DNA strand and ligation of the remaining nick completes the repair process.
RAD54 is a key factor in the RAD52 DNA repair pathway. RAD54, RAD51, and RAD52 belong to the Saccharomyces cerevisiae RAD52 epistasis group (RAD50–RAD57, RAD24, RAD59, meiotic recombination [MRE]11, and x-ray sensitivity [XRS]2). Mutants show a wide range of recombination deficiencies including a defect in double-strand–break repair (DSBR) through homologous recombination. Consistent with a defect in DSBR, RAD54 double-knockout embryonic stem cells display a reduced frequency in homologous recombination and are sensitive to ionizing radiation, mitomycin C, and the alkylating agent methyl methane-sulfonate, but not to UV light (23). RAD51 and its mouse counterpart are functional homologues of the Escherichia coli recombination protein RecA, which forms nucleoprotein filaments with single-stranded DNA and mediates homologous DNA pairing and strand exchange. RAD54 is likely to be involved in this process since it was found to physically interact with RAD51 in S. cerevisiae (34).
PARP, a zinc-finger DNA binding protein, is involved in single-stranded break detection. In response to these breaks, the catalytic domain of PARP catalyses the covalent attachment of poly-ADP ribose to itself and to nuclear proteins. Its catalytic activity requires free DNA ends. Upon binding, PARP is thought to recruit DNA-repair components. A subsequent automodification of the enzyme negatively interferes with DNA binding. Because of these features, PARP appears to act as a “nick sensor”. This function is consistent with the increased sensitivity of PARP-deficient mice to γ-irradiation and MNU treatment (24).
The AAG is like other DNA-glycosylases involved in the initial step of base-excision repair (BER), the detection and processing of a modified base (25). Removal of the damaged base generates an AP (apurinic or apyrimidinic) site. The AP site is processed by the sequential action of AP-endonuclease/deoxyribophosphodiesterase or AP-lyase/3′ repair diesterase, leaving a single-stranded gap, which in mammalian cells is filled by the DNA polymerase β (35). Alternatively, proliferating cell nuclear antigen–dependent longer patch repair synthesis may contribute to the BER pathway (36). Ligation of the remaining nick is thought to be performed by DNA ligase II. AAG mutant mice are viable and do not show an overt phenotype.
Materials and Methods
Mice.
The background, genotyping, and characterization of XPA, CSB, MSH2, RAD54, PARP, and AAG (our manuscript submitted) mutant mice have been published elsewhere (20–24a). Since all mice had to be imported from outside laboratories, there was an infection risk for our animal facility at the University of Cologne (Cologne, Germany). Given this problem and the limited space in our quarantine facility, the availability of the mice was limited and they had to be analyzed soon after arrival. Nonimmunized wild-type and CSB, MSH2, XPA, and PARP mutant mice were analyzed at an age of 10–30 wk. RAD54 and AAG mutant mice were immunized with 50 μg NP-CG alumn in 100 μl PBS 10 d before analysis and killed at an age of 10 and 11 wk, respectively. For the analysis of hypermutation in XPDTTDIBEL mutant mice, which carry the same mutation in XPD as the TTDIBEL patient (20a), an immunized mouse (10 wk) and a nonimmunized mouse (11 wk) were used. Data from immunized or nonimmunized mice are presented together, since immunization did not affect the frequency and pattern of Vλ1 mutations in memory B cells.
Isolation of Single B220+, Igμ+, Igδ +, Vλ1+ Naive B Cells and B220+, Igμ−, Igδ −, Vλ1+ Memory B Cells from Spleen.
Single cell suspensions were obtained by mincing the spleen through a nylon mesh (cell strainer; Falcon; Becton Dickinson, Franklin Lakes, NJ). Upon lysis of the erythrocytes, white blood cells were washed twice with 10 ml PBA (PBS, 0.5% BSA, and 0.05% sodium azide). After washing, memory B cells were enriched by means of magnetic cell depletion using IgM–, thymidine (Thy)- 1– or CD5–, and MAC-1–specific beads according to the procedure of the manufacturer (Miltenyi Biotec GmbH, Bergisch Gladbach, Germany). The memory B cell–enriched cell population was stained with saturating amounts of anti-IgM FITC (clone R33-24-12), anti-IgD FITC (clone 1.3), anti-Thy1.2 FITC (clone CFO-1), anti–MAC-1 FITC (clone M1/70.15.11), anti-B220 APC (clone RA3-6B2), and anti-λ1 PE (clone LS136) as described (37). After washing twice in PBA, lymphocytes were sorted in the presence of propidium iodide (PI) on a FACStar® Plus flow cytometer (Becton Dickinson, Mountain View, CA). Cells were gated electronically by means of forward scatter, side scatter, B220+ (APC+), and Igμ−, Igδ−, Thy-1−, Mac-1− (FITC−) staining. From those single, viable (PI−), Vλ1+ (PE+) cells were sorted into PCR tubes (Roth, Karlsruhe, Germany) containing 20 μl Taq buffer (20 mM Tris-HCl, pH 8.4, 50 mM KCl, and 2.5 mM MgCl2; GIBCO BRL, Gaithersburg, MD) and 33 ng/μl 5S E. coli rRNA (Boehringer Mannheim, Indianapolis, IN). Since Vλ1+ memory B cells are very infrequent (∼0.01% of all splenic B cells), no statements on the numbers of memory B cells present in the various experimental animals can be made. Naive B cells were sorted by gating on viable B220+, Igμ+, δ+, Vλ1+ cells. Cells were put immediately on dry ice and stored at −80°C.
Amplification of Rearranged λ1 Genes from Single Cells.
Single cells in 20 μl of Taq buffer were incubated with 0.5 μg/1 μl proteinase K (Boehringer Mannheim) for 40 min at 55°C. After heat inactivation of the protease (8 min at 95°C), specific amplification of rearranged λ1 genes was achieved by using a seminested PCR strategy. The first round of amplification was carried out in the same reaction tube in a 50-μl volume containing 20 mM Tris-HCl, pH 8.4; 50 mM KCl; 2.5 mM MgCl2; 400 μM each dATP, dGTP, dCTP, and dTTP (Pharmacia Biotech, Piscataway, NJ), 40 nM Vλ1 primer (5′-GGGTATGCAACAATGCGCATCTTGTC-3′), 40 nM Jλ1 primer (5′-CACGGACAGGATCCTAGGACAGTCAG TTTGGT-3′), and 5U Taq DNA polymerase. The polymerase was added after the first denaturation step. The temperature cycle program consisted of one cycle at 95°C for 2 min, 88°C for 3 min, 60°C for 2 min, 72°C for 2 min, followed by 28 cycles of 94°C for 1 min, 60°C for 1 min, and 72°C for 90 s. The final extension time was 10 min, and thereafter the reaction was cooled to 15°C.
Of the first round of PCR, a 1.5 μl sample was used for the second, seminested amplification in a 50-μl volume using the same buffer but containing 140 nM of a nested Vλ1 primer (5′-GCGAAGAGAAGCTTGTGACTCAGGAATCTGCA-3′), 140 nM of the Jλ1 primer used in the first round of PCR, and 5 U of Taq DNA polymerase (GIBCO BRL). The cycle program consisted of one step at 92°C for 1 min, followed by 30 cycles of 92°C for 1 min, 63°C for 1 min, and 72°C for 90 s. Again, the final extension time was 10 min and thereafter the reaction was cooled to 15°C.
Sequencing of λ1 PCR Products.
PCR products were separated from primers on a 1% TAE agarose gel (SeaKem GTG agarose; Biozym, Hessich Oldendorf, Germany) and visualized under long wave UV light. DNA was isolated from agarose gel slices by centrifugation through a 0.22-μm molecular filter (SpinX tubes; Costar Corp., Cambridge, MA). Ethanol-precipitated DNA was resolved in 25 μl water and 50–150 ng of template was used for dye-terminated automatic sequencing (PE Applied Biosystems, Foster City, CA) in combination with either the nested Vλ1 primer or the Jλ1 primer.
Results and Discussion
Ex vivo Analysis of Hypermutation by Single Cell PCR.
To analyze somatic hypermutation efficiently in any mouse strain, we established a seminested PCR approach to amplify and sequence functionally rearranged Vλ1 genes from single cells. The method is based on the availability of the Vλ1-specific monoclonal Ab LS136 (38) that allows the identification and isolation of single B cells expressing a functionally rearranged Vλ1 gene (Fig. 1). An equivalent monoclonal Ab specific for the translational product of a unique V gene in both IgH allotypes is not available. In addition, gene conversion, which might to some extent contribute to the diversification of rearranged VH genes (39), does not occur in Vλ1 genes (40). Therefore, the analysis of hypermutation in rearranged Vλ1 genes will focus on the primary molecular mechanism of somatic hypermutation, the introduction of untemplated point mutations into rearranged Ig heavy and light chain gene segments.
Figure 1.


Enrichment and sorting of single Vλ1+ memory B cells for amplification and sequencing of the rearranged Vλ1 gene. Cells, before and after magnetic activated cell sorting (MACS) were stained as described in the Materials and Methods section. The enriched memory B cells (CD45R+, IgM−, IgD−) were gated. From those, single, viable (PI−), Vλ1+ memory B cells (*, the population) were sorted on a FACStar®. PCR products were run on a 1% TAE gel; lanes 1 and 2 are negative controls (non–B cells), lanes 3–10 are naive (IgM+, IgD+), Vλ1+ B cells, and lanes 11–48 are the products of the Vλ1+ memory B cells. Numbers above the gates indicate percentages. The analysis from a wild-type mouse is shown.
The single cell PCR approach omits cloning of the PCR products and thus excludes the potential formation of recombination products between PCR intermediates generated during amplification of genomic DNA or cDNA from sorted populations. It is also essentially not obscured by errors of the Taq polymerase since the amplificates are directly sequenced. Using this ex vivo approach it is possible to efficiently analyze hypermutation parameters like mutation frequency, base exchange pattern, ratio of transitions versus transversions, and distribution in any mouse strain.
Frequency, Distribution, and Base Exchange Pattern of Somatic Hypermutation in DNA Repair–deficient Mouse Strains.
A possible role of NER in somatic hypermutation was analyzed in XPA, XPD, and CSB mutant mice. Consistent with the role of CSB in TCR, CSB-deficient mice lack TCR activity (21) and accordingly form an ideal system to test the gratuitous TCR-based hypermutation model (6, 10, 11). This model proposes a mutator that is loaded during transcription initiation and carried into the gene by the elongating transcription complex. Next, mutator-mediated stalling of RNA polymerase II, in the absence of DNA lesions (41), is thought to recruit factors involved in NER. A small single-stranded DNA fragment of the transcribed strand is excised by the NER machinery, and the fill in of the single stranded gap by an unknown error-prone DNA polymerase is thought to introduce the mutation. The mutation rate is determined by the error rate of the DNA polymerase.
In line with these considerations, triplex-forming oligonucleotides were found to inhibit transcription and lead to mutations 5′ of a transcriptional block in mammalian cells. Mutagenesis was dependent on NER since it was not detectable in XPA- or CSB-deficient cells, respectively (42). The mechanism by which the triplex-mediated transcription inhibition not only triggers repair but also induces mutagenesis was extrapolated from the model of Hanawalt (41), which proposes that the TCR pathway of a cell may trigger “gratuitous repair” when transcription stalls at natural pause sites, even in the absence of a classical DNA lesion. These data raise the possibility that a transcriptional block might initiate the hypermutation process. A transcriptional block, for example in the V gene intron, mediated by an as yet unknown cis motif(s), could explain the reduced mRNA and protein levels of Ig in germinal center B cells (43, 44). In the frame of these speculations, hypermutation should not occur in XPA-, XPD-, or CSB-deficient mice.
However, with regard to distribution of point mutations along the rearranged gene (Fig. 2), strand bias of hypermutation, as reflected by a preferential mutation of A, G, and C rather than T bases (Fig. 3), frequency and ratio of transitions versus transversions (Table 1), the pattern of hypermutation appears to be normal in memory B cells of XPA-, XPD-, and CSB-mutant mice and hypermutation is absent in naive B cells (0/7 naive B cells carrying mutations in XPA-mutant mice, 0/5 in XPD-mutant mice, 1/28 in CSB-mutant mice, and 2/61 in wild-type mice; data not shown). Naive B cells were included in this analysis because of the possibility that the mutator is constitutively expressed throughout B cell development is efficiently counteracted by a specific DNA repair pathway in naive B cells and becomes only apparent in germinal center B cells. The slightly decreased mutation frequency in XPDTTDIBEL mice (0.7% in XPD versus 1.0–1.6% in other mice) might relate to the fact that relatively young mice were analyzed in this case (Table 1), since both the proportion of B cells that express mutated V-genes and the average number of nucleotide substitutions carried by the mutant V genes can increase considerably with age (2). Hypermutation thus occurs in the absence of NER, in accord with the finding of normal hypermutation in XPB, XPD, XPV, and CSA patients (45, 46). This strongly argues against the gratuitous TCR-based model, but does not exclude a physical link between hypermutation and transcription. The latter is based on the findings that hypermutation is most efficient in the presence of both Ig transcriptional enhancers (47), displays a strand bias that is based on the preference to mutate A, G, or C rather than T (2), is restricted to a 1.5-kb window downstream of the leader intron (48–51), and can be reinitiated by placing an additional Igκ promoter immediately 5′ to the constant portion of the Igκ chain gene, which leads to hypermutation in the rearranged VJ region and the Cκ region but not in the spacer region between these elements (11). However, the apparent dependency of the mutator on an active locus might merely reflect the accessibility of the rearranged Ig locus for the mutator. This possibility calls for the analysis of other transcription-independent DNA repair pathways in hypermutation.
Figure 2.
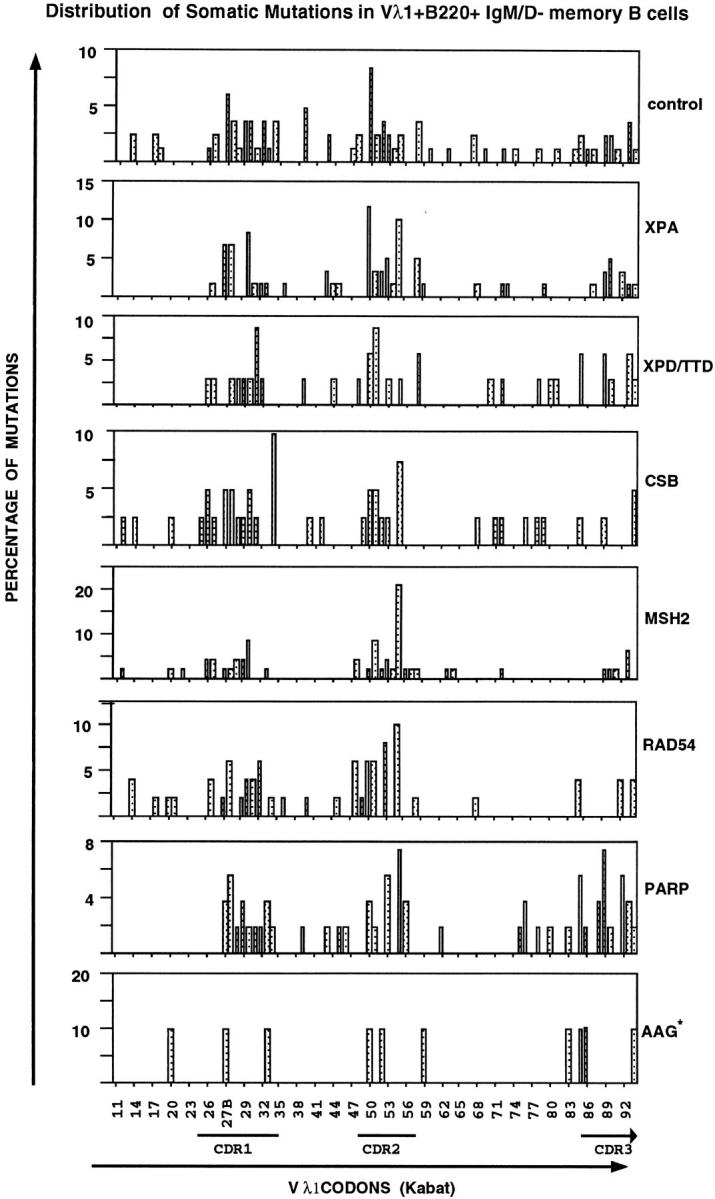
Distribution of point mutations found in memory B cells from XPA, XPD TTDIBEL, CSB, MSH2, RAD54, PARP, and AAG mutant mice. The point mutations found in the Vλ1 locus of memory B cells are indicated as percentage of mutations versus 87 Vλ1 codons (11–94, numbering according to Kabat, reference 56). *, a small data base of point mutations in the AAG-mutant mice. Irrespective of the mouse strain (right), a typical clustering of mutations within the CDR regions was found. The published hot spots in Vλ1 (57): Ser30 II, Asn31 III, Gly50 II, Ala55 I ανδ II, and Leu90 III were confirmed in this study. Additional hot spots are at position Ala27B I, Val27C III, Gly50 II, Thr51 I, Ala89 II, and Ser93 II and III. The Roman numbers of the hot spots indicate the nucleotide position in the codons.
Figure 3.
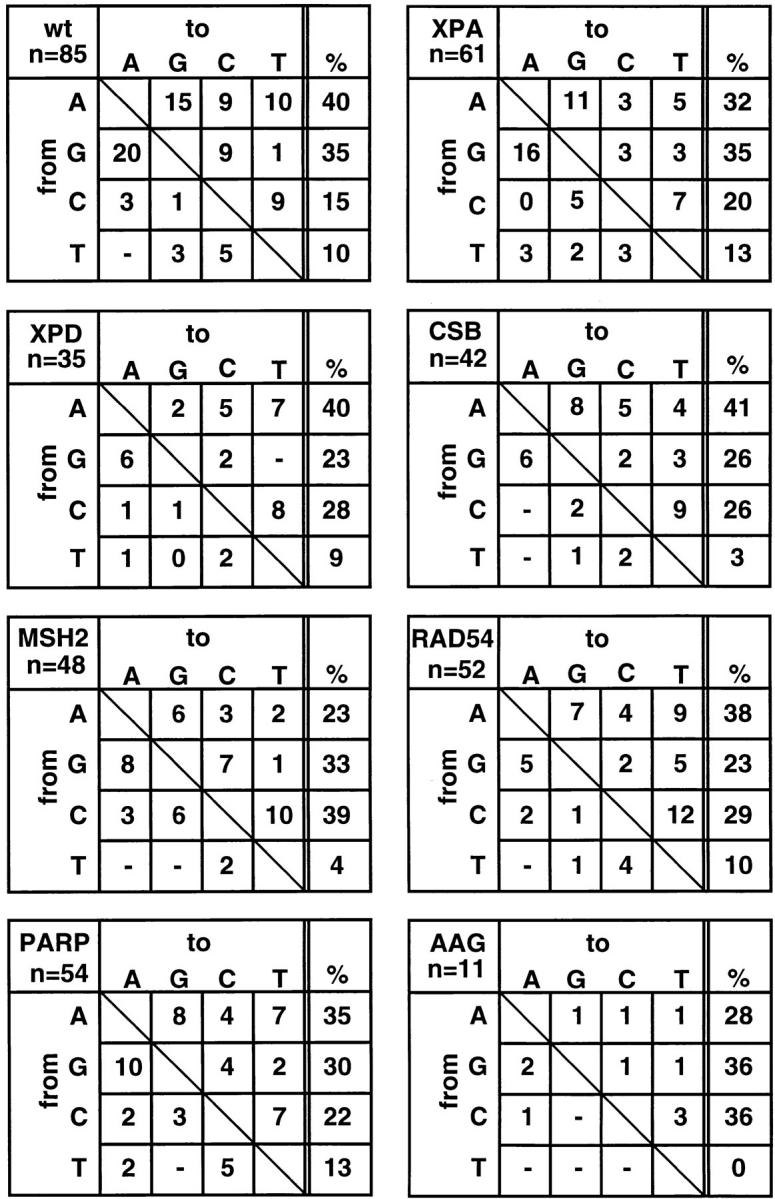
Base exchange patterns in the Vλ1 genes of memory B cells from XPA-, XPD TTDIBEL-, CSB-, MSH2-, RAD54-, PARP-, and AAG-mutant mice. Note that the typical strand bias of hypermutation, which is defined by a preferential mutation of A, G, and C rather than T bases (AGC > T) is evident in all mice. n, the number of mutations found.
Table 1.
Mutation Analysis of V21 Genes from Memory B Cells from XPA-, XPDTTDIBEL-, CSB-, MSH2-, RAD54-, PARP-, and AAG-mutant Mice
| Genotype | Deficiency | Remark | Age | No. of mutated sequences/total no. sequences | Mutation Frequency | Mutation range | Mutation frequency in mutated cells | Transition/ transversions | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| wk | ||||||||||||||||
| Wild type | — | — | 12, 20 | 27/42 (64%) | 0.78% (85/10,962) | 1–11 | 1.2% (85/7,047) | 1.4 | ||||||||
| XPA | NER (GGR) | Complete | ||||||||||||||
| knockout | 30 | 16/30 (53%) | 0.78% (61/7,830) | 1–8 | 1.4% (61/4,176) | 1.5 | ||||||||||
| XPD | NER | XPD mutation of | ||||||||||||||
| transcription | a TTD patient | 10, 11 | 19/40 (48%) | 0.33% (35/10,440) | 1–3 | 0.7% (35/4,959) | 1.1 | |||||||||
| CSB | NER (TCR) | K337 stop* | 30 | 16/20 (80%) | 0.80% (42/5,220) | 1–5 | 1.0% (42/4,176) | 1.5 | ||||||||
| MSH2 | MMR | Complete | ||||||||||||||
| knockout | 20 | 20/30 (66%) | 0.66% (52/7,830) | 1–6 | 1.0% (52/5,220) | 1.2 | ||||||||||
| RAD54 | DSBR | Complete | ||||||||||||||
| knockout | 10 | 13/22 (59%) | 0.91% (53/5,742) | 1–7 | 1.6% (53/3,393) | 1.2 | ||||||||||
| PARP | Nick | Complete | ||||||||||||||
| sensing | knockout | 12 | 16/33 (48%) | 0.65% (56/8,613) | 1–7 | 1.3% (56/4,176) | 1.3 | |||||||||
| AAG* | BER | Complete | ||||||||||||||
| knockout | 11 | 06/14 (43%) | 0.46% (17/3,654) | 1–5 | 1.0% (17/1,599) | 1.0‡ | ||||||||||
| Naive B cells | Internal | |||||||||||||||
| from all mice | PCR control in | |||||||||||||||
| each experiment | 3/130 (2%) | 0.03% (11/33,930) | 3–4 |
The CSB protein is not detectable.
A small data base.
The MSH2-deficient mouse strain allowed us to test the MMR-based model of somatic hypermutation (8). A temporal and/or local inhibition of MMR would increase the mutation rate. First, because of an increased frequency of recombination between identical and related sequences, and second, because of a defective repair of mismatches that occur during DNA replication or DNA repair. This model has an interesting evolutionary aspect. Both gene conversion and hypermutation, two processes known to diversify the Ig repertoire in chickens (conversion), sheep (hypermutation), and rabbits (conversion and hypermutation) (52–54) would be increased in the absence of MMR. In addition, a temporal and/or local lack of MMR could explain how a mismatch that has been introduced by the mutator can be maintained until segregation of the daughter cells. Alternatively, the mutator might “abuse” mismatch recognition proteins to fix the introduced mismatches by a temporal and/or local inhibition of subsequent MMR steps. However, in the absence of MSH2-mediated MMR throughout B cell development, the mutation frequency in memory B cells of MSH2-deficient mice was neither increased (as expected from the MMR inhibition model) nor decreased (as expected from the MMR abuse model), and the distribution of mutations along the rearranged locus were normal (Fig. 2 and Table 1). An excess of mutations at germline G·C rather than A·T pairs was found (Fig. 3). This is in accord with Phung et al., who suggested that MMR, although not required for the occurrence of somatic mutations, may influence its pattern (55). In another study, high levels of mutations were also found in variable genes from mice deficient for the mismatch repair protein PMS2. However, genes from PMS2-deficient mice showed a higher frequency of tandem mutations (Gearhart, P., personal communication). In MSH2-deficient memory B cells, the frequency of doublet mutations appears normal, i.e., one doublet mutation out of 54 mutations as compared with one triplet and two doublet mutations out of 85 mutations in wild-type memory B cells. The difference between the two mutants might relate to the fact that PMS2 lies downstream of not only MSH2 but also MSH3, which is known to bind preferentially to extrahelical loops. Considering a constitutively active mutator that is efficiently counteracted by MMR in naive B cells, a lack of MMR throughout B cell development might have caused hypermutation in naive B cells. However, seven out of seven naive B cells analyzed were unmutated (data not shown).
A possible role of RAD54 in somatic hypermutation was addressed in RAD54-mutant mice. The RAD54 mutants are deficient in DSBR through homologous recombination (23). One could imagine that pairing of homologous Ig alleles can take place in germinal center B cells. Based on the unique DNA sequence at the V(D)J recombination site, which lacks a homologous stretch of DNA on the other allele, pairing would be interrupted at this site. To resolve the Holiday junction(s), a resolvase would have to introduce nick(s) into the V(D)J region. A subsequent error-prone repair process, which might involve a nuclease activity or displacement synthesis, would introduce mutations in the recombinant strand. However, a lack of RAD54 did not influence hypermutation in memory B cells (Figs. 2 and 3, Table 1) and was absent in naive B cells (data not shown). Also V(D)J recombination and class switching were found to be normal in RAD54-mutant mice (23). Additional experiments are required to verify, whether other components of the S. cerevisiae RAD52 epistasis group (RAD50–57, RAD24, and x-ray sensitivity [XRS]2) or other recombinational repair genes are involved in the molecular mechanism underlying somatic hypermutation.
Hypermutation is characterized by nucleotide exchanges, which likely involve “nicks” of the DNA. Nicks exist as intermediates in DNA repair and DNA recombination, are intermediates in most hypermutation models, and were proposed to be actively introduced to initiate somatic hypermutation (12). To test whether the “nick-sensing activity” of PARP might be functionally involved in hypermutation, e.g., by recruiting the mutator, we analyzed hypermutation in PARP-deficient mice. Again, hypermutation occurred normally in memory B cells from PARP-deficient mice (Figs. 2 and 3, Table 1) and did not occur in the 13 naive B cells analyzed (data not shown).
In the course of this study, AAG-deficient mice became available and were included in the analysis, as AAG is one of the enzymes involved in BER, another mechanism of DNA repair that could potentially be involved in somatic hypermutation. Hypermutation appears to be unaffected in AAG-deficient memory B (Figs. 2 and 3, Table 1) and naive B cells (5/5 unmutated). Although our data base comprises only 11 mutations (six clonally related mutations were excluded), the base exchange patterns are within the expected range. In addition, hypermutation has been found to be normal in DNA polymerase β–deficient memory B cells (Texido, G., and K. Rajewsky, unpublished data). These data argue against a connection between BER and hypermutation.
In conclusion, single B220+, Igμ+, Igδ+, Vλ1+ naive B cells and B220+, Igμ−, Igδ−, Vλ1+ memory B cells were isolated ex vivo from XPA, XPD, CSB, MSH2, RAD54, PARP, and AAG mutant mice and used to amplify and sequence rearranged Igλ1 genes. The results indicate that NER does not control somatic hypermutation, strongly arguing against the gratuitous TCR-based models of somatic hypermutation. Furthermore MMR, as well as RAD54-dependent DSBR- and AAG-mediated BER are not required for the hypermutation process. Although not addressed in detail, the presence of Vλ1+, Igμ−, Igδ− memory B cells in all the mutant mice analyzed in addition indicates, that Ig class switching takes place in these animals.
Acknowledgments
The authors are grateful to R. Zoebelein and C. Göttlinger for technical help and Drs. J.H.J. Hoeijmakers, J. Bachl, K.S. Campbell, R. Jessberger, and R. Torres for comments and critical reading.
This work was supported by a European Molecular Biology Organization (EMBO) long-term postdoctoral fellowship to H. Jacobs, the Deutsche Forschungsgemeinschaft through Sonderforschungsbereich 243, the Land Nordrhein-Westfalen, by the Netherlands Organization for Scientific Research (PGN 901-01-093), a fellowship of the Royal Netherlands Academy of Arts and Sciences to G. Weeda, and grants from the Dutch Cancer Society (projects EUR 94-763 and 94-858).
Abbreviations used in this paper
- AAG
3-alkyladenine DNA-glycosylase or 3-methyladenine DNA-glycosylase
- AP
apurinic or apyrimidinic
- BER
base-excision repair
- CS
Cockayne syndrome
- DSBR
double-strand– break repair
- GGR
global genome repair
- MMR
mismatch repair
- MSH
mut S homologue
- NER
nucleotide-excision repair
- PARP
poly (ADP-ribose) polymerase
- PI
propidium iodide
- PMS
postmeiotic segregation
- RAD
radiation sensitivity
- TCR
transcription-coupled repair
- Thy1
CD90
- TTD
trichothiodystrophy
- XP
xeroderma pigmentosum complementation group
References
- 1.Rajewsky K. Clonal selection and learning in the antibody system. Nature. 1996;381:751–758. doi: 10.1038/381751a0. [DOI] [PubMed] [Google Scholar]
- 2.Neuberger MS, Milstein C. Somatic hypermutation. Curr Opin Immunol. 1995;7:248–254. doi: 10.1016/0952-7915(95)80010-7. [DOI] [PubMed] [Google Scholar]
- 3.McKean D, Huppi K, Bell M, Staudt L, Weigert M. Generation of antibody diversity in the immune response of BALB/c mice to influenza virus hemagglutinin. Proc Natl Acad Sci USA. 1984;81:3180–3184. doi: 10.1073/pnas.81.10.3180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Berek C, Milstein C. The dynamic nature of the antibody repertoire. Immunol Rev. 1988;105:5–26. doi: 10.1111/j.1600-065x.1988.tb00763.x. [DOI] [PubMed] [Google Scholar]
- 5.Allen D, Cumano A, Dildrop R, Kocks C, Rajewsky K, Rajewsky N, Roes J, Sablitzky F, Siekevitz M. Timing, genetic requirements and functional consequences of somatic hypermutation during B-cell development. Immunol Rev. 1987;96:5–22. doi: 10.1111/j.1600-065x.1987.tb00506.x. [DOI] [PubMed] [Google Scholar]
- 6.Storb U. The molecular basis of somatic hypermutation of immunoglobulin genes. Curr Opin Immunol. 1996;8:206–221. doi: 10.1016/s0952-7915(96)80059-8. [DOI] [PubMed] [Google Scholar]
- 7.Maizels N. Somatic hypermutation: how many mechanisms diversify V region sequences? . Cell. 1995;83:9–12. doi: 10.1016/0092-8674(95)90227-9. [DOI] [PubMed] [Google Scholar]
- 8.Reynaud C-A, Quint L, Bertocci B, Weill J-C. Introduction: what mechanism(s) drive hypermutation. Semin Immunol. 1996;8:125–129. doi: 10.1006/smim.1996.0016. [DOI] [PubMed] [Google Scholar]
- 9.Tumas-Brundage K, Vora KV, Giusti AM, Manser T. Characterization of cis-acting elements required for somatic hypermutation of murine antibody V genes using conventional transgenic and transgene homologous recombination approaches. Semin Immunol. 1996;8:141–150. doi: 10.1006/smim.1996.0018. [DOI] [PubMed] [Google Scholar]
- 10.Storb U, Peters A, Klotz E, Rogerson B, Hackett J., Jr The mechanism of somatic hypermutation studied with transgenic and transfected target genes. Semin Immunol. 1996;8:131–140. doi: 10.1006/smim.1996.0017. [DOI] [PubMed] [Google Scholar]
- 11.Peters A, Storb U. Somatic hypermutation of immunoglobulin genes is linked to transcription initiation. Immunity. 1996;4:57–65. doi: 10.1016/s1074-7613(00)80298-8. [DOI] [PubMed] [Google Scholar]
- 12.Brenner S, Milstein C. Origin of antibody variation. Nature. 1966;211:242–243. [PubMed] [Google Scholar]
- 13.Gearhart PJ. Generation of immunoglobulin variable region gene diversity. Immunol Today. 1982;3:107–112. doi: 10.1016/S0167-5699(82)80026-1. [DOI] [PubMed] [Google Scholar]
- 14.Maizels N. Might gene conversion be the mechanism of somatic hypermutation of mammalian Ig genes? . Trends Genet. 1989;5:4–8. doi: 10.1016/0168-9525(89)90004-8. [DOI] [PubMed] [Google Scholar]
- 15.Manser T. The efficiency of antibody maturation: can the rate of B cell division be limiting? . Immunol Today. 1990;11:305–308. doi: 10.1016/0167-5699(90)90124-r. [DOI] [PubMed] [Google Scholar]
- 16.Steele E, Pollard J. Hypothesis: somatic hypermutation via the error prone DNA-to-RNA-to-DNA information loop. Mol Immunol. 1987;24:667–673. doi: 10.1016/0161-5890(87)90049-6. [DOI] [PubMed] [Google Scholar]
- 17.Rogerson B, Hackett J, Peters A, Haasch D, Storb U. Mutation pattern of immunoglobulin transgenes is compatible with a model of somatic mutation in which targeting of the mutator is linked to the direction of DNA replication. EMBO (Eur Mol Biol Organ) J. 1991;10:4331–4341. doi: 10.1002/j.1460-2075.1991.tb05011.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cleaver JE. It was a very good year for DNA repair. Cell. 1994;76:1–4. doi: 10.1016/0092-8674(94)90165-1. [DOI] [PubMed] [Google Scholar]
- 19.Gellert M. A new view of V(D)J recombination. Genes Cells. 1996;1:269–275. doi: 10.1046/j.1365-2443.1996.22023.x. [DOI] [PubMed] [Google Scholar]
- 20.de Vries A, van Oostrom MTM, Hofhuis FMA, Dortant PM, Berg RJW, de Gruijl FR, Wester PW, van Kreijl CF, Capel PJA, van Steeg H, Verbeek SJ. Increased susceptibility to ultraviolet-B and carcinogens of mice lacking the DNA excision repair gene XPA. Nature. 1995;377:169–173. doi: 10.1038/377169a0. [DOI] [PubMed] [Google Scholar]
- 20a.De Boer, J., J. de Wit, H. van Steeg, R.J.W. Berg, H. Morreau, P. Visser, M. Duran, C.F. van Kreijl, F.R. de Gruijl, J.H.J. Hoeijmakers, and G. Weeda. 1998. A mouse model for the basal transcription/DNA repair syndrome trichothiodystrophy. Mol. Cell. In press. [DOI] [PubMed]
- 21.van der Horst GTJ, van Steeg H, Berg RJW, van Gool AJ, de Wit J, Weeda G, Morreau H, Beems RB, van Kreijl CF, de Gruijl FR, et al. Defective transcription-coupled repair in Cockayne syndrome B mice is associated with skin cancer predisposition. Cell. 1997;89:425–435. doi: 10.1016/s0092-8674(00)80223-8. [DOI] [PubMed] [Google Scholar]
- 22.de Wind N, Dekker M, Berns A, Radman M, te Riele H. Inactivation of the mouse Msh2 gene results in mismatch repair deficiency, methylation tolerance, hyperrecombination, and predisposition to cancer. Cell. 1995;82:321–330. doi: 10.1016/0092-8674(95)90319-4. [DOI] [PubMed] [Google Scholar]
- 23.Essers J, Hendriks RW, Swagemakers SMA, Troelstra C, de Wit J, Bootsma D, Hoeijmakers JHJ, Kanaar R. Disruption of mouse RAD54 reduces ionizing radiation resistance and homologous recombination. Cell. 1997;89:195–204. doi: 10.1016/s0092-8674(00)80199-3. [DOI] [PubMed] [Google Scholar]
- 24.Ménissier de Murcia J, Niedergang C, Trucco C, Ricoul M, Dutrillaux B, Mark M, Javier F, Oliver, Masson M, Dierich A, Le Meur M, et al. Requirement of poly(ADP-ribose) polymerase in recovery from DNA damage in mice and in cells. Proc Natl Acad Sci USA. 1997;94:7303–7307. doi: 10.1073/pnas.94.14.7303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24a.Engelward BP, Dreslin A, Christensen J, Huszar D, Kurahara C, Samson L. Repair deficient 3-methyladenine DNA glycosylase homozygous mutant mouse cells have increased sensitivity to alkylation induced chromosome damage and cell killing. EMBO (Eur Mol Biol Organ) J. 1996;15:945–952. [PMC free article] [PubMed] [Google Scholar]
- 25.Friedberg, E.C., G.C. Walker, and W. Siede. 1995. DNA Repair and Mutagenesis. American Society for Microbiology Press, Washington. 698 pp.
- 26.Park C-H, Mu D, Reardon JT, Sancar A. The general transcription-repair factor TFIIH is recruited to the excision repair complex by the XPA protein independent of the TFIIE transcription factor. J Biol Chem. 1995;270:4896–4902. doi: 10.1074/jbc.270.9.4896. [DOI] [PubMed] [Google Scholar]
- 27.Nakane H, Takeuchi S, Yuba S, Saijo M, Nakatsu Y, Murai H, Nakatsuru Y, Ishikawa T, Hirota S, Kitamura Y, et al. High incidence of ultraviolet-B– or chemical-carcinogen–induced skin tumours in mice lacking the xeroderma pigmentosum group A gene. Nature. 1995;377:165–168. doi: 10.1038/377165a0. [DOI] [PubMed] [Google Scholar]
- 28.Weeda G, Eveno E, Donker I, Vermeulen W, Chevallier-Lagente O, Taieb A, Stary A, Hoeijmakers JH, Mezzina M, Sarasin A. A mutation in the XPB/ERCC3 DNA repair transcription gene, associated with trichothiodystrophy. Am J Hum Genet. 1997;60:320–329. [PMC free article] [PubMed] [Google Scholar]
- 29.Vermeulen W, van Vuuren AJ, Chipoulet M, Schaeffer L, Appeldoorn E, Weeda G, Jaspers NGJ, Priestley A, Arlett CF, Lehmann AR, et al. Cold Spring Harbor Symp Quant Biol. 1994;59:317–329. doi: 10.1101/sqb.1994.059.01.036. [DOI] [PubMed] [Google Scholar]
- 30.Troelstra C, van Gool A, de Wit J, Vermeulen W, Bootsma D, Hoeijmakers JH. ERCC6, a member of a subfamily of putative helicases, is involved in Cockayne's syndrome and preferential repair of active genes. Cell. 1992;71:939–953. doi: 10.1016/0092-8674(92)90390-x. [DOI] [PubMed] [Google Scholar]
- 31.Drummond JT, Li GM, Longley MJ, Modrich P. Isolation of an hMSH2-p160 heterodimer that restores DNA mismatch repair to tumor cells. Science. 1995;268:1909–1912. doi: 10.1126/science.7604264. [DOI] [PubMed] [Google Scholar]
- 32.Habraken Y, Sung P, Prakash L, Prakash S. Binding of insertion/deletion DNA mismatches by the heterodimer of yeast mismatch repair proteins MSH2 and MSH3. Curr Biol. 1996;6:1185–1187. doi: 10.1016/s0960-9822(02)70686-6. [DOI] [PubMed] [Google Scholar]
- 33.Acharya S, Wilson T, Gradia S, Kane MF, Guerrette S, Marsischky GT, Kolodner R, Fishel R. hMSH2 forms specific mispair-binding complexes with hMSH3 and hMSH6. Proc Natl Acad Sci USA. 1996;93:13629–13634. doi: 10.1073/pnas.93.24.13629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jiang H, Xie Y, Houston P, Stemke-Hale K, Mortenson UH, Rothstein R, Kodadek T. Direct association between the yeast Rad51 and Rad54 recombination proteins. J Biol Chem. 1996;271:33181–33186. doi: 10.1074/jbc.271.52.33181. [DOI] [PubMed] [Google Scholar]
- 35.Sobol RW, Horton JK, Kühn R, Gu H, Singhal RK, Prasad R, Rajewsky K, Wilson SH. Requirement of mammalian DNA polymerase-beta in base-excision repair. Nature. 1996;379:183–186. doi: 10.1038/379183a0. [DOI] [PubMed] [Google Scholar]
- 36.Frosina G, Fortini P, Rossi O, Carrozino F, Raspaglio G, Cox LS, Lane DP, Abbondanolo A, Dogliotti E. Two pathways for base excision repair in mammalian cells. J Biol Chem. 1996;271:9573–9578. doi: 10.1074/jbc.271.16.9573. [DOI] [PubMed] [Google Scholar]
- 37.Texidó G, Jacobs H, Meiering M, Kühn R, Roes J, Müller W, Gilfillan S, Fujiwara H, Kikutani H, Yoshida N. Somatic hypermutation occurs in B cells of terminal deoxynucleotidyl transferase–, CD23–, interleukin-4–, IgD– and CD30–deficient mouse mutants. Eur J Immunol. 1996;26:1966–1969. doi: 10.1002/eji.1830260843. [DOI] [PubMed] [Google Scholar]
- 38.Reth M, Imanishi-Kari T, Rajewsky K. Analysis of the repertoire of anti-(4-hydroxy-3-nitrophenyl)acetyl (NP) antibodies in C 57 BL/6 mice by cell fusion. II. Characterization of idiotypes by monoclonal anti-idiotope antibodies. Eur J Immunol. 1979;9:1004–1013. doi: 10.1002/eji.1830091216. [DOI] [PubMed] [Google Scholar]
- 39.Xu B, Selsing E. Analysis of sequence transfers resembling gene conversion in a mouse antibody transgene. Science. 1994;265:1590–1593. doi: 10.1126/science.8079173. [DOI] [PubMed] [Google Scholar]
- 40.Ford JE, Lieber MR. Analysis of individual immunoglobulin l light chain genes amplified from single cells is inconsistent with the variable region gene conversion in germinal-center B cell somatic mutation. Eur J Immunol. 1994;24:1816–1822. doi: 10.1002/eji.1830240814. [DOI] [PubMed] [Google Scholar]
- 41.Hanawalt PC. Transcription-coupled repair and human disease. Science. 1994;266:1957–1958. doi: 10.1126/science.7801121. [DOI] [PubMed] [Google Scholar]
- 42.Wang G, Seidman MM, Glazer PM. Mutagenesis in mammalian cells induced by triple helix formation and transcription-coupled repair. Science. 1996;271:802–803. doi: 10.1126/science.271.5250.802. [DOI] [PubMed] [Google Scholar]
- 43.Lui Y-J, Johnson GD, Gordon J, MacLennan ICM. Germinal centres in T-cell-dependent responses. Immunol Today. 1992;13:17–21. doi: 10.1016/0167-5699(92)90199-H. [DOI] [PubMed] [Google Scholar]
- 44.Close PM, Pringle JH, Ruprai AK, West KP, Lauder I. Zonal distribution of immunoglobulin-producing cells within the germinal centre: an in situ hybridization and immunohistochemical study. J Pathol. 1990;162:209–216. doi: 10.1002/path.1711620306. [DOI] [PubMed] [Google Scholar]
- 45.Wagner SD, Elvin JG, Norris P, McGregor JM, Neuberger MS. Somatic hypermutation of Ig genes in patients with xeroderma pigmentosum (XP-D) Int Immunol. 1996;8:701–705. doi: 10.1093/intimm/8.5.701. [DOI] [PubMed] [Google Scholar]
- 46.Kim N, Kage K, Matsuda F, Lefranc M-P, Storb U. B lymphocytes of xeroderma pigmentosum or Cockayne syndrome patients with inherited defects in nucleotide excision repair are fully capable of somatic hypermutation of immunoglobulin genes. J Exp Med. 1997;186:413–419. doi: 10.1084/jem.186.3.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Betz A, Milstein C, Gonzáles-Fernández A, Pannell R, Larson T, Neuberger M. Elements regulating somatic hypermutation of an immunoglobulin k gene: critical role for the intron enhancer/matrix attachment region. Cell. 1994;77:239–248. doi: 10.1016/0092-8674(94)90316-6. [DOI] [PubMed] [Google Scholar]
- 48.Gearhart, P.J., and N.S. Levy. 1991. Kinetics and molecular model for somatic mutation in immunoglobulin variable genes. In Somatic Hypermutation in V-Regions. E.J. Steele, editor. CRC Press, Boca Raton. 1:29–47.
- 49.Both G, Taylor L, Pollard J, Steele E. Distribution of mutations around rearranged heavy chain antibody variable-region genes. Mol Cell Biol. 1990;10:5187–5196. doi: 10.1128/mcb.10.10.5187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rada C, Gonzales-Fernandez A, Jarvis JM, Milstein C. The 5′ boundary of somatic hypermutation in a Vk gene is in the leader intron. Eur J Immunol. 1994;24:1453–1457. doi: 10.1002/eji.1830240632. [DOI] [PubMed] [Google Scholar]
- 51.Rogerson B. Mapping the upstream boundary of somatic mutations in rearranged immunoglobulin transgenes and endogenous genes. Mol Immunol. 1994;31:83–98. doi: 10.1016/0161-5890(94)90081-7. [DOI] [PubMed] [Google Scholar]
- 52.Reynaud C, Anquez V, Grimal H, Weill J-C. A hyperconversion mechanism generates the chicken light chain preimmune repertoire. Cell. 1987;48:379–388. doi: 10.1016/0092-8674(87)90189-9. [DOI] [PubMed] [Google Scholar]
- 53.Weinstein PD, Anderson AO, Mage RG. Rabbit IgH sequences in appendix germinal centers: VH diversification by gene conversion-like and hypermutation mechanisms. Immunity. 1994;1:647–659. doi: 10.1016/1074-7613(94)90036-1. [DOI] [PubMed] [Google Scholar]
- 54.Becker R, Knight K. Somatic diversification of Ig heavy chain VDJ genes: evidence for somatic gene conversion in rabbits. Cell. 1990;63:987–997. doi: 10.1016/0092-8674(90)90502-6. [DOI] [PubMed] [Google Scholar]
- 55.Phung QH, Winter DB, Cranston A, Tarone RE, Bohr VA, Fishel R, Gearhart PJ. Increased hypermutation at G and C nucleotides in immunoglobulin variable genes from mice deficient for the MSH2 mismatch repair protein. J Exp Med. 1998;187:1745–1751. doi: 10.1084/jem.187.11.1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kabat EA, Wu TT. Identical V region amino acid sequences and segments of sequences in antibodies of different specificities. Relative contribution of VH and VL genes, minigenes, and complementarity determining regions to binding of anti body binding sites. J Immunol. 1991;147:1709–1719. [PubMed] [Google Scholar]
- 57.González-Fernández A, Gupta SK, Pannell R, Neuberger MS, Milstein C. Somatic mutation of immunoglobulin λ chains: a segment of the major intron hypermutates as much as the complementarity-determining region. Proc Natl Acad Sci USA. 1994;91:12614–12618. doi: 10.1073/pnas.91.26.12614. [DOI] [PMC free article] [PubMed] [Google Scholar]